

Abstract
Transcription polymerases can exhibit an unusual mode of regenerating certain RNA templates from RNA, yielding systems that can replicate and evolve with RNA as information carrier. Two classes of pathogenic RNAs (Hepatitis delta virus in animals and viroids in plants) are copied by host transcription polymerases. Using in vitro RNA replication by the transcription polymerase of T7 bacteriophage as an experimental model, we identify hundreds of new replicating RNAs, define three mechanistic hallmarks of replication (subterminal de novo initiation, RNA shape-shifting and interrupted rolling circle synthesis) and describe emergence from DNA seeds as a mechanism for the origin of novel RNA replicons. These results inform models for the origins and replication of naturally occurring RNA genetic elements and suggest a means by which diverse RNA populations could be propagated as hereditary material in cellular contexts.
Graphical Abstract
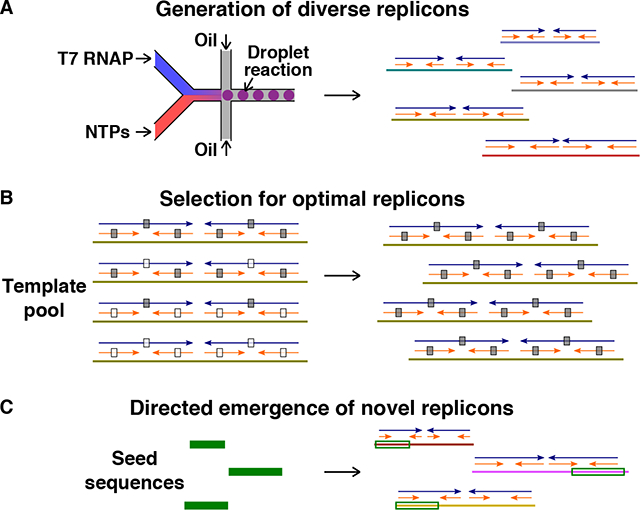
Origins of RNAs replicated by a DNA-dependent RNA polymerase (T7 RNAP). (A) A diversity of RNA replicons was obtained from no-template-added T7 RNAP reactions set up as aqueous droplets in oil using microfluidics. Replicons consistently exhibited a structural framework of 2-way repeats (blue arrows) and 4-way repeats (orange arrows). NTPs = nucleoside triphosphates. (B) Replicons with paired-2-way- and paired-4-way- repeats amplified more efficiently and outcompeted other RNAs (gray boxes: paired nucleotides, white boxes: unpaired nucleotides). (C) Novel replicons can originate from DNA seeds via DNA-templated transcription, 2-way- and 4-way- repeat generation, and Darwinian selection.
Introduction
Although genetic information is commonly encoded in DNA and transmitted via DNA-templated DNA replication, RNA can also be transferred as hereditary material via RNA-templated RNA replication. Two classes of protein-catalyzed RNA replication systems have been described. In the first, specialized RNA-dependent RNA polymerases replicate the genomes of RNA viruses such as influenza and dengue. In the second, cellular enzymes normally involved in the transcription of DNA to RNA can copy certain RNAs such as plant viroids and human Hepatitis delta virus. The diversity of RNAs using DNA-dependent RNA polymerases for replication and the underlying molecular mechanisms have not been fully explored.
Rationale
Five RNA sequences that can be replicated in vitro by bacteriophage T7 DNA-dependent RNA polymerase (T7 RNAP) have previously been described. The origins of these replicating RNAs and requirements for replication by T7 RNAP are unclear. We applied next-generation sequencing, microfluidics and bioinformatics to address (i) how a DNA-dependent RNA polymerase can replicate RNA, (ii) which RNA templates are efficiently replicated, and (iii) how replicating RNAs originate.
Results
We set up a series of T7 RNAP reactions with no explicitly added templates. These reactions yielded RNA replicons with different sequences but a consistent structural framework defined by a 2-way repeat (a long inverted repeat throughout the RNA length) and a 4-way repeat (a shorter inverted repeat embedded within each arm of the 2-way repeat). We showed that 2-way- and 4-way- repeats are required for efficient RNA replication by T7 RNAP, suggesting that RNAs with 2-way- and 4-way- repeats arise in no-template-added reactions by “survival of the fittest” in the test tube. The requirement of 2-way repeats further implies that RNAs form a long hairpin structure whereas the 4-way repeat requirement suggests that RNAs change structure during replication— what we refer to as RNA shape-shifting. From experiments with chemically synthesized RNA templates, we also identified a non-templating 3’-nucleotide extension as a critical requirement for the initiation of replication. This requirement suggests that initiation of new RNA products occurs internally within the template sequence, which we call subterminal de novo initiation. We additionally found that RNA templates with defined 5’ and 3’ ends can be used processively for multiple rounds of RNA synthesis. Such a mechanism, which we term interrupted rolling circle synthesis, yields RNA products consisting of multiple repeats of template sequence.
RNAs synthesized in no-template-added T7 RNAP reactions could have been replication products of pre-existing sequences or products of a de novo process. To distinguish between these two possibilities, we isolated hundreds of RNA replicons by scaling up our experimental throughput using microfluidics. Analysis of our large replicon repertoire led us to hypothesize that replicating RNAs can originate de novo through partial instruction from DNA seeds. In support of this hypothesis, we demonstrated the formation of novel replicating RNAs from a DNA seed pool of our own choosing. We observed that RNA replicons consisted of DNA seed sequence information that had been duplicated and reduplicated to yield the characteristic 4-way repeat pattern, suggesting a specific cascade of steps of polymerase activity and molecular evolution leading to the origin of replicating RNAs.
Conclusions
Our results inform models for the origins and replication of naturally occurring RNA genetic elements and suggest a means by which diverse RNA populations could serve as hereditary material in cellular contexts.
Transcription polymerases (DNA-dependent RNA polymerases) mediate information transfer from DNA to RNA across the tree of life. In addition to their expected activity, the linear amplification of RNA from DNA templates, transcription polymerases can exponentially replicate certain RNA templates; this has been demonstrated in vitro for transcription polymerases from Escherichia coli (1, 2) and bacteriophage T7 (3–6). By RNA replication, we mean a template-regenerating process that includes full-length copying of an RNA template, with the resultant RNA copy then serving as template for new synthesis of full-length RNA copies. Importantly, such an RNA replication process does not involve DNA.
Historically, the transcription polymerase of T7 bacteriophage (T7 RNAP) has served as a model enzyme for its DNA-dependent RNA polymerase activity (7). T7 RNAP also provides a paradigm for investigating RNA replication by transcription polymerases at the molecular level (3–5). Of note, a chloroplastic transcription polymerase similar to T7 RNAP may be the enzyme that replicates avocado sunblotch viroid (ASBVd) (8). To date, five distinct RNA sequences that can be replicated by T7 RNAP have been described (4, 5), all of them capable of forming long-hairpin secondary structures. The origins of the RNAs replicated by T7 RNAP are unclear. Konarska and Sharp (3, 4) speculated that replicating RNA templates may have been pre-existing RNA contaminants in T7 RNAP preparations, whereas Biebricher and Luce (5) proposed that replicating RNAs arise as a result of molecular evolution in T7 RNAP reactions.
By combining next-generation sequencing, microfluidics and bioinformatics with classical biochemistry approaches, we set out to address three questions: (i) how does a DNA-dependent RNA polymerase replicate RNA? (ii) how diverse is the family of RNAs that can be replicated by a transcription polymerase? and (iii) what are the origins of RNAs replicated by a transcription polymerase?.
Emergence of diverse but structurally similar replicating RNAs from no-template-added reactions
We set up a series of T7 RNAP reactions in parallel using aliquots of the same reagents (fig. S1A). Each reaction contained a high concentration (2 uM) of T7 RNAP. No nucleic acid template was added to the reactions, with the reaction composition (3) otherwise typical for T7 RNAP. After incubation at 37°C for ~24 hours, each reaction contained large amounts of synthesized RNA. The relative gel migration of synthesized RNA products varied from reaction-to-reaction (fig. S1B), indicating the presence of distinct RNAs in each reaction. These data are consistent with the findings of Biebricher and Luce (5).
We next analyzed the synthesized sequences for a set of 24 no-template-added T7 RNAP reactions conducted in parallel. Dominant reaction products were sequenced using an RNA-Seq protocol that we optimized for efficient reverse transcription of structured RNAs (fig. S2). Upon unsupervised sequence classification of the reaction products, we observed that each reaction yielded one or more clusters of RNA sequences. Each such cluster—henceforth referred to as an RNA species—was itself a heterogenous population of closely related sequences. For each RNA species, we chose a canonical, abundant sequence that could serve as a “reference” for the information content of the RNA species.
A small number (1 to 3) of RNA species was predominant in each of the 24 sequenced pools (Fig. 1A, table S1; predominant defined here as relative abundance >5% within a sequenced pool). Reference sequences for the predominant RNA species differed from each other (Fig. 1B), although some reactions (e.g., reactions 11 and 22) yielded reference sequences that were related. Furthermore, three of the reference sequences (12.1, 14.1 and 24.1) were related to Y RNA, which was previously characterized as an RNA replicated by T7 RNAP (4).
Fig. 1. Diverse but structurally similar RNAs can be isolated from no-template-added T7 RNAP reactions.
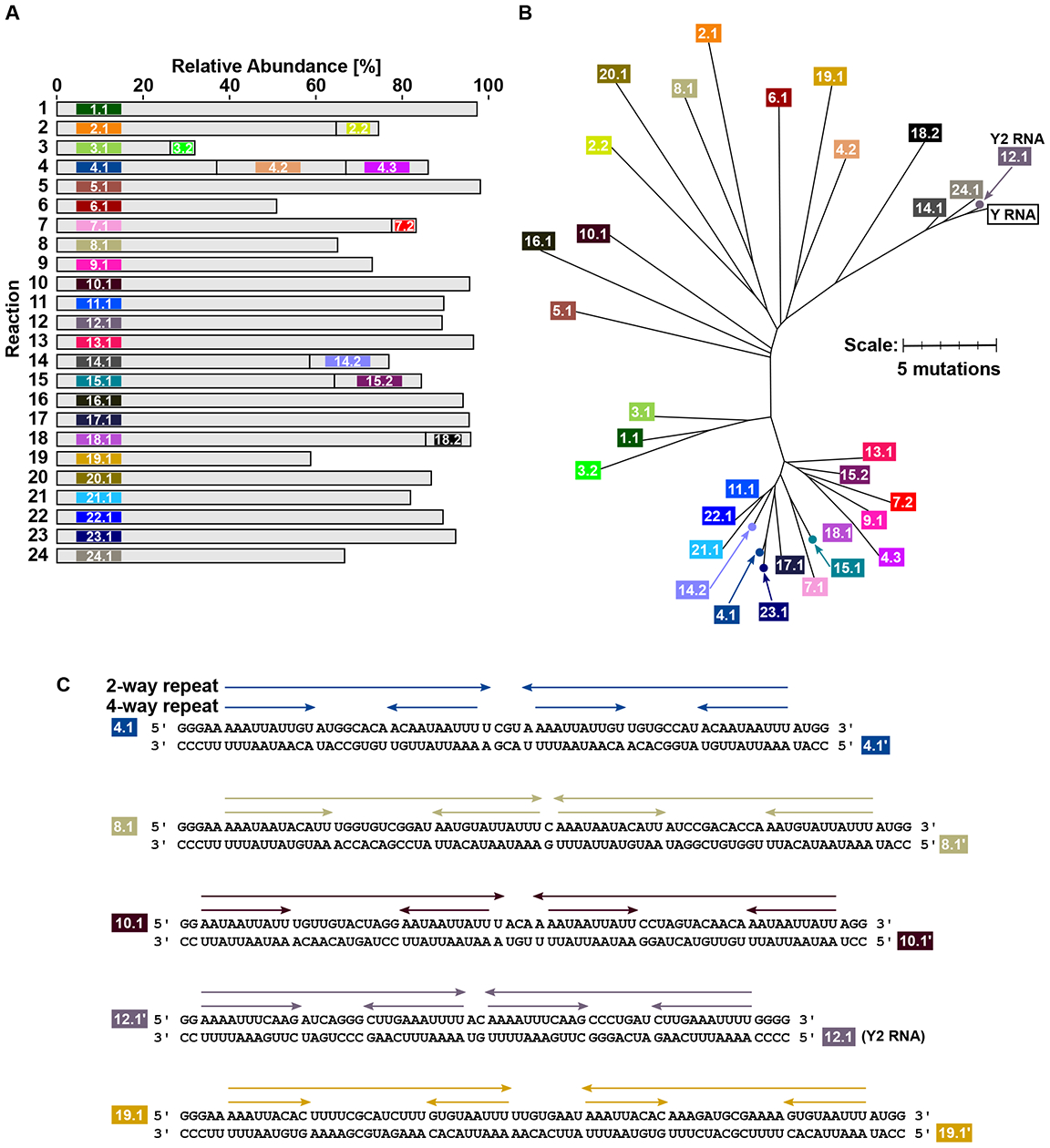
(A) Twenty-four no-template-added reactions with high concentration of T7 RNAP were set up in parallel, followed by high-throughput sequencing of the RNAs synthesized in each reaction. Sequenced pools were dominated by one to three RNA species. We use the term “RNA species” to refer to a heterogenous population of closely related RNA sequences. RNA species constituting >5% of any sequenced pool are shown. Colors for RNA species were chosen to qualitatively depict sequence relatedness between species [see panel (B)]. Each RNA species was assigned a numeric label based on its source reaction. (B) Differences between sequences of RNA species from (A). Relatedness of species to Y RNA, a previously characterized sequence that can be replicated by T7 RNAP (4), is also shown. We refer to species 12.1 as Y2 RNA elsewhere in the text because of its sequence similarity to Y RNA. (C) Structural similarity of RNA species from (A), as exemplified by the five species shown. Arrows denote 2-way- and 4-way- repeats. Arrows of the same length pointing in opposite directions denote complementary repeats. Prime (‘) = reverse complement. Panels (A)-(C) follow the same color coding for RNA species.
Most RNA reference sequences were between 60 to 80 nucleotides in length (fig. S1C), consistent with the migration patterns observed on denaturing gels. As our RNA-Seq protocol is strand-specific (e.g., see sequencing of chemically synthesized RNA oligos in fig. S7), we further analyzed the strand orientations of RNA sequences within each RNA species. Most RNA species showed comparable counts of (i) reads with the same strand orientation as the species reference sequence and (ii) reads with a strand orientation complementary to the species reference sequence (fig. S1D). Of note, RNA replication would be expected to yield sequences with both strand orientations.
Although distinct in sequence content, the RNA species shared structural features: (i) A “2-way repeat” configuration characterized by an inverted repeat throughout the RNA length, suggesting possible formation of a long hairpin structure, and (ii) A “4-way repeat” configuration entailing a shorter inverted repeat embedded within each arm of the 2-way repeat. Fig. 1C shows representative examples, with quantification of repeats across all RNA species shown in fig. S1E. The 2-way- and 4-way- repeat configurations were also noted for the previously described RNAs that can be replicated by T7 RNAP (4, 5). The capability of no-template-added T7 RNAP reactions to yield novel RNA sequences with the 2-way- and 4-way- repeat patterns was observed in experiments conducted independently at Stanford University and at the University of Texas Medical Branch, Galveston.
To determine whether the RNA species from no-template-added reactions can be sustainably replicated by T7 RNAP, we assessed growth of several distinct RNA species in parallel upon dilution into fresh T7 RNAP reactions. A clear sequence correspondence was evident between the RNA species used as spike-in templates in the reactions and the resulting products (fig. S3), suggesting that the RNAs were replicating. Importantly, to test templated RNA replication in this experiment (and also in the ensuing work), we used a low reaction concentration of T7 RNAP and checked that no-template-added controls conducted in parallel at the low T7 RNAP concentration did not yield any products detectable by gel electrophoresis. In concordance with previous reports [e.g. (5)], we note that T7 RNAP reaction concentration provides a means to experimentally distinguish between (i) RNA replication starting from a defined RNA template, which we assayed at low T7 RNAP concentration, and (ii) an enzymatic capability to synthesize replicating RNAs unique to a reaction without added template, which we assayed at high T7 RNAP concentration.
3’ end sequence requirements for RNA replication
Although regeneration of RNA species upon dilution into fresh T7 RNAP reactions suggested an ongoing templated synthesis process, it remained possible that the RNA species we were analyzing were not themselves templates but rather byproducts of more complex reactions. To establish replication from defined RNA templates, we probed a series of chemically synthesized RNAs for replication by T7 RNAP. In describing the templates tested, we use the nomenclature of Konarska and Sharp (4), who referred to the complementary strands of replicating RNAs as the G strand and C strand. The G strand sequence has two G nucleotides at the 5’ end and two G nucleotides at the 3’ end, while the C strand sequence has two C nucleotides at the 5’ end and two C nucleotides at the 3’ end. We initially tested replication of chemically synthesized G and C strand sequences for the RNA species 12.1 from Fig. 1. Henceforth, we will refer to this RNA species as Y2 RNA because of its sequence similarity to Y RNA, an RNA replicated by T7 RNAP that was previously characterized by Konarska and Sharp (4). Sequences of the Y2 RNA G and C strands are shown in fig. S6. Synthetic Y2 RNA G and C strands failed to efficiently instruct RNA synthesis. Mixing the two strands, to assess template activity of the RNA duplex between the G and C strands, did not increase RNA synthesis.
In considering possible features that may define active templates, we initially focused our attention on 3’ end sequences. Compared to the previously proposed replicating RNA 3’ end sequences (…GG-3’ for one strand, …CC-3’ for complementary strand) (4, 5), the Y2 RNA species we had isolated contained a diversity of 3’ sequence additions from one to a few nucleotides in length. More generally, 3’ nucleotide additions [a known feature of T7 RNAP activity, e.g. (9, 10)] were frequent in the RNA species we had isolated from no-template-added, high concentration T7 RNAP reactions (fig. S4). To mimic the 3’ nucleotide additions, we added an extra nucleotide to the 3’ ends of the Y2 RNA G and C strands. Upon adding an extra nucleotide to the 3’ end either enzymatically (Fig. 2A, quantification of gel intensities in fig. S5) or chemically (fig. S6A), the amounts of T7 RNAP reaction products increased dramatically. These results demonstrate a requirement of 3’ nucleotide additions to G and C strand sequences for efficient RNA synthesis.
Fig. 2. 3’ nucleotide additions to G and C strand templates are required for efficient RNA synthesis by T7 RNAP.
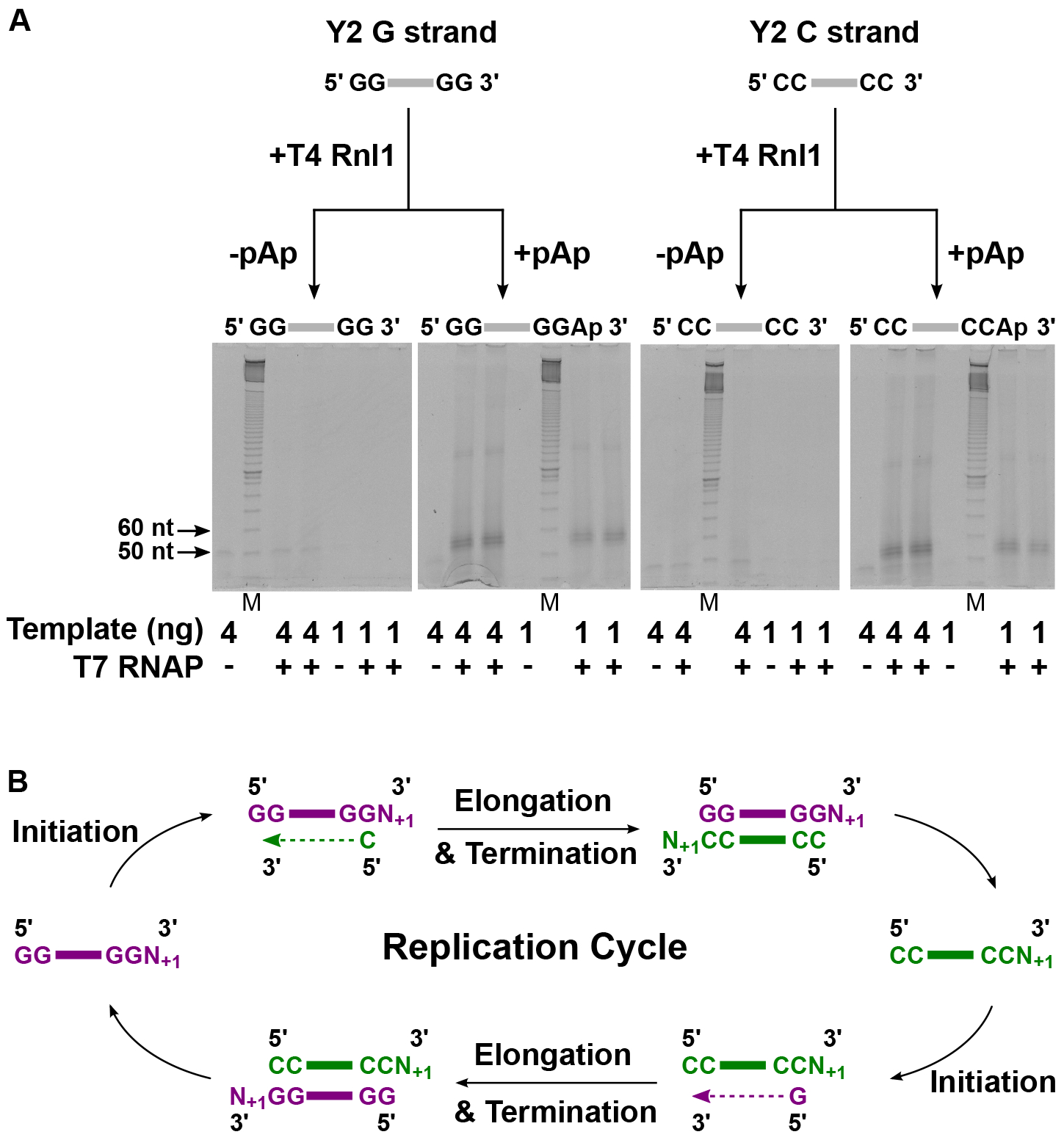
(A) Gel-based assay showing increased T7 RNAP reaction products after T4 RNA ligase 1 (T4Rnl1)-catalyzed addition of pAp (adenosine 3’,5’-diphosphate) to the Y2 RNA G and C strands. M = marker (denatured 10 base-pair DNA ladder), nt = nucleotides, ng = nanograms. All gels were processed in parallel. (B) Increased RNA synthesis by T7 RNAP upon addition of 3’ nucleotide extensions to templates and the lack of copying of the 3’ nucleotide extensions during RNA product generation (figs. S6C and S7), suggest a subterminal de novo initiation model. In this model, T7 RNAP de novo initiates at a template position upstream of the 3’ nucleotide extension, and regenerates template molecules by adding 3’ nucleotide extensions to RNA products. N+1 denotes one or a few extra nucleotides at the 3’ end. Purple = G strand with 3’ nucleotide extension, Green = C strand with 3’ nucleotide extension.
To characterize T7 RNAP reaction products generated from chemically synthesized RNA templates with 3’ nucleotide extensions, we compared template and product pools by RNA-Seq. The product sequences corresponded to the input template sequences, consistent with templated RNA replication. Template pools contained, as expected, sequences of a single strand orientation with a specific 3’ nucleotide extension (fig. S7). In contrast, product pools contained sequences of both strand orientations with a diversity of 3’ nucleotide extensions (fig. S7). The generated 3’ end sequence diversity in the product pools provides evidence for 3’ nucleotide additions by T7 RNAP during RNA-templated RNA synthesis. Furthermore, sequence comparison of template and product pools enables a direct molecular readout of the RNA replication cycle involving both strands. For example, starting from a particular chemically synthesized RNA template of G strand orientation, we observed synthesis of complementary C strand products, followed by these products serving as templates for synthesis of G strand products containing diverse 3’ nucleotide extensions that were not present in the starting template pool (fig. S7A). Similar results were obtained when reactions were initiated using a chemically synthesized RNA template of C strand orientation (fig. S7B). Therefore, defined G and C strand sequences with 3’ nucleotide extensions serve as templates for RNA replication (fig. S7C).
Our results, in particular the lack of copying of the added 3’ nucleotide (Supplementary Text), support a “subterminal de novo initiation” model for RNA replication by T7 RNAP (Fig. 2B). In this model, T7 RNAP de novo initiates upstream of the 3’ nucleotide extension rather than at the 3’ end. After 5’ → 3’ copying of the RNA template, T7 RNAP adds a 3’ nucleotide extension to the RNA product. In effect, the 3’ nucleotide addition confers the appropriate 3’ end for the RNA product to subsequently serve as an efficient template, while maintaining the chain length of the replicating RNA species.
Replicating RNAs as sequence ensembles
To investigate the sequence heterogeneity observed within RNA species upon RNA-Seq of no-template-added T7 RNAP reactions, we examined sequence variants present in replicating RNA populations with respect to reference G and C strands. We found that sequence variants on the two RNA strands were complementary and that complementary variants occurred at similar frequencies (fig. S8). As an example of such complementarity, for the RNA species shown in fig. S8A, G → A variation on the G strand at position 44 (from the 5’ end) was observed at a frequency of ~1.1%, while C → U variation on the complementary C strand at position 21 (from the 5’ end; this is position 44 from the C strand 3’ end) was detected at a similar frequency of ~1.3%. As our RNA-Seq protocol is strand-specific (e.g., see sequencing of chemically synthesized RNA oligos in fig. S7), complementary variation on the two strands shows that RNA templates with sequence variants can be replicated. An RNA species replicated by T7 RNAP thus functions as an ensemble of multiple replication-competent sequences rather than simply as a pair of complementary, replicating sequences. Viroids and RNA viruses are well-known to similarly form heterogeneous populations consisting of multiple replication-competent sequences [e.g., (11)].
RNA structure requirements for replication
To assess the significance of the structural features present in replicating RNA sequences (Fig. 1C), we performed high-throughput mutagenesis of the 2-way- and 4-way- repeat configurations. Specifically, we designed a series of degenerate libraries; each library was made by randomizing a subset of base identities at a distinct set of 5 or 6 positions in either X RNA, an RNA replicated by T7 RNAP previously characterized by Konarska and Sharp (3, 4), or Y2 RNA (fig. S9A). Each library thus contained 1024 (45) or 4096 (46) RNA sequence variants. To test the 4-way repeat requirement, we randomized four potentially base pairing positions in the 4-way repeat. To test the 2-way repeat requirement, we randomized two potentially base pairing positions in parts of the 2-way repeat that were non-overlapping with the 4-way repeat. We performed T7 RNAP replication reactions with the degenerate libraries to enrich for efficiently replicating RNAs, sequenced RNA populations before and after replication, and examined the replicated populations to determine whether complementary bases were present at the randomized positions (Fig. 3).
Fig. 3. 2-way- and 4-way- repeat configurations are required for efficient replication of X and Y2 RNA.
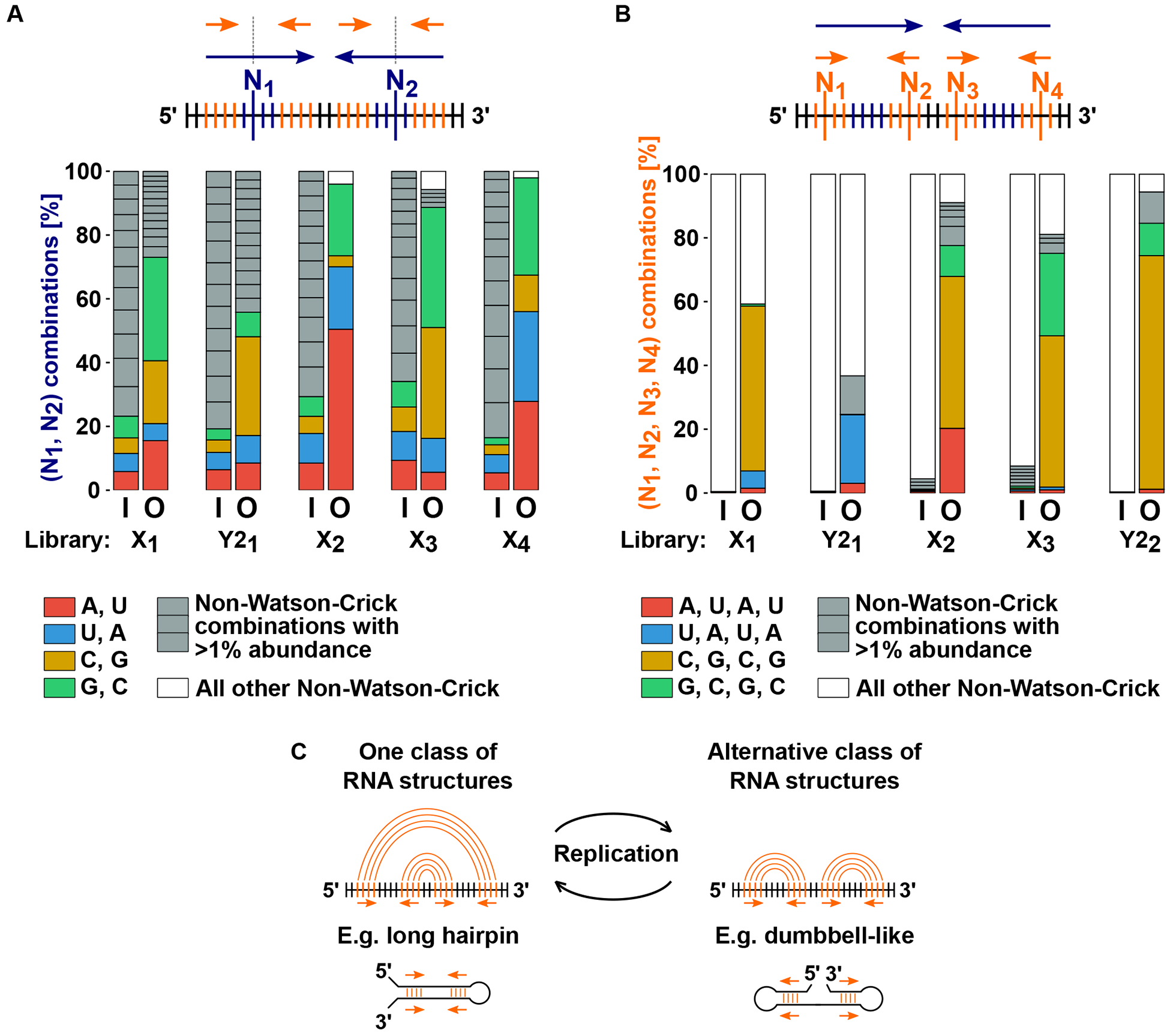
Six degenerate libraries (X1-X4, Y21-Y22) were constructed by randomizing the base identities at a subset of sequence positions in either X RNA or Y2 RNA. Degenerate libraries were used as templates in T7 RNAP reactions, and RNA populations before replication [input “I” on x axes of bar graphs in panels (A) and (B)] and after replication [output “O” on x axes] were sequenced to test whether Watson-Crick base combinations were enriched at randomized positions after replication. (A) 2-way repeat requirement was tested by randomizing bases at two potentially base pairing positions (“N1” and “N2”) in parts of the 2-way repeat (blue arrows) that are non-overlapping with the 4-way repeat (orange arrows). (B) 4-way repeat requirement was tested by randomizing bases at four potentially base pairing positions (“N1” through “N4”) in the 4-way repeat. Color coding of bar graphs in panels (A) and (B): different Watson-Crick base combinations are shown in unique colors; abundant (>1%) non-Watson-Crick base combinations are shown individually in gray; infrequent (<1%) non-Watson-Crick base combinations are summed together and shown in white. (C) Shape-shifting model. 2-way repeat requirement provides evidence for a long hairpin RNA secondary structure whereas 4-way repeat requirement suggests the formation of an alternative RNA secondary structure during replication.
At the positions used to test the 2-way repeat requirement, the combinations represented after RNA replication were dominated by Watson-Crick base-pairs (Fig. 3A). At the positions used to test the 4-way repeat requirement, the most abundant RNA sequences had one of the four possible 4-way Watson-Crick base combinations—(A,U,A,U), (U,A,U,A), (G,C,G,C) or (C,G,C,G) (Fig. 3B). It should be noted that not all Watson-Crick base combinations were replicated with similar efficiency for any given degenerate library. But for each set of positions used to test the 2-way- or 4-way- repeat requirements, we detected at least two abundant Watson-Crick base combinations after replication (Figs. 3A and 3B). We also constructed a degenerate library in which we randomized the base identities at only two of the four potentially base pairing positions in the 4-way repeat. After templated replication of this library, the most abundant RNA sequences only had the specific 4-way Watson-Crick base combination that was expected given the identity of the two non-randomized bases in the 4-way repeat (fig. S9B).
We conclude from these experiments that both the 2-way- and 4-way- repeats are required for efficient replication of X and Y2 RNA by T7 RNAP. Based on the role of the 2-way repeat, we suggest that a long hairpin RNA structure is required for replication by T7 RNAP. A long hairpin structure may thermodynamically allow the separation of complementary strands and could thus promote generation of active single-stranded templates for continued replication (12).
The functional requirement of the 4-way repeat suggests that the capacity to change secondary structure (“shape-shift”) is required for an RNA template to be efficiently replicated by T7 RNAP (Fig. 3C).
Interrupted rolling circle mechanism for RNA concatemer synthesis
RNA concatemers—RNA chains consisting of multiple, full-length repeats of template sequence—have been identified as intermediates during replication of viroids and Hepatitis delta virus (13, 14). A ladder of RNA concatemers (dimers, trimers, tetramers etc.) also forms during RNA replication by T7 RNAP (3). To investigate mechanisms of RNA concatemer formation, we analyzed the sequences of RNA dimers obtained from T7 RNAP reactions starting with diverse pools of chemically synthesized RNA monomer templates. For terminology, we define an “RNA monomer” as comprising a single repeat of full-length template RNA sequence and an “RNA dimer” as comprising two repeats. We considered two types of mechanisms for RNA dimer formation templated by RNA monomers (Fig. 4A): uni-templated and bi-templated. In a uni-templated mechanism, the same monomer template molecule instructs synthesis twice to form the dimer. In a bi-templated mechanism, two different monomer template molecules, which may still have the same sequence, instruct synthesis of each half of the dimer.
Fig. 4. T7 RNAP can use a template molecule processively to instruct multiple rounds of RNA synthesis.
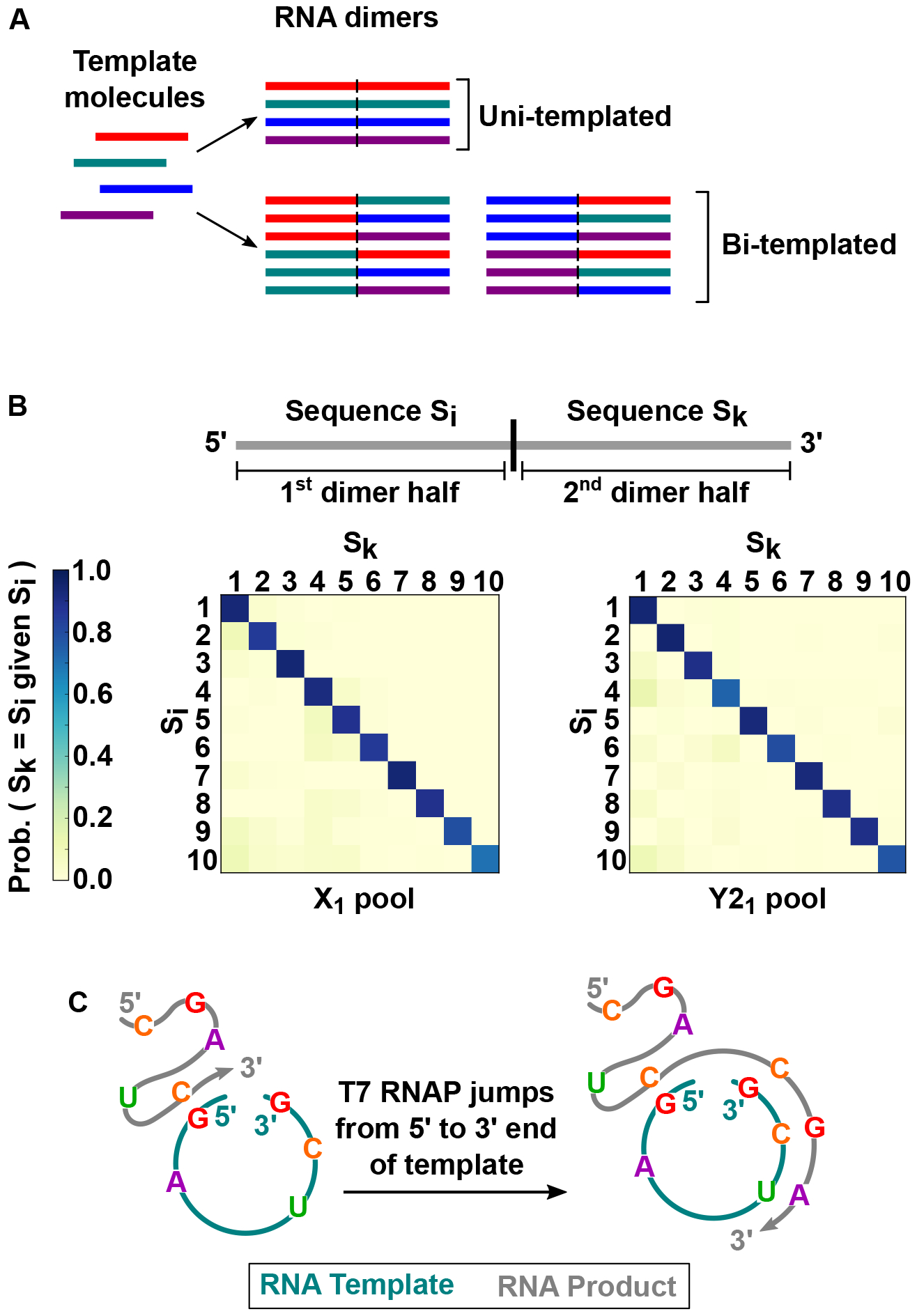
(A) Two possible mechanisms for RNA dimer generation, with both halves of a dimer synthesized using the same monomer template molecule in uni-templated synthesis but using two different monomer molecules in bi-templated synthesis. (B) Uni-templated synthesis is the dominant mechanism for RNA dimer generation. Two diverse monomer template pools, X1 and Y21, with base randomization at six positions in X RNA and Y2 RNA respectively, were used as templates for dimer synthesis followed by high-throughput sequencing of RNA dimers. Plots show the sequence agreement between the six randomized positions in the first dimer half (Si) and in the second dimer half (Sk), for the ten most abundant RNA templates present in the sequenced pools. No mismatches were allowed in sequence agreement calculations. Prob = Probability. (C) Model for uni-templated synthesis is an interrupted rolling circle mechanism. In this model, the RNA template is positioned such that T7 RNAP can jump from the template 5’ to 3’ end, and synthesize RNA products that are longer than monomers.
The presence of a diversity of monomer sequences in the same T7 RNAP reaction was a key aspect of the experiments we designed to elucidate the RNA dimer formation mechanism (fig. S10) (15). We performed RNA-Seq on dimers from two starting monomer template pools called X1 and Y21. These pools were also used in earlier experiments (Fig. 3) and were constructed by randomizing a subset of base identities at six positions each in X RNA and Y2 RNA, respectively. We expected that uni-templated synthesis would result in the two halves of RNA dimers containing the same six-base combination at the positions with initially randomized bases. In contrast, bi-templated synthesis would be expected to lead to relatively rare agreement of the six-base combination between the two dimer halves, with the concentration of a six-base combination in the reaction pool determining the proportion of dimers containing that six-base combination in both dimer halves. We found strong sequence agreement between the six-base combinations of both dimer halves for the vast majority of dimer sequences. In two experimental replicates with the X1 pool, 87% and 88% of the dimers had the same sequence in each of the two dimer halves. Similarly, two experimental replicates with the Y21 pool resulted in 88% of dimers with the same sequence in the first and second dimer halves. We obtained similar results when we analyzed the sequence concordance between dimer halves for each individual RNA template (Fig. 4B and fig. S11). These results suggest that uni-templated synthesis is the dominant mechanism for formation of RNA dimers (also see Supplementary Text).
As predicted by a uni-templated synthesis mechanism, we also found that sequence variants located outside the intentionally randomized six bases were concordant between RNA dimer halves (figs. S12 and S13). Of note, the concordance of sequence variants between the two dimer halves provides direct and independent evidence for active replication of RNA templates with sequence variation compared to the reference sequence (see also fig. S8).
How does T7 RNAP use the same template molecule processively to instruct multiple rounds of RNA synthesis? We propose that after reaching the 5’ end of a replicating RNA template during RNA synthesis, T7 RNAP can jump (16) from the 5’ end to the 3’ end of the template without dissociation of the RNAP-template-product complex. Continued RNA synthesis after the jump appends a new copy of the template to the existing RNA product. We refer to this mechanism as interrupted rolling circle synthesis (Fig. 4C).
We further examined the junction sequences between the two RNA dimer halves to assess whether the proposed jumping of T7 RNAP is associated with any sequence signatures. A diversity of sequences was found at the dimer junction. The junction sequences qualitatively resembled the 3’ end sequences of RNA monomers (including the extra nucleotide additions) followed by the 5’ end sequences of RNA monomers. Further, as would be expected for RNA dimer synthesis from linear monomer templates, the sequence at the junction for a particular dimer molecule was not necessarily the same as the 3’ end sequence of that dimer (fig. S14). Two additional pieces of evidence suggest that the monomer templates instructing dimer synthesis were linear rather than circular: (i) we obtained RNA dimers starting with monomer RNAs bearing ends (5’-OH and 3’-OH) that are chemically incompatible for ligation; and (ii) Konarska and Sharp found that explicitly circularized X or Y RNA were not replicated efficiently (4).
Origin of T7 RNAP-RNA replicons by molecular evolution from DNA seeds
The variability observed in the sequences of replicating RNAs between no-template-added reactions raises several fundamental questions regarding the origin of replicating RNAs. Do distinct replicating RNAs originate in each reaction or are pre-existing replicating RNAs amplified? If new replicating RNAs do originate in each reaction, are they assembled from single nucleotides or is their formation partly templated?
We conjectured that obtaining many additional sequences of RNA replicons may provide insight into the origin of replicating RNAs. We thus developed a microfluidic assay to conduct high-throughput no-template-added reactions (fig. S15). By splitting our usual 10 μl reaction volume into ~170 thousand isolated drop reactions (each drop was ~60 picoliters), we expected to capture a higher diversity of replicons that would otherwise be lost because of domination by the most efficiently replicated RNAs in bulk reactions. We analyzed the RNA contents of the no-template-added, high concentration T7 RNAP reactions in drop format by aggregating ~105 drops at a time, and found, as expected, numerous RNA species (table S5) that had different sequences but similar structures to what was observed in the earlier test tube reactions. Examples of RNA species obtained from the aggregated drop reactions are shown in Fig. 5C, fig. S18 and fig. S20C.
Fig. 5. DNA-seeded origin of RNAs replicated by T7 RNAP.
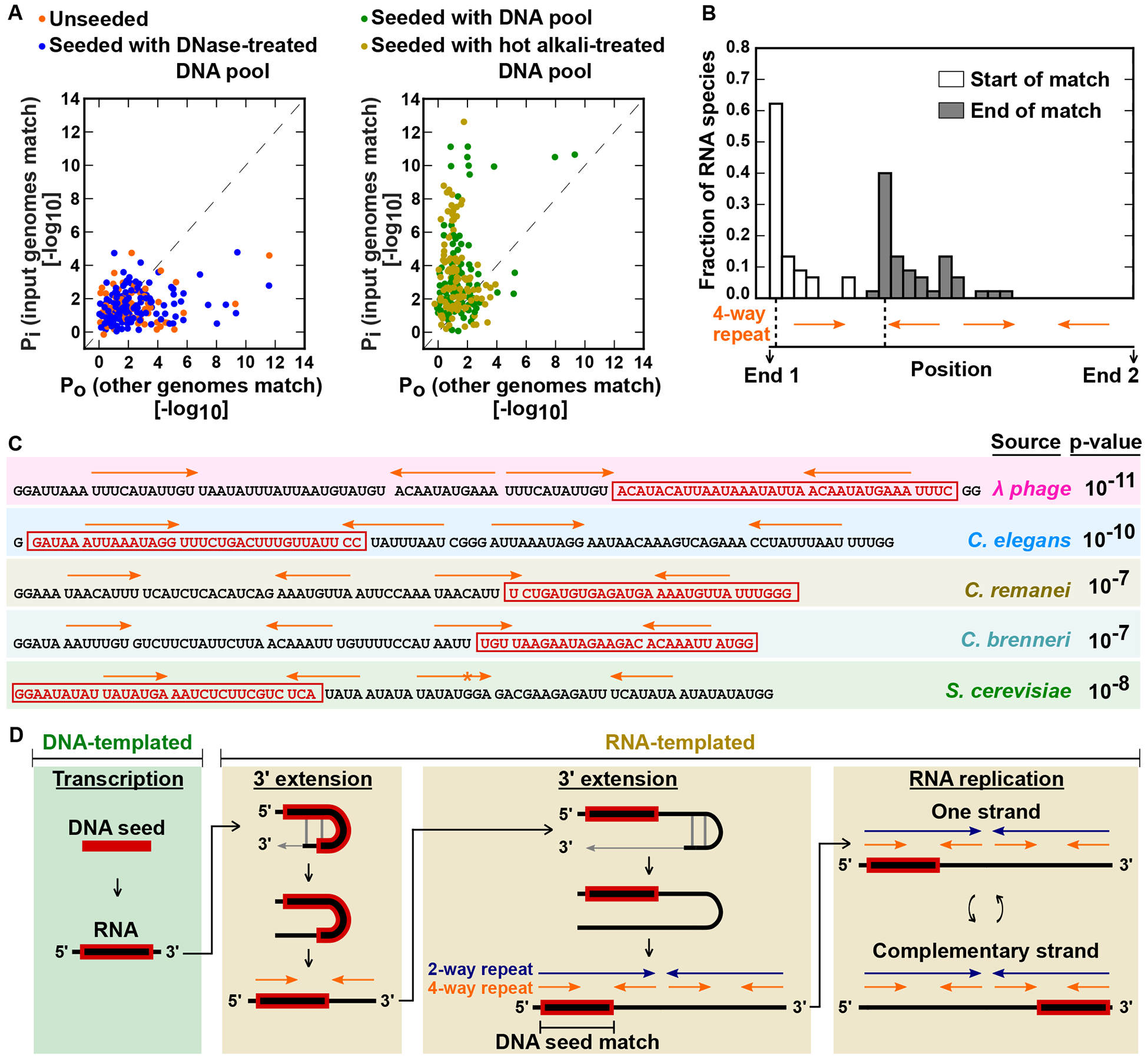
(A) Test of hypothesis that RNA replicons can originate through partial instruction from DNA seeds. T7 RNAP reactions seeded with a complex DNA pool (including DNA from λ phage, C. elegans, C. remanei, C. brenneri and S. cerevisiae) were compared to two negative controls (“unseeded” and “seeded with DNase-treated DNA pool”) and a positive control (“seeded with hot alkali-treated DNA pool”). Scatter plots show alignment significance of RNA species (individual points) to input DNA pool (y axis; Pi refers to the probability of finding a match to the input genomes by random chance) and to all available RefSeq genome assemblies excluding those in our DNA pool (x axis; Po refers to the probability of finding a match to these other genomes by random chance). In support of a DNA-seeded origin, RNA species with strong sequence matches to input DNA seeds (upper left regions of scatter plots) were specifically observed for reactions seeded with the DNA pool or the hot alkali-treated DNA pool compared to the two negative controls. (B) Histogram shows relative locations of seed matches and 4-way repeats for RNA species derived from our designed DNA pool. (C) Examples of RNA species that originated from different sources in our designed DNA pool. Best match to a source genome is shown in a red box. 4-way repeats are shown as orange arrows, with orange asterisks indicating sequence disagreements between 4-way repeat units. Candidate G strand sequences are shown for RNA species. p-values are based on alignment to a database consisting of sequences in our DNA pool. (D) Proposed model for the origin of replicating RNAs involving DNA-templated transcription, 2-way- and 4-way- repeat generation, and Darwinian selection of efficient replicators.
Within the large repertoire of RNA species we compiled using drop reactions, a subset of the RNAs contained sequence stretches that matched perfectly to known biological sources. Matches were commonly found to humans and to biological materials or organisms found in proximity to humans. From one set of no-template-added drop reactions, where we had included bovine serum albumin (BSA) to aid drop-reaction generation, we isolated RNA sequences similar to a replicating RNA sequence T7rp1 reported previously by Biebricher and Luce (5). Interestingly, T7rp1, and also the RNA sequences we isolated, strongly matched a sequence found in the genomes of cow and yak (fig. S19). These results suggested that replicating RNAs could evolve from residual nucleic acids present in the high concentration T7 RNAP reactions. We note here that we never synthesized or handled any of the replicating RNA sequences reported by Biebricher and Luce, indicating that the reagents used for drop-reaction generation, rather than the laboratory environment, were likely to be the initial source of the T7rp1-like sequences we isolated. As in the case of drop reactions, we also found novel RNA species that matched known genomes upon sequencing more no-template-added reactions set up in tubes. For example, fig. S20A shows an RNA species matching the human genome and an RNA species matching the genome of Lactococcus lactis, a bacterium commonly found in cheese (17).
We hypothesized that the RNAs replicated by T7 RNAP can originate through partial instruction from DNA seeds. We first focused on DNA seeds as a possibility, rather than the alternative possibility of RNA seeds, because the detected sequence homologies in replicating RNAs were represented throughout the genomes of matching organisms rather than in specific, highly transcribed regions. To test the hypothesis that replicating RNAs can originate from DNA seeds, we assessed whether T7 RNAP could catalyze the emergence of new replicating RNAs from a complex DNA seed pool of our own choosing (fig. S20B). The seed pool we used was a mixture of well-characterized model system genomes [three nematode species (Caenorhabditis elegans, Caenorhabditis remanei and Caenorhabditis brenneri), yeast (Saccharomyces cerevisiae strain S288C), bacteriophage lambda and a laboratory plasmid]. We chose these particular sources of DNA because they represent a range of genome sizes and sequence complexities. An additional consideration was that the chosen DNA sources did not significantly match any of the replicating RNAs that we had previously isolated. With the exception of lambda DNA, which was isolated from purified phage, the DNA seeds were derived from cellular sources. Hence, we extensively treated the DNA seeds with RNase A and RNase I before use. After treating with RNases and combining the DNA seeds from all the chosen sources, we split the seed pool into three equal parts. One part underwent no further treatment, the second part was treated with DNase and the third part was heated with alkali (0.2 N sodium hydroxide at 70°C for 1 hour) to further hydrolyze any possible remaining RNA (hot alkali treatment also provided an assessment of seed activity from denatured DNA).
We conducted high concentration T7 RNAP reactions in drop and tube format for four experimental conditions in parallel: (i) unseeded; (ii) seeded with DNA pool that we had prepared; (iii) seeded with DNase-treated DNA pool; and (iv) seeded with hot alkali-treated DNA pool. For each experimental condition, we performed RNA-Seq on aggregated drop and tube reactions. From comparable reaction volumes and sequencing depths, the number of replicating RNAs identified per reaction was 53 ± 22 (mean ± standard deviation) for eight aggregated drop reactions and 7 ± 5 for six tube reactions (Supplementary Text, table S5). We then used BLAST (18) to align the replicating RNAs obtained from all four conditions to the expected sequences present in our designed DNA pool. As a control, we also aligned the replicating RNAs to all complete genomes available in the RefSeq Genomic database, excluding the genomes present in our designed DNA pool (19). Of the four experimental conditions examined, only the “seeded with DNA pool” and “seeded with hot alkali-treated DNA pool” conditions yielded replicating RNAs that were derived from our designed DNA pool (Fig. 5A, Fig. 5C, fig. S20C). Significant matches specific to our DNA pool were absent in two negative controls—the “unseeded” and “seeded with DNase-treated DNA pool” conditions (Fig. 5A). We conclude that the RNAs replicated by T7 RNAP can originate from DNA seeds.
To investigate the molecular mechanism by which replicating RNAs arise from DNA seeds, we compared the location of the matching seed in a replicating RNA sequence to the positions of the 4-way repeat units for that sequence. We found that seed matches started at either end of replicating RNAs and extended up to the second 4-way repeat unit that is encountered from the start of the match (Fig. 5B). These data are consistent with mechanisms for the formation of replicating RNAs which at minimum include the steps shown in Fig. 5D: (i) transcription of a DNA seed to RNA, (ii) one round of self-templated 3’ extension of RNA to acquire a second 4-way repeat unit, and (iii) a second round of self-templated 3’ extension of RNA to acquire the full 2-way- and 4-way- repeat configurations (20, 21). Once an RNA with 2-way- and 4-way- repeats forms, it could quickly become predominant in its reaction compartment through Darwinian selection, as RNA templates with 2-way- and 4-way- repeats are especially optimal for replication by T7 RNAP (Fig. 3). Such a model for replicon emergence explains how different sequences with 2-way- and 4-way- repeats come to dominate no-template-added T7 RNAP reactions set up in parallel (Fig. 1, fig. S1B).
Discussion
RNA replication by transcription polymerases could serve as a means for the propagation of RNA-based genetic information in cellular contexts. To define the landscape of RNA replication by transcription polymerases at a molecular level, we investigated the diversity, mechanisms and origins of RNAs replicated by a model transcription polymerase—T7 RNAP. We have described subterminal de novo initiation, RNA shape-shifting and interrupted rolling circle synthesis as three underlying mechanisms for RNA replication by T7 RNAP.
Subterminal de novo initiation exemplifies a hallmark of RNA replication that is shared between numerous viral RNA-dependent RNA polymerase (RdRp) systems (22) and the T7 RNAP system. A possible advantage of initiation at a subterminal position, upstream of 3’ nucleotide extensions, is suggested by experiments with the RdRp of bacteriophage Qβ showing that a 3’ nucleotide extension can stabilize interactions at the polymerase active site for more efficient de novo initiation (23).
We speculate that changes in RNA secondary structure during replication—what we call RNA shape-shifting—may promote faster separation of complementary strands (24), more efficient unwinding of the structured RNA template by T7 RNAP, and/or formation of a T7 RNAP elongation complex-like structure for processive RNA synthesis [with a dumbbell-shaped RNA template making contacts with T7 RNAP both upstream and downstream of the active site (25, 26)].
Interrupted rolling circle synthesis provides a mechanism for RNA concatemer generation using linear templates. To assess a potential role of this mechanism in viroid replication, we examined published data for two viroids—ASBVd (27) and peach latent mosaic viroid (PLMVd) (28). We found that for these two viroids, an interrupted rolling circle model provides a more parsimonious explanation for data on initiation and termination during RNA synthesis than the current rolling circle model involving circular templates (see Supplementary Text).
Our work on the origins of replicating RNAs provides new insights into the rich history of mysterious products emerging from in vitro no-template-added reactions for both DNA and RNA polymerases [e.g. (29, 30)]. A key question was whether such reactions can evidence molecular evolution or are the observed products a result of amplification of pre-existing templates. Ascertaining the involvement of a pre-existing template was challenging because a replication cycle triggered by a single template molecule (which would be below detection limits) could have resulted in the observed products. Emergence of novel RNA replicons from a complex DNA seed pool of our own choosing (Fig. 5) shows that T7 RNAP reactions can witness DNA-seeded origin and evolution of replicating RNAs rather than just amplification of pre-existing templates.
Our results also provide a basis to understand previous observations by Konarska and Sharp (4) and by Biebricher and Luce (5) regarding RNA synthesis in no-template-added T7 RNAP reactions. We agree with Konarska and Sharp’s interpretation that a replicon may be a pre-existing contaminant in T7 RNAP reactions when there is consistent amplification of that replicon in several no-template-added reactions set up in parallel. However, when different replicon sequences amplify in reactions set up in parallel, Biebricher and Luce’s hypothesis of molecular evolution of replicons can be experimentally probed. While Biebricher and Luce suggested that RNA replicons are assembled in the absence of template, they also stated that partial instruction by oligo- or poly- nucleotides cannot be ruled out in explaining how RNA replicons arise (5). Our findings offer a different perspective towards these hypotheses underlying RNA replicon emergence. We demonstrated that the origin of a subset of replicon sequences can be traced to partial instruction from DNA seeds. The remaining replicon sequences could also possibly have originated from DNA seeds, but it is difficult to assign a definitive source for these replicons (Supplementary Text). Furthermore, we cannot rule out the possibility that a replicon with no significant matches to known genomes, originated in the absence of any initial template.
Just as new replicating RNAs originate from distinct DNA seeds in our T7 RNAP reactions, so may emergence of new RNA replicons be ongoing in nature, independent of other pre-existing RNA replicons. Our work thus provides an experimental window into how replicating RNAs such as viroids or Hepatitis delta might originate via host transcription polymerase activities. Of note, derivation from host nucleic acids is one of several hypotheses that have been put forth for the origins of viroids and Hepatitis delta (31–33).
Our study further suggests some biotechnological uses of RNA replication by transcription polymerases. A particularly appealing application of the T7 RNAP-RNA replication system may be amplification of hairpin RNAs for knocking down expression of genes of interest. Of relevance for this application, RNAs replicated by T7 RNAP are capable of forming extended hairpin structures. Also pertinent in this regard, our data on the diversity of RNA replicon sequences (Figs. 1 and 5) and on mutagenesis of particular replicons (Fig. 3) suggest the feasibility of designing novel replicating RNAs that encode short sequence homologies to genes of interest. We note that in particular cases, replication of user-defined sequences may require strategies such as compartmentalization and phenotypic selection to mitigate competitive amplification of other RNA templates that are either pre-existing or capable of forming during replication [e.g. (34–37)].
Methods Summary
General protocols used throughout our study are listed in this section. For supplementary protocols specific to certain figures, see Supplementary Materials.
Reagents
Key reagents/equipment used in this study are listed in table S6.
DNA and RNA oligonucleotide synthesis
Oligos were purchased from IDT and are listed in table S7.
Polyacrylamide gels
Samples were loaded on denaturing gels after adding an equal volume of Gel Loading Buffer II and denaturing at 95°C for >=2 minutes. Gels were pre-run for at least 30 minutes before sample loading. Gels were stained in a 1:5000–1:10,000 dilution of SYBR Gold stock reagent (dilution in 1x TBE) for 15–30 minutes covered with aluminium foil on a rocker. Gels were imaged using an AlphaImager HP system (ProteinSimple). Two 10 nucleotide ladders were used as markers on denaturing gels: (i) TrackIt 10 bp DNA ladder and (ii) 20/100 ladder mixed with a set of DNA ultramers to get a 10 nucleotide ladder from 20–200 nucleotides. The ladders were also dissolved in Gel Loading Buffer II and denatured at 95°C prior to gel loading.
For display purposes, for each of the gel images shown in fig. S1B, fig. S3, fig. S6 and fig. S17, a constant gamma correction (ɣ=0.3) was applied uniformly across the entire image using MATLAB (Natick, MA). For display purposes in Fig. 2A, a constant gamma correction (ɣ=0.3) and a constant increase in brightness were applied uniformly across the entire set of gel images using MATLAB. For display purposes in fig. S2, a constant gamma correction with ɣ=3.33 was applied uniformly to the denaturing gel images (first three gels from top to bottom) and with ɣ=1.0 was applied uniformly to the PCR gel image (bottommost gel) using AlphaView software (ProteinSimple). Gel images that are shown for side-by-side comparison (Fig. 2A, fig. S3 and fig. S6A) were all taken at the same exposure.
T7 RNAP-RNA replication reactions
High concentration T7 RNAP was either prepared in-house using a protocol (38) previously used to purify crystallography-grade T7 RNAP (39), or purchased as a special order from New England Biolabs (NEB). High concentration T7 RNAP was stored at −80°C. Commercially available low concentration T7 RNAP preps (from either NEB or Agilent) were stored either at −20°C or −80°C. Unless otherwise stated, buffer composition of T7 RNAP reactions was: 40 mM Tris-HCl (pH 8), 80 mg/ml PEG 8000, 20 mM MgCl2, 5 mM DTT, 1 mM spermidine, 0.01% (v/v) Triton X-100, and 4 mM each of ATP, CTP, GTP and UTP (3). Before use, buffers were sterile filtered using a 0.2 micron syringe filter. In experiments where several conditions were tested in parallel, the same stocks of buffers, NTPs and T7 RNAP were used for all conditions. Table S8 details reaction conditions for the various sets of T7 RNAP reactions in our study. Gel filtration (GF) buffer (50 mM Tris-HCl at pH 8, 200 mM NaCl, 2 mM EDTA, 5% glycerol and 2 mM DTT) was used for storage and dilution of the in-house isolated T7 RNAP. To minimize formation of protein aggregates, we recommend diluting T7 RNAP by no more than 10-fold at a time. It is important to incubate high concentration T7 RNAP reactions at 37°C quickly after setup (see Supplementary Text regarding temperature dependence of high concentration T7 RNAP reactions). We also assessed how addition of DNase influences RNA synthesis in no-template-added reactions (see Supplementary Text). We further note that while the reactions described in Fig. 1 were incubated for ~1 day, subsequent experiments showed that turbidity and substantial RNA synthesis for no-template-added, high concentration T7 RNAP reactions set up in tubes can also be observed at much earlier time points (e.g. at ~4–5 hours into incubation at 37°C).
Gel extraction from polyacrylamide gels
Excised gel fragments were transferred to autoclaved, nuclease-free 0.6 ml tubes that had small cross-shaped incisions at the bottom. The 0.6 ml tubes were contained in 1.5 ml siliconized tubes. Gel fragments were shredded by centrifugation. 300–400 μl of RNA elution buffer (300 mM sodium acetate at pH 5.3, 1 mM EDTA) or DNA elution buffer (300 mM sodium chloride, 10 mM Tris-HCl at pH 8, 1 mM EDTA) (40) was added to shredded gel pieces. The specific elution buffer used depended on the nature of nucleic acid to be extracted (e.g. RNA elution buffer was used for extracting replicating RNA populations and for extracting ligated RNA during RNA-Seq library preparation; DNA elution buffer was used for extracting cDNA and for extracting DNA oligos such as the reverse transcription primer used for RNA-Seq library preparation). Shredded gel with elution buffer added was briefly vortexed and frozen at −80°C for 15 minutes, followed by rocking overnight at 4°C (for RNA) or at room temperature (for DNA). Gel was then sedimented by centrifugation, and the supernatant transferred to a new 1.5 ml siliconized tube. To ensure maximal recovery of nucleic acids, gel was further washed in 100 μl of elution buffer and centrifuged. The resultant supernatant was combined with the supernatant obtained from the previous gel centrifugation step. After a final centrifugation of the pooled supernatants to sediment any remaining gel pieces, the recovered solution was ethanol precipitated with 2.5 volumes of 100% ethanol.
RNA-Seq protocol (see also fig. S2)
The basic workflow for the RNA-Seq protocol used in this study is based on previous work by our lab and others [e.g. see protocol “RNA-seq1” in (41) and references therein; see also (40)]. We made several modifications for efficient capture of replicating RNAs. In particular, we optimized full-length cDNA synthesis because under standard reverse transcription conditions with commonly available enzymes, no full-length cDNAs were detectable by SYBR Gold gel staining (though bands corresponding to particular truncated cDNA fragments were clearly observed). The problem of inefficient reverse transcription of the RNAs replicated by T7 RNAP was also reported previously (5). Sequencing of chemically synthesized RNA oligos (e.g. AF-NJ-223 and AF-NJ-224) served as a positive control for our protocol. There are four main steps in our RNA-Seq protocol:
3’ ligation of ssDNA adapter to RNA: A 20 μl reaction was set up for each sample = 7.6 μl RNA + 2 μl 100% DMSO + 6 μl 50% PEG 8000 + 2 μl 10x T4 RNA ligase buffer + 0.4 μl 100 μM AF-NJ-269 (or AF-JA-34) + 2 μl T4 RNA ligase 2, truncated, K227Q (400 units). Ligation reactions were incubated at 16°C in a thermal cycler for 18 hours-19 hours 10 minutes (depending on the sample, see table S9). Reactions were heat-inactivated at 65°C for 20 minutes. Ligation products were gel extracted and resuspended in 0.5x TE (pH 7.4). Note that AF-NJ-269 and AF-JA-34 have 8 and 6 degenerate nucleotides at the 5’ end, respectively, which serve as molecular identifiers (UMIs) in downstream bioinformatic analyses.
-
Reverse transcription: 8 μl of the ligated RNA was heated at 95°C for 3 minutes in a thermal cycler, followed by snap cooling on ice for 3 minutes [see Table 1 in (42)]. Next, added to each reaction (on ice) was 4 μl 5x First Strand Buffer, 1 μl RNase OUT (40 units), 1 μl 0.1 M DTT, 1 μl dNTPs (10 mM stock concentration of each dNTP), 0.64 μl 72 ng/μl gel-extracted AF-JA-126 (concentration quantified by Qubit ssDNA kit, Thermo Fisher #Q10212) and 0.36 μl water, followed by 4 μl (800 units) of SuperScript III. Of note, the 95°C denaturation-snap cooling step and a higher concentration of SuperScript III (compared to standard protocols) were key optimizations for increasing yield of full-length cDNAs.
After SuperScript III addition, reactions were immediately placed in a thermal cycler with a pre-heated lid and incubated at 50°C for 2 hours 30 minutes. [After cDNA synthesis, RNA can be hydrolyzed by treatment with sodium hydroxide (final concentration 0.2 N) at 70°C for 15 minutes.] cDNA products were gel extracted and resuspended in RNase-free water.
A no-template reaction was set up in parallel each time the reverse transcription protocol was performed; no products were detected for these no-template controls.
Circularization of cDNA: 5.5 μl of the cDNA was heated at 95°C for 3 minutes in a thermal cycler, followed by snap cooling on ice for 3 minutes. Either CircLigase reaction components or CircLigase II reaction components were then added to each reaction on ice [CircLigase reaction components: 1 μl 10x circLigase buffer + 0.5 μl 50 mM MnCl2 + 2 μl 5M Betaine + 0.5 μl 1 mM ATP + 0.5 μl circLigase enzyme (50 units); CircLigase II reaction components: 1 μl 10x circLigase II buffer + 0.5 μl 50 mM MnCl2 + 2 μl 5M Betaine + 1 μl circLigase II enzyme (100 units)]. Reactions were immediately placed in a thermal cycler with a pre-heated lid and incubated at 60°C for 1–2 hours (depending on the sample, see table S9), followed by heat inactivation at 80°C for 10 min.
- PCR: Illumina TruSeq HT indices and adapter sequences were appended using PCR. We set up 30 μl PCR reactions consisting of: 15 μl 2x Phusion Master Mix + 0.3 μl 100 μM Primer 1 + 0.3 μl 100 μM Primer 2 + 1 μl circularized cDNA (reaction contents from step 3 directly used) + 13.4 μl nuclease-free water. For each sample, we set up several PCR reactions with differing PCR cycle numbers, and selected for sequencing the reaction with the least number of cycles that yielded the expected product band on an ethidium bromide-stained 3.5%−4% agarose gel. The PCR conditions were:
- 98°C, 30 seconds
- For n cycles, where n is variable, perform:
- 98°C, 10 seconds
- 60°C, 10 seconds
- 72°C, 20 seconds-60 seconds (depending on the sample, see table S9)
-
10°C, holdPCR amplified RNA-Seq libraries were gel-extracted using the MinElute gel extraction kit (Qiagen #28604), and quantified using the Qubit dsDNA HS kit.
All samples were sequenced on the Illumina MiSeq platform.
Note that gel electrophoresis following each of the steps of 3’ ligation, reverse transcription and PCR provided a visual assessment of reaction efficiencies for each sample we sequenced. During sample loading on gels, samples were always separated by at least one gel lane (which was either left empty or contained a size marker) to minimize cross-contamination. Relevant parameters of library preparation for samples in our study are described in table S9.
For experiments where we compared template sequences with product sequences for a T7 RNAP-RNA replication reaction, gel cuts for the template and product pools were made at similar sizes during RNA-Seq library preparation.
Droplet Microfluidics
We used standard methods in soft lithography (43) to fabricate all microfluidic devices using a 10:1 base-to-curing agent ratio from the Sylgard 184 Silicone Elastomer kit (Dow Corning). Inlet and outlet holes were made using a 1 mm biopsy punch (Miltex), and the PDMS devices were plasma bonded to glass slides in a cleanroom.
We used a standard flow-focusing geometry with a Y-junction mixer to generate droplets (fig. S15). The height of our droplet generation channels was 27 microns. Three syringe pumps (Kent Scientific) were used to inject the three fluid streams into our device at fixed flow rates. The aqueous droplet phase consisted of a mixture of two aqueous reagent streams which were combined at a Y-junction upstream of the flow-focusing nozzle. One aqueous reagent stream was used to flow in NTPs, PEG 8000 and any DNA/RNA template, and the other stream was used to flow in all other reagents for T7 RNAP reactions. The continuous oil phase consisted of HFE-7500 containing 2% wt/wt 008-FluoroSurfactant. We used Aquapel (Pittsburgh, PA) to render the channels hydrophobic to prevent droplet wetting of the walls. Following Aquapel treatment, we carefully wrapped the droplet generation devices in aluminum foil and autoclaved on a gravity cycle. Autoclaved channels were kept wrapped in aluminum foil until use. In cases where multiple experimental conditions were tested in parallel, a separate autoclaved channel was used for each condition.
We used a flow rate of 0.1 ml/hr for each of the two aqueous reagent streams (0.2 ml/hr combined flow rate) and a flow rate of 0.4 ml/hr for the continuous oil phase. We used a high-speed camera (Phantom v7.3) mounted on an inverted microscope with a 4× objective to continuously monitor droplet generation and also to record videos of the droplet formation process at 40,000 fps for measurement of droplet size. For the latter, we measured the time it took to form a single drop and calculated the droplet size based off of the combined aqueous phase flow rate of 0.2 ml/hr. We did this for multiple drops to obtain a distribution of droplet size.
Once the droplet size stabilized (after the first few minutes of drop generation), we serially collected droplets in 0.2 ml PCR tubes for assay purposes.
Bioinformatic Analysis
We have deposited code used in our study in a public GitHub repository (named JainEtAl_T7rnaReplication). A brief description of the deposited code can be found in table S10. Other software that was additionally used for analysis includes the ViennaRNA suite (44), Phylip (45), Interactive Tree of Life web interface (46), Trimmomatic (47), BWA (48) and Samtools (49). For sequence alignment of replicating RNAs, we used the classical Needleman-Wunsch (50) and Smith-Waterman algorithms (51).
Supplementary Material
Acknowledgments:
We thank U. Phan, K. Artiles, L. Wahba, C. Froekjaer-Jensen, E. Cenik, M. McCoy, J. Arribere, L. Hansen, P. Geiduschek, K. Kirkegaard, V. Walbot, Z. Bryant, D. Herschlag, R. Lehman, M. Guzman, K. Perez, M. Shoura, D. Jeong, S. Silas, V. Topkar, P. Greenside, J. Garcia, D. Yue and C. Penedo for help and feedback. We thank P. Sharp and M. Konarska for discussions regarding their prior work on the T7 RNAP-RNA replication system. We further thank the UC Davis Veterinary Genetics Laboratory for cattle genotyping of our T7 RNAP preps.
Funding: This work was supported by NIH grants R01GM37706 (A.Z.F.), R35GM130366 (A.Z.F.), R01AI134611 (Y.W.Y.), R35GM122579 (R.D.) and T32GM008412 (L.R.B.); a Stanford Graduate Fellowship (N.J.); NSF DBI-1548297 Center for Cellular Construction (L.R.B. and S.K.Y.T.); National Science Centre, Poland grant UMO-2016/21/P/NZ1/01085 (M.R.S.); and a Jane B. Kempner Postdoctoral Fellowship (M.R.S). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152.
Footnotes
Competing interests: N.J., A.Z.F., L.R.B., S.K.Y.T., Y.W.Y. and M.R.S. are inventors on a provisional patent application (number 62/872,540) filed by Stanford University that covers methods, compositions and kits for amplifying and generating RNA replicons.
Data and materials availability: Sequencing data are publicly available through NCBI SRA (PRJNA563092). Code is publicly available through GitHub (repository name JainEtAl_T7rnaReplication) and was archived on 25th September 2019 using Zenodo (52). Functionality of the code is briefly described in table S10.
Supplementary Materials:
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at https://science.sciencemag.org/content/368/6487/eaay0688. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Biebricher CK, Orgel LE, An RNA that Multiplies Indefinitely with DNA-Dependent RNA Polymerase: Selection from a Random Copolymer. Proc. Natl. Acad. Sci 70, 934–938 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wettich A, Biebricher CK, RNA Species that Replicate with DNA-Dependent RNA Polymerase from Escherichia coli. Biochemistry. 40, 3308–3315 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Konarska MM, Sharp PA, Replication of RNA by the DNA-dependent RNA polymerase of phage T7. Cell. 57, 423–431 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Konarska MM, Sharp PA, Structure of RNAs replicated by the DNA-dependent T7 RNA polymerase. Cell. 63, 609–618 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Biebricher CK, Luce R, Template-free generation of RNA species that replicate with bacteriophage T7 RNA polymerase. EMBO J. 15, 3458–3465 (1996). [PMC free article] [PubMed] [Google Scholar]
- 6.Kakimoto Y, Fujinuma A, Fujita S, Kikuchi Y, Umekage S, Abnormal rapid non-linear RNA production induced by T7 RNA polymerase in the absence of an exogenous DNA template. AIP Conf. Proc 1649, 113–115 (2015). [Google Scholar]
- 7.Steitz TA, The structural basis of the transition from initiation to elongation phases of transcription, as well as translocation and strand separation, by T7 RNA polymerase. Curr. Opin. Struct. Biol 14, 4–9 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Navarro J-A, Vera A, Flores R, A Chloroplastic RNA Polymerase Resistant to Tagetitoxin Is Involved in Replication of Avocado Sunblotch Viroid. Virology. 268, 218–225 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Gholamalipour Y, Karunanayake Mudiyanselage A, Martin CT, 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-Seq analyses. Nucleic Acids Res. 46, 9253–9263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarcar SN, Miller DL, A specific, promoter-independent activity of T7 RNA polymerase suggests a general model for DNA/RNA editing in single subunit RNA Polymerases. Sci. Rep 8 (2018), doi: 10.1038/s41598-018-32231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domingo E, Sabo D, Taniguchi T, Weissmann C, Nucleotide sequence heterogeneity of an RNA phage population. Cell. 13, 735–744 (1978). [DOI] [PubMed] [Google Scholar]
- 12.Priano C, Kramer FR, Mills DR, Evolution of the RNA Coliphages: The Role of Secondary Structures during RNA Replication. Cold Spring Harb. Symp. Quant. Biol 52, 321–330 (1987). [DOI] [PubMed] [Google Scholar]
- 13.Flores R, Gas M-E, Molina-Serrano D, Nohales M-Á, Carbonell A, Gago S, De la Peña M, Daròs J-A, Viroid Replication: Rolling-Circles, Enzymes and Ribozymes. Viruses. 1, 317–334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai MMC, RNA Replication without RNA-Dependent RNA Polymerase: Surprises from Hepatitis Delta Virus. J. Virol 79, 7951–7958 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fire A, Xu SQ, Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. U. S. A 92, 4641–4645 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou W, Reines D, Doetsch PW, T7 RNA polymerase bypass of large gaps on the template strand reveals a critical role of the nontemplate strand in elongation. Cell. 82, 577–585 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginzburg C, Il formaggio e i vermi. Il cosmo di un mugnaio del ‘500 (Turin: Einaudi, 1976). [Google Scholar]
- 18.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, Basic local alignment search tool. J. Mol. Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Pruitt K, Brown G, Tatusova T, Maglott D, The Reference Sequence (RefSeq) Database (National Center for Biotechnology Information (US), 2012; https://www.ncbi.nlm.nih.gov/books/NBK21091/). [Google Scholar]
- 20.Cazenave C, Uhlenbeck OC, RNA template-directed RNA synthesis by T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A 91, 6972–6976 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaher HS, Unrau PJ, T7 RNA Polymerase Mediates Fast Promoter-Independent Extension of Unstable Nucleic Acid Complexes. Biochemistry. 43, 7873–7880 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Kao CC, Singh P, Ecker DJ, De Novo Initiation of Viral RNA-Dependent RNA Synthesis. Virology. 287, 251–260 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Takeshita D, Tomita K, Molecular basis for RNA polymerization by Qβ replicase. Nat. Struct. Mol. Biol 19, 229–237 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Bartel D, 5 Re-creating an RNA Replicase. Cold Spring Harb. Monogr. Arch 37 (1999) (available at https://cshmonographs.org/index.php/monographs/article/view/5108/4205). [Google Scholar]
- 25.Tahirov TH, Temiakov D, Anikin M, Patlan V, McAllister WT, Vassylyev DG, Yokoyama S, Structure of a T7 RNA polymerase elongation complex at 2.9 Å resolution. Nature. 420, 43–50 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Yin YW, Steitz TA, Structural Basis for the Transition from Initiation to Elongation Transcription in T7 RNA Polymerase. Science. 298, 1387–1395 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Navarro J-A, Flores R, Characterization of the initiation sites of both polarity strands of a viroid RNA reveals a motif conserved in sequence and structure. EMBO J. 19, 2662–2670 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delgado S, de Alba ÁEM, Hernández C, Flores R, A Short Double-Stranded RNA Motif of Peach Latent Mosaic Viroid Contains the Initiation and the Self-Cleavage Sites of Both Polarity Strands. J. Virol 79, 12934–12943 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sumper M, Luce R, Evidence for de novo production of self-replicating and environmentally adapted RNA structures by bacteriophage Qbeta replicase. Proc. Natl. Acad. Sci 72, 162–166 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zyrina NV, Antipova VN, Zheleznaya LA, Ab initio synthesis by DNA polymerases. FEMS Microbiol. Lett 351, 1–6 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Brazas R, Ganem D, A Cellular Homolog of Hepatitis Delta Antigen: Implications for Viral Replication and Evolution. Science. 274, 90–94 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Salehi-Ashtiani K, Lupták A, Litovchick A, Szostak JW, A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science. 313, 1788–1792 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Diener TO, Viroids: “living fossils” of primordial RNAs? Biol. Direct 11, 15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills DR, Engineered recombinant messenger RNA can be replicated and expressed inside bacterial cells by an RNA bacteriophage replicase. J. Mol. Biol 200, 489–500 (1988). [DOI] [PubMed] [Google Scholar]
- 35.Mindich L, in Advances in Virus Research, Margniorosch K, Murphy FA, Shatkin AJ, Eds. (Academic Press, 1999; http://www.sciencedirect.com/science/article/pii/S0065352708603553), vol. 53, pp. 341–353. [DOI] [PubMed] [Google Scholar]
- 36.Urabe H, Ichihashi N, Matsuura T, Hosoda K, Kazuta Y, Kita H, Yomo T, Compartmentalization in a Water-in-Oil Emulsion Repressed the Spontaneous Amplification of RNA by Qβ Replicase. Biochemistry. 49, 1809–1813 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Matsumura S, Kun Á, Ryckelynck M, Coldren F, Szilágyi A, Jossinet F, Rick C, Nghe P, Szathmáry E, Griffiths AD, Transient compartmentalization of RNA replicators prevents extinction due to parasites. Science. 354, 1293–1296 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Grodberg J, Dunn JJ, ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol 170, 1245–1253 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin YW, Steitz TA, The Structural Mechanism of Translocation and Helicase Activity in T7 RNA Polymerase. Cell. 116, 393–404 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS, Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science. 324, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arribere JA, Cenik ES, Jain N, Hess GT, Lee CH, Bassik MC, Fire AZ, Translation readthrough mitigation. Nature. 534, 719–723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkinson KA, Merino EJ, Weeks KM, Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc 1, 1610 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Xia Y, Whitesides GM, Soft Lithography. Annu. Rev. Mater. Sci 28, 153–184 (1998). [Google Scholar]
- 44.Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL, ViennaRNA Package 2.0. Algorithms Mol. Biol 6, 26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Felsenstein J, PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics. 5, 164–166 (1989). [Google Scholar]
- 46.Letunic I, Bork P, Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res, doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Durbin R, Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Needleman SB, Wunsch CD, A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol 48, 443–453 (1970). [DOI] [PubMed] [Google Scholar]
- 51.Smith TF, Waterman MS, Identification of common molecular subsequences. J. Mol. Biol 147, 195–197 (1981). [DOI] [PubMed] [Google Scholar]
- 52.nimitjainFireLab, nimitjainFireLab/JainEtAl_T7rnaReplication: JainEtAl_T7rnaReplication_CodeRepository_v1.0_20190925, Zenodo (2019); https://zenodo.org/badge/latestdoi/210945803
- 53.Krupp G, Unusual promoter-independent transcription reactions with bacteriophage RNA polymerases. Nucleic Acids Res. 17, 3023–3036 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moody MD, Burg JL, DiFrancesco R, Lovern D, Stanick W, Lin-Goerke J, Mahdavi K, Wu Y, Farrell MP, Evolution of Host Cell RNA into Efficient Template RNA by Q.beta. Replicase: The Origin of RNA in Untemplated Reactions. Biochemistry. 33, 13836–13847 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T, Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63–e63 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seidl CI, Lama L, Ryan K, Circularized synthetic oligodeoxynucleotides serve as promoterless RNA polymerase III templates for small RNA generation in human cells. Nucleic Acids Res. 41, 2552–2564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner SD, Yakovchuk P, Gilman B, Ponicsan SL, Drullinger LF, Kugel JF, Goodrich JA, RNA polymerase II acts as an RNA-dependent RNA polymerase to extend and destabilize a non-coding RNA. EMBO J. 32, 781–790 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehmann E, Brueckner F, Cramer P, Molecular basis of RNA-dependent RNA polymerase II activity. Nature. 450, 445–449 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Wassarman KM, Saecker RM, Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science. 314, 1601–1603 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Gildehaus N, Neußer T, Wurm R, Wagner R, Studies on the function of the riboregulator 6S RNA from E. coli: RNA polymerase binding, inhibition of in vitro transcription and synthesis of RNA-directed de novo transcripts. Nucleic Acids Res. 35, 1885–1896 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murchie AIH, Lilley DMJ, The mechanism of cruciform formation in supercoiled DNA: initial opening of central basepairs in salt-dependent extrusion. Nucleic Acids Res. 15, 9641–9654 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng GX, Sinden RR, Effect of base composition at the center of inverted repeated DNA sequences on cruciform transitions in DNA. J. Biol. Chem 263, 5356–5361 (1988). [PubMed] [Google Scholar]
- 63.England TE, Uhlenbeck OC, 3′-Terminal labelling of RNA with T4 RNA ligase. Nature. 275, 560 (1978). [DOI] [PubMed] [Google Scholar]
- 64.Guo H, Ingolia NT, Weissman JS, Bartel DP, Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 466, 835–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanders R, Mason DJ, Foy CA, Huggett JF, Evaluation of Digital PCR for Absolute RNA Quantification. PLOS ONE. 8, e75296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furrows SJ, Ridgway GL, ‘Good laboratory practice’ in diagnostic laboratories using nucleic acid amplification methods. Clin. Microbiol. Infect 7, 227–229 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.