

Abstract
Background
The aggressive clinical behavior of poorly differentiated and anaplastic thyroid cancers (PDTC and ATC) has proven challenging to treat, and survival beyond a few months from diagnosis is rare. Although 30%–60% of these tumors contain mutations in the BRAF gene, inhibitors designed specifically to target oncogenic BRAF have shown limited and only short-lasting therapeutic benefits as single agents, thus highlighting the need for improved treatment strategies, including novel combinations.
Methods
Using a BRAFV600E-driven mouse model of ATC, we investigated the therapeutic efficacy of the combination of BRAF inhibition and oncolytic herpes simplex virus (oHSV). Analyses of samples from tumor-bearing mice were performed to immunologically characterize the effects of different treatments. These immune data were used to inform the incorporation of immune checkpoint inhibitors into triple combination therapies.
Results
We characterized the immune landscape in vivo following BRAF inhibitor treatment and detected only modest immune changes. We, therefore, hypothesized that the addition of oncolytic virotherapy to BRAF inhibition in thyroid cancer would create a more favorable tumor immune microenvironment, boost the inflammatory status of tumors and improve BRAF inhibitor therapy. First, we showed that thyroid cancer cells were susceptible to infection with oHSV and that this process was associated with activation of the immune tumor microenvironment in vivo. Next, we showed improved therapeutic responses when combining oHSV and BRAF inhibition in vivo, although no synergistic effects were seen in vitro, further confirming that the dominant effect of oHSV in this context was likely immune-mediated. Importantly, both gene and protein expression data revealed an increase in activation of T cells and natural killer (NK) cells in the tumor in combination-treated samples. The benefit of combination oHSV and BRAF inhibitor therapy was abrogated when T cells or NK cells were depleted in vivo. In addition, we showed upregulation of PD-L1 and CTLA-4 following combined treatment and demonstrated that blockade of the PD-1/PD-L1 axis or CTLA-4 further improved combination therapy.
Conclusions
The combination of oHSV and BRAF inhibition significantly improved survival in a mouse model of ATC by enhancing immune-mediated antitumor effects, and triple combination therapies, including either PD-1 or CTLA-4 blockade, further improved therapy.
Keywords: oncolytic viruses; immunotherapy; drug therapy, combination; T-lymphocytes
Background
Poorly differentiated and anaplastic thyroid cancers (PDTC and ATC) are rare, but highly aggressive, forms of thyroid cancer which have few effective treatment options. The BRAFV600E mutation is the most common somatic mutation (approximately 60%) driving this disease, and several studies have suggested a link between BRAF mutation status and tumor aggression.1–4 Although BRAF inhibitors (BRAFi) may yield short-term clinical benefits,5–7 monotherapy-targeted BRAF inhibition has been disappointing overall in clinical trials for PDTC/ATC.8 Improved efficacy has been observed with combined inhibition of BRAF and MEK kinases, a combination that also enhanced antitumor activity in mouse models.9 Based on the encouraging results from a phase II trial (NCT02034110), combined dabrafenib/trametinib has been approved by the Food and Drug Administration (FDA) for BRAF-mutant ATC since 2018.10 However, experience from thyroid and other tumors types suggests that patients receiving single agent or combined kinase inhibitor therapy will almost invariably develop resistance to treatment. Therefore, it is important to explore other combination strategies incorporating a BRAFi backbone with the goal of achieving improved, durable treatment outcomes for patients with PDTC/ATC.
In BRAF-mutant melanoma, the role of BRAF/MEK inhibitor combination therapy continues to evolve in the context of successful immunotherapy, particularly targeting the CTLA-4 and the PD-1/PD-L1 axis.11–15 Such sequential or concurrent combinations now need to take into account our increasing understanding that targeted small molecules can themselves be immunomodulatory and alter an immunosuppressive tumor milieu into one with more immune-active characteristics, thus supporting effective treatment with immune checkpoint blockade.12 However, using murine models, we and others have detected only modest immune-mediated activity of BRAFi monotherapy in thyroid cancer.16 17 Nevertheless, combining immunotherapy with targeted small molecules is a logical approach in aggressive thyroid cancer and has been the subject of limited exploration to date in both the preclinical and early clinical setting.
Several studies using immunocompetent murine models of thyroid cancer have shown effective results using either BRAF inhibition or the multikinase (VEGFR, FGFR, PDGFR, RET) inhibitor lenvatinib together with immune checkpoint PD-1/PD-L1 blockade.16–18 However, in these preclinical models, despite significantly prolonged survival and reduced tumor growth, mice were not cured of their disease, suggesting that there remains the opportunity to test additional drugs in combination strategies. Immunohistochemical analysis of patient tumor samples shows that 23%–81.3% of ATC and 25% of PDTC express PD-L1, although the data supporting a link between PD-L1 expression and disease aggression are conflicting.19 To date, there is no information from large clinical trials in aggressive thyroid cancer treated with immunotherapies blocking the PD-1/PD-L1 axis, although a phase II trial (NCT02688608, clinicaltrials.gov) using pembrolizumab in patients with ATC is ongoing. A case study of a patient with ATC treated with vemurafenib together with nivolumab showed a remarkable response with compete radiographic and clinical remission,20 and a further phase Ib/II clinical study (NCT02501086, clinicaltrials.gov) combining lenvatinib and pembrolizumab in solid cancers, including thyroid cancers, is recruiting. Furthermore, a recent study investigated if resistance to kinase inhibitors could be overcome by pembrolizumab, but this showed limited benefit in patients with ATC, and the authors concluded that better treatment strategies incorporating immunotherapy should be explored.21
Given the paucity of effective treatment options for this patient group, even with small molecule/standard immunotherapy combinations, we designed a study to investigate if immunovirotherapy with oncolytic herpes simplex virus (oHSV) would improve therapeutic responses in BRAF-mutant thyroid cancer treated with BRAFi. oHSV, encoding GM-CSF, has proven successful in melanoma trials and T-VEC (talimogene laherparepvec) has been an FDA-approved agent for melanoma since 2015.22 Different genetically modified versions of HSV have been shown to be effective in vitro and in xenograft models of thyroid cancer.23–27 We hypothesized that the addition of oHSV to BRAF inhibition in thyroid cancer would create a favorable tumor immune microenvironment and boost the inflammatory status of tumors, enhancing the cytotoxic and/or immunogenic effects of BRAFi alone. We used immunocompetent murine models of ATC driven by the BRAFV600E oncogene and confirmed limited efficacy with BRAF inhibition as monotherapy. The addition of oHSV to BRAFi significantly improved therapy via natural killer (NK) and T cell-mediated immune mechanisms, and the combination treatment was associated with increase gene expression of CTLA-4 and PD-L1. Subsequently, triple therapy using anti-CTLA-4 or anti-PD-1 checkpoint blockade further improved outcome, resulting in long-term regression of tumors in almost all mice.
Methods
Animal models and cell lines
Using Cre recombinase/loxP technology, we expressed BRAFV600E together with Trp53R172H or PTEN deletion in the thyrocytes of C57Bl/6 mice.28–30 Cre recombinase was under the TPO promoter and recombination started from E14.5.31 Mice were genotyped using genomic DNA prepared from ear biopsies and PCRs were performed using primers for BRAF (5′ GCCCAGGCTCTTTATGAGAA 3′, 5′ AGTCAATCATCCACAGAGACCT 3′ and 5′ GCTTGGCTGGACGTAAACTC 3′), Cre recombinase (5′ TGCCACGACCAAGTCACAGCAATG 3′ and 5′ AGAGACGGAAATCCATCGCTCG 3′), Trp53 (5′ CTTGGAGACATAGCCACACTG 3′, 5′ AGCTAGCCACCATGGCTTGAGTAAGTCTGCA 3′ and 5′ TTACACATCCAGCCTCTGTGG 3′) and PTEN (5′ CTCCTCTACTCCATTCTTCCC 3′ and 5′ ACTCCCACCAATGAACAAAC 3′). The murine primary cell lines TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4 were established from thyroid tumors from TPO-Cre;BrafV600E;Trp53R172H mice (TBP) and TPO-Cre;LSL-BrafV600E;PTEN+/fl (TBPt) mice, respectively. Tumors were dissociated by mincing and enzymatic digestion in Hank’s balanced salt solution with 0.5 mg/mL Collagenase type I-S (Sigma-Aldrich), 0.4 mg/mL Dispase II protease (Sigma-Aldrich) and 4% trypsin (0.25% in Tris saline) for 1 hour at 37°C with gentle shaking and repeated, gentle pipetting. After filtering through a 70 µM cell strainer, dissociated cells were plated on standard cell culture plates in Dulbecco’s modified Eagle’s medium DMEM with 10% heat-inactivated fetal bovine serum (FBS) (Gibco), 60 µg/mL penicillin, 100 µg/mL streptomycin and 0.1 mg/mL Primocin (InvivoGen). Four or five subcultures were done every 0.5–1.5 hours, transferring the medium with cells still not attached in order to perform a partial purification. Most purified subcultures were chosen by genotyping the mutated Braf-floxed allele derived from the Cre-Lox recombination technology28 by PCR and western blotting showing expression of BRAFV600E protein. All cell lines were regularly tested for mycoplasma using eMyco Plus Mycoplasma PCR Detection Kit (iNtRON Biotechnology). Human (8505 c, C643) and murine (TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4) thyroid cancer cell lines were used in this study. The murine melanoma cell line 4434 (a gift from Richard Marais, CRUK Manchester Institute) was used as positive control for the BRAF PCR. Human cells were cultured RPMI 1640 medium and murine cells in DMEM, supplemented with 10% heat-inactivated FBS and 60 µg/mL penicillin, 100 µg/mL streptomycin and 0.1 mg/mL primocin (InvivoGen). Human cell lines were authenticated by short tandem repeat analysis using GenePrint 10 System (Promega) following the manufacturer’s instructions and using in-house sequencing facilities. TC1 cells, a modified mouse lung epithelial cell line transformed with HPV-16 E6 and E7 and oncogenic HRAS, were a kind gift from Professor Tzyy-Choou Wu (Johns Hopkins University, Baltimore, Maryland, USA) and Professor Eric Deutsch (Institute Gustave Roussy, Villejuif, France).
Viruses, compounds
The BRAF inhibitor PLX4720 was purchased from 3wayPharm and dissolved in dimethyl sulfoxide (DMSO). RP1 is a herpes simplex virus type 1 that was provided from Replimune. The titer of RP1 was determined by plaque-forming assays using Vero cells. Two versions of RP1 viruses were used in these studies: RP1-19 (oHSV) encoding murine granulocyte/macrophage-colony stimulating factor (GM-CSF) and RP1-24 (oHSV-GFP) encoding green fluorescent protein (GFP).
Histology and immunohistochemistry
Mouse tumors were formalin-fixed, processed and subsequently stained with H&E.
Proliferation assays, virus infection assays, crystal violet survival assays and western blotting
For proliferation assays, 2000 cells were plated into 96-well plates and, after overnight incubation, treated with serial dilutions of oHSV or vehicle control (phosphate-buffered saline (PBS)) in triplicate as indicated. After 72 hours of incubation, viability was measured using the CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega) following the manufacturer’s instructions. Relative survival was normalized to controls and fitted a sigmoidal dose–response (variable slop) curve using GraphPad Prism. For virus infection assays, repeated at least twice, 600,000 cells were plated into 6-well plates and treated with oHSV-GFP or vehicle control (PBS) as indicated. After 24 or 48 hours, images were taken using Evos FL Cell Imaging System (Thermo Fisher Scientific) and cells were collected, stained with viability dye eFluor 780 (eBioscience) for 30 min and fixed with 2% PFA overnight at 4°C. GFP quantification was analyzed by flow cytometry as described below. For crystal violet survival assays, 100,000 cells were seeded into 24-well plates or 600,000 cells into 6-well plates and treated with BRAF inhibitor (PLX4720) or vehicle control (DMSO) and oHSV or vehicle control (PBS) as indicated. Cell survival was determined using crystal violet staining as previously described.32 The crystal violet cell viability assay is commonly used on adherent cells for visualization of live cells by staining DNA and protein in a deep purple color. The different shades of violet correlate to numbers of alive cells. Dead cells are washed off and create non-stained acellular dead zones. For western blotting, lysates were obtained directly on the culture surface with Cold Spring Harbor buffer (1% NP40) containing protease (Complete, Roche) and phosphatase inhibitors (phosphoSTOP, Roche) and subjected to protein quantification (BCA; Thermo Scientific) prior to western blotting. The following antibodies were used: BRAF V600E (RM8) (RevMab Bioscience), phosphorylated ERK1/2 (Thr183 and Tyr185) (Sigma M8159), ERK2 (Santa Cruz C-14), α-tubulin (Sigma T5168).
Ex vivo restimulation of splenocytes
Spleens were excised from euthanized mice, kept on ice-cold PBS and dissociated in vitro to single cell suspensions through a 0.7 μm cell strainer with cold media IMDM (Gibco) with 2 mM L-Glutamine, 5% FBS, 24 µg/mL penicillin and 40 µg/mL streptomycin. Red blood cells were lysed incubating for 2 min with cold ACK buffer (Thermo Fisher) at room temperature. A final cell suspension was cultured in IMDM medium (Gibco) with 50 U/mL recombinant human interleukin 2 (IL-2), 2 mM L-Glutamine, 5% FBS, 24 µg/mL penicillin and 40 µg/mL streptomycin. In parallel, the same thyroid tumor cells TBP-B79 used in the model of the experiment (as described in in vivo experiments) and a non-related cancer cell line TC1 were treated with 1 µM of BRAF inhibitor (PLX4720) or vehicle control (DMSO) and multiplicity of infection (MOI) =1 of virus or vehicle control (PBS) for 48 hours, stimulated with 100 ng/mL of recombinant murine interferon gamma (IFNγ) (First Link UK) for 24 hours and washed prior the ex vivo stimulation of the splenocytes. After 1 hour of incubation, 1 µL/mL of protein transport inhibitor (BD GolgiPlug, Biosciences) was added to the ex vivo stimulation, and after 6 hours more of incubation, cells were collected, stained and analyzed by flow cytometry as described in the next section. Two-way analysis of variance (ANOVA), Tukey’s multiple comparison test was performed using Prism Software (GraphPad).
Cell isolation from tissues and flow cytometry analysis
Tumors and tumor-draining lymph nodes (tdLNs) were dissected from experimental mice. Tumors were mechanically dissociated with scissors and enzymatic digested with RPMI 0.5 mg/mL Collagenase type I-S (Sigma-Aldrich), 0.4 mg/mL Dispase II protease (Sigma-Aldrich), 0.2 mg/mL DNase I (Roche) and 4% trypsin (0.25% in Tris saline) for 30 min at 37°C with gentle shaking. Resulting cell suspension was passed through a 70 µm cell strainer with RPMI 5% FBS and 5 mM EDTA. tdLNs were directly mushed through a 70 µm cell strainer with RPMI 5% FBS. Resulting samples were divided in equals parts for each antibody panel set into 96-well round bottom plates and incubated with a mouse Fc blocker (BD Pharmingen) 2.5 µg/mL for tumor samples and 1 µg/mL for lymph node samples at 4°C for 10 min. After, cells were extracellularly stained with relevant antibodies (online supplementary table) in FACS Buffer (PBS 5% FBS) for 1 hour at 4°C, permeabilized when necessary with Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for 30 min at 4°C and intracellularly stained with relevant antibodies (online supplementary table 1) in permeabilization buffer 10% FACS Buffer for 30 min at 4°C. All samples were fixed with IC fixation buffer (eBioscience) for 20 min at room temperature and analyzed the following day using LRSII flow cytometer (BD Bioscience), using 123count eBeads Counting Beads (Invitrogen) for the quantification. Resulting data were analyzed using FlowJo software, and statistical analysis was performed using non-parametric Kruskal-Wallis test/Dunn’s multiple comparisons using Prism Software (GraphPad).
jitc-2020-000698supp001.pdf (69.8KB, pdf)
Gene expression
Tumor-bearing animals were treated as indicated and on treatment day 11, mice were sacrificed and tumors were dissected and placed in RNAlater RNA stabilization reagent (Qiagen). After disruption and homogenization of the whole tumors in lysis buffer Buffer RLT (Qiagen) using Precellys 24 homogenizer (Bertin Technologies), RNA was isolated using RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. RNA concentration was determined by NanoDrop, and 80–120 ng of each sample was used for the analysis using the nCounter PanCancer Immune Profiling Panel (NanoString Technologies). Raw NanoString data were preprocessed using R package NanoStringNorm (V.1.2.1).33 Genes were assessed for differential abundance using voom (TMM normalization) with R package limma (V.3.34.9).34 Genes with log2 fold change >1 and adjusted p value<0.1 were considered significant. NanoString analyses were performed in R statistical programming language (V.3.4.4). Visualizations were generated using in-house plotting libraries implemented in R.
In vivo experiments
For orthotopic injections, TBP-B79 cells (5×105 cells) were injected unilaterally into the left thyroid gland of C57Bl/6 mice that were 6–8 weeks old (Charles River) using surgical methods and a Hamilton syringe. After inoculation of cells for 8–10 days, mice were randomized and treatment started. Mice received 40 mg/kg of BRAF inhibitor (PLX4720) or vehicle control (5% DMSO in water) by oral gavage daily. For virus injections, tumors were exposed using surgical methods and the virus was injected in a single injection into the tumor using a Hamilton syringe. A survival experiment was performed by utilizing humane endpoints (daily monitoring, >20% wt loss, palpable tumors, respiratory distress, poor general condition). For subcutaneous injections, TBP-B79 or TBP-67 cells (3×106 cells) were injected in the right flank of C57Bl/6 females that were 6–8 weeks old. Treatments commenced when tumors reached a tumor volume of 75–100 mm3. BRAF inhibitor (40 mg/kg) was given daily throughout the experiment by oral gavage. 5×105 pfu of virus was injected in 30–50 μL intratumorally for a total of three injections as shown in schematic pictures of each figure. 150 μg of anti-CTLA-4 (clone 9H10) and 200 μg anti-PD-1 (clone RMP1-14) were given using intraperitoneal injections two to three times per week. Tumors were measured twice weekly and tumor volumes were calculated using the formula: length × width × height (mm)× 0.5236. One-way analysis of variance (ANOVA) on area under curve (AUC) was performed using Prism Software (GraphPad) to compare tumor growth curves. For survival experiments, tumors were allowed to reach 15 mm in any dimension and then sacrificed. The Kaplan-Meier survival curves were compared using the log-rank (Mantel-Cox) test using Prism Software (GraphPad).
Immune cell depletion experiments
Tumor-bearing animals were treated with BRAFi, oHSV and depletion antibodies. One hundred and fifty micrograms of in vivo depletion antibodies was injected via intraperitoneal injection every second day, starting 1 day prior to BRAF inhibition treatment, with a 2-day break between the second and third dose. A total of 11 injections were given. The following antibodies were used: anti-CD8 (clone 2.43), anti-CD4 (clone GK1.5), anti-NK1.1 (clone PK136) and isotype control (clone LTF-2) (InVivoMab, BioXCell). The depletion experiment was performed one time (n=6 mice per group).
Results
Establishment and characterization of BRAF-mutant murine thyroid cancer models and the immunological effects of single modality BRAF inhibition or oncolytic virus treatment
In order to mimic BRAF-mutant human advanced thyroid cancer in immunocompetent mouse models, we used Cre recombinase technology to express BRAFV600E and Trp53R172H or loss of PTEN selectively in the thyrocytes of C57Bl/6 mice. Consistent with previously published studies, we showed that these transgenic animals developed thyroid tumors within 6–8 weeks (figure 1A). Thyroid cancer cell lines derived from these tumors were validated as BRAF mutant using PCR and western blotting (figure 1B, C). Furthermore, using western blotting and proliferation assays, we showed that these cell lines responded to BRAF inhibitors in vitro (figure 1D, E). Importantly, these thyroid cancer cell lines engrafted in immunocompetent C57Bl/6 mice both orthotopically (figure 1F) and subcutaneously, and formed tumors that histologically resembled ATC. The tumors presented with morphology essentially identical to ATC in human patients composed of sheets of markedly atypical cells with large, pleomorphic, ovoid to spindled vesicular nuclei with clumped, uneven chromatin, and large nucleoli, and with numerous apoptotic bodies (figure 1G). On daily treatment with a BRAFi, mice bearing orthotopic TBP-B79 tumors survived significantly longer than vehicle-treated controls; however, all mice had to be culled within 30 days due to disease progression (figure 1H). To address the immune consequences of BRAFi treatment in this model, we performed flow cytometry analysis of orthotopic tumors, which revealed limited immune effects from treatment (online supplementary figure 1A). Four days of daily BRAF inhibition did not increase numbers of T cells or the CD8:Treg ratio within tumors (data not shown), nor did this treatment result in a significant increase in proliferation (as assessed by Ki67) of effector CD4+ T cells (Teff), regulatory CD4+ T cells (Treg) or CD8+ T cells in tumors (online supplementary figure 1B). Granzyme B (GzmB) positivity was used as a measurement of T cell functional activity, but this again did not significantly change (online supplementary figure 1C). Taken together, these data led us to hypothesize that the positive effects of BRAF inhibition on tumor growth could be further improved by boosting antitumor immunity in the context of BRAF inhibitor cytotoxicity, using oncolytic virotherapy. Therefore, we next tested whether oHSV could stimulate the immune tumor microenvironment (TME) within orthotopic thyroid tumors (online supplementary figure 1D). A single intratumoral injection of oHSV increased both the proliferation and activation of CD8+ T cells, supporting the application of combined oHSV/BRAFi therapy as a potentially immunogenic, as well as directly cytotoxic, strategy for thyroid cancer (online supplementary figure 1E and F).
Figure 1.
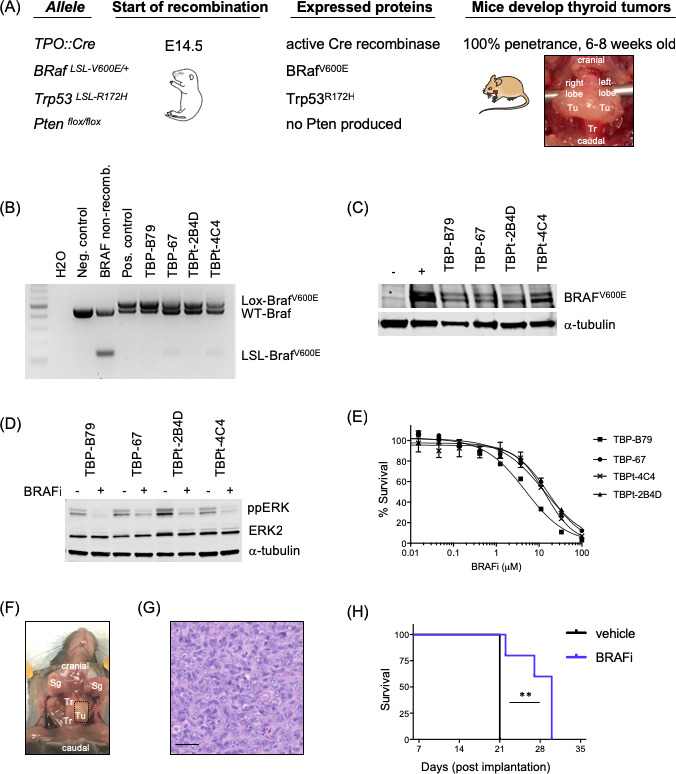
Establishment and characterization of murine thyroid cancer models. (A) Using transgenic mice on a C57Bl/6 strain background, we developed a thyroid cancer mouse model driven by oncogenic BRAF (BRAFV600E). These mice were further bred with mice carrying Trp53 mutation or PTEN deletion to develop aggressive anaplastic thyroid cancer (ATC). Site-specific expression of transgenes was obtained by Cre recombinase/loxP technology and a thyroid-specific promoter, thyroid peroxidase, TPO:Cre and recombination started at E14.5. Mice carrying activated transgenes developed tumors exclusively in the thyroid lobes. These tumors were dissected and grown into thyroid cancer cell lines in vitro and used for further in vitro analysis or for in vivo orthotopic or subcutaneous injections into immunocompetent C57Bl/6 mice. Generated cell lines were subsequently named based on which mutations were induced in the corresponding mice: TPO:BRAF:Trp53=TBP and TPO:BRAF:Pten=TBPt. Photograph showing representative whole thyroid tumor of TPO:BRAF:Trp53 mouse. Tu, tumor; Tr, trachea. (B) Locus recombination of the BRAFV600E transgene in established murine thyroid cancer cell lines was confirmed by PCR. The murine melanoma BRAF wild-type cell line 1014 was used as negative control, and the murine melanoma BRAFV600E mutant cell line 4434 was used as positive control of the Cre recombinase rearranged LSL-BrafV600E allele. Non-recombined DNA from a BRAFV600E mouse served as control for band sizes of wild-type BRAF (WT-Braf) and the LSL-BrafV600E allele.28 (C) Established murine thyroid cancer cell lines (TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4) were confirmed to express oncogenic BRAFV600E by western blot analysis. Lysates from human C643 (BRAF wild-type) and murine 4434 (BRAF V600E mutant) cells were used as negative and positive controls, respectively. Tubulin served as loading control. (D) Immunoblots for ppERK, total ERK and α-tubulin in murine thyroid cancer cell lines (TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4) following treatment with DMSO control or BRAFi (1 µm, 4 hours). (E) Proliferation dose–response curves for murine thyroid cancer cell lines (TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4) treated with BRAFi over a period of 72 hours. Cell survival was measured with CellTitreGlo and normalized to DMSO-treated controls. (F) Representative photograph of unilateral thyroid tumor (dotted box) following injection of the thyroid cancer cell line TBP-B79 into the left thyroid lobe, just adjacent to the trachea of a C57Bl/6 mouse and left to grow for 4 weeks. Tu, tumor; Tr, trachea; Sg, salivary gland. (G) H&E stained photomicrograph of TBP-B79 tumor growing in the thyroid of a C57Bl/6 mouse. Scale bar=50 µm. This shows hypercellular tumor, composed of sheets of markedly atypical cells with large, pleomorphic, ovoid to spindled vesicular nuclei with clumped, uneven chromatin, and large nucleoli. Apoptotic bodies are numerous. The morphology here is essentially identical to ATC in human patients, which typically manifests histologically as sheets of markedly atypical spindle cells with the nuclear features described. (H) The thyroid cancer cell line TBP-B79 was injected unilaterally into the left thyroid lobe of C57Bl/6 mice. After tumors were established, mice were treated by oral gavage with vehicle or BRAF inhibitor (40 mg/kg) daily throughout the experiment. Kaplan-Meier survival graph displaying the survival of the animals treated with vehicle control or BRAF inhibitor (survival=palpable measurable neck mass of maximum 10 mm combined with clinical signs of disease). Statistical analysis: survival curve comparison using log-rank (Mantel-Cox) test. (**p=0.0027) was performed using Prism Software (GraphPad). BRAFi, BRAF inhibitor.
jitc-2020-000698supp002.pdf (116.6KB, pdf)
jitc-2020-000698supp005.pdf (46.6KB, pdf)
Combining oHSV and BRAFi enhances antitumor activity in vivo, but not in vitro
Subsequently, we set out to test if the combination of the two different treatments would act synergistically. Using both human and murine thyroid cancer cell lines, we observed tumor cell infection and cell killing by oHSV in vitro in a dose-dependent manner (figure 2A–C). However, using two murine cell lines, the addition of BRAFi to oHSV in vitro did not further increase the effect of the virus alone (figure 2D, E). Despite the lack of synergistic effects in vitro, we decided to test the combination in vivo in immunocompetent mouse models, since our hypothesis was that oHSV might improve therapy by enhancing the immunogenicity of tumor cell death, rather than the direct cytotoxicity of BRAF inhibition. Since oHSV is administered intratumorally and we were limited to one oHSV injection into the orthotopic model (due to animal welfare constraints in the context of a tumor seeded in the neck), we used a subcutaneous tumor model for these, and subsequent, experiments. Furthermore, subcutaneously implanted tumors also allow easier monitoring and measurement of tumor growth in therapy experiments. Importantly, we compared the immune cell infiltrate in response to short-term (2 days) oHSV treatment in tumors seeded orthotopically or in the flank, and found no significant differences between them. Using the TBP-B79 model in immunocompetent mice, tumor-bearing animals were treated as outlined in figure 3A. Combination treatment with oHSV and BRAF inhibition delayed tumor growth and increased median survival, which was 22.5 days (vehicle), 25 days (oHSV), 28 days (BRAFi) and 37.5 days (oHSV/BRAFi) (figure 3B, C). Consistent with the lack of synergistic effects of combination treatment in vitro, on measurement by ex vivo plaque assays, we did not retrieve increased amounts of virus from oHSV/BRAFi-treated tumors relative to the dose injected, or compared with those treated with oHSV only (data not shown). This indicates that the enhanced therapy seen with the combination in vivo was unlikely due to increased viral replication, and more likely to be immune-mediated. To explore further the mechanisms underlying combination therapy, we assessed whether single modality or combination treatment resulted in adaptive T cell priming, by restimulating splenocytes from treated mice ex vivo with TBP-B79 cells, or an irrelevant tumor cell line control. Restimulation of splenocytes from oHSV/BRAFi-treated mice with TBP-B79 induced significant intracellular IFNγ in responder CD8+ splenic T cells, consistent with priming of a specific antitumor cytotoxic T cell response following combination therapy (figure 3D).
Figure 2.
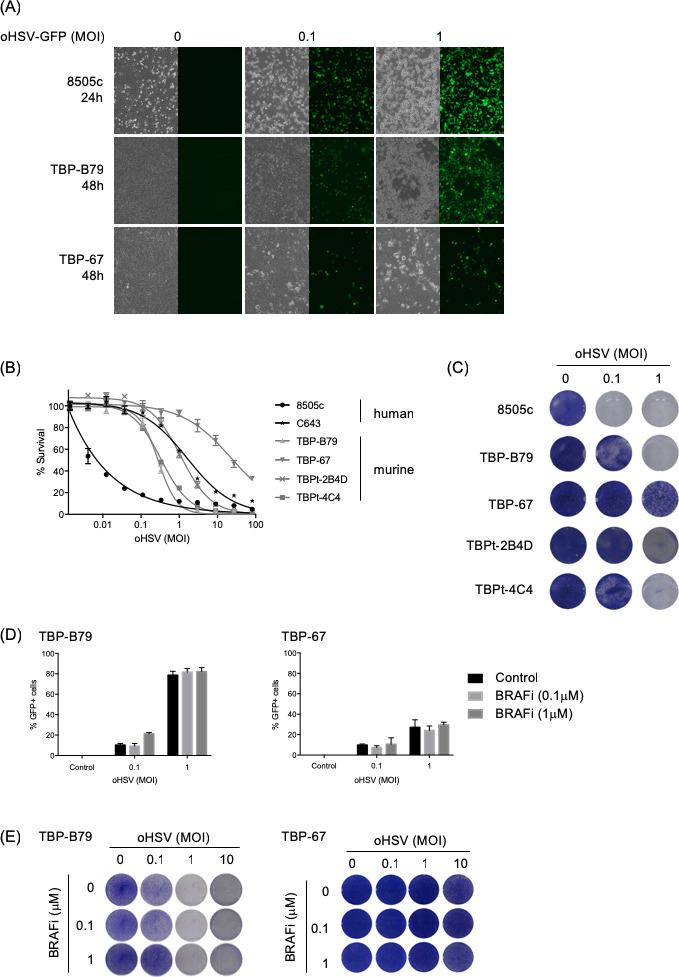
In vitro effects of oHSV on human and murine thyroid cancer cell lines. (A) Human (8505 c) and murine (TBP-B79 and TBP-67) thyroid cancer cell lines were infected with oHSV-GFP at various MOIs and cells were imaged for virus replication (visualized by GFP signal) at indicated time points post-infection. Corresponding bright-field images show cell morphology changes following virus infection. (B) Human (8505 c and C643) and murine (TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4) thyroid cancer cell lines were infected with 1:3 dilutions of oHSV starting at MOI 100 or left uninfected as controls and cell survival was measured 72 hours later using CellTitreGlo assay. (C) Human (8505 c) and murine (TBP-B79, TBP-67, TBPt-2B4D and TBPt-4C4) thyroid cancer cell lines were infected with oHSV (MOI 0.1 or 1) or left uninfected and cells were fixed and stained with crystal violet 48 hours later. (D) Murine thyroid cancer cell lines (TBP-B79 and TBP-67) were infected with oHSV-GFP (MOI 0.1 or 1) or left uninfected, and subsequently treated with BRAF inhibitor (BRAFi) (0.1 or 1 µM). Cells were analyzed for GFP positivity (virus infection) using flow cytometry 48 hours post-treatment. (E) Murine thyroid cancer cell lines (TBP-B79 and TBP-67) were infected with oHSV at various MOI and treated with BRAFi at indicated concentrations. Cells were fixed and stained with crystal violet 48 hours later. Each experiment was performed a minimum of two times with similar results. BRAFi, BRAF inhibitor; GFP, green fluorescent protein; oHSV, oncolytic herpes simplex virus.
Figure 3.
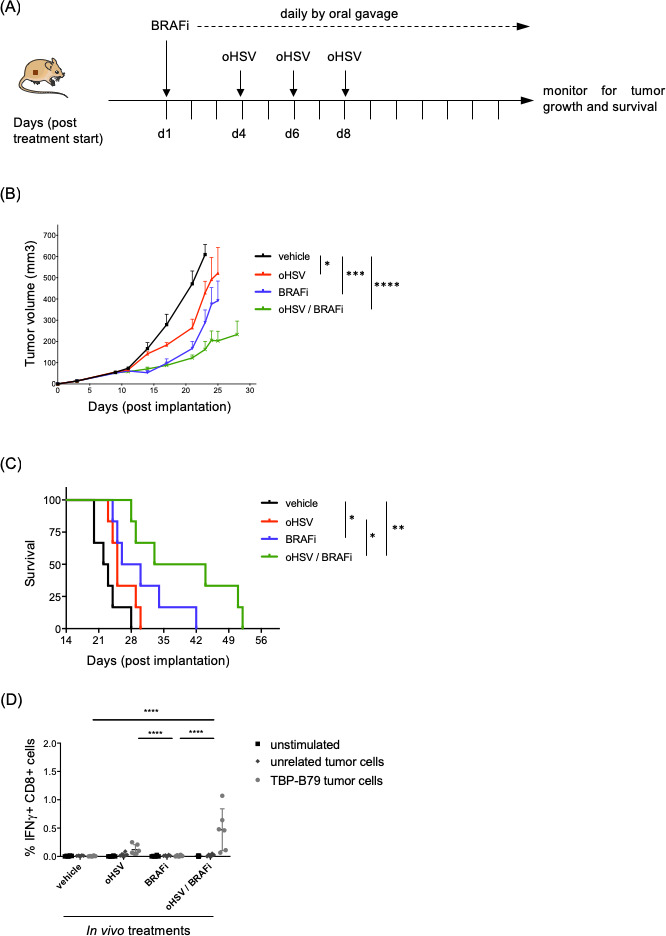
Combination treatment with oHSV/BRAFi delays tumor growth and increases survival in an immunocompetent mouse model of thyroid cancer. (A) C57Bl/6 mice (n=6/group) were subcutaneously implanted with murine thyroid cancer cell line TBP-B79 in the right flank (3×106 cells/mouse) and mice were treated with single agent BRAFi (40 mg/kg daily by oral gavage), oHSV (5×105 pfu × 3 by intratumoral injection) or both agents in combination, starting on day 10 post implantation of cells. (B) Tumor growth curves with data presented as mean±SEM. Tumor growth curves were compared using ordinary one-way analysis of variance (ANOVA) on AUC (area under curve) between day 0 and day 23 using Prism Software (GraphPad)=vehicle vs oHSV: *Vehicle vs BRAFi: ***Vehicle vs oHSV/BRAFi: ****oHSV vs BRAFi: ns, oHSV vs oHSV/BRAFi: *BRAFi vs oHSV/BRAFi: ns. (C) Kaplan-Meier survival graph displaying the survival of the animals treated with different therapies or vehicle control (survival=a tumor measures 15 mm in any dimension). Median survival: 22.5 days (vehicle), 25 days (oHSV), 28 days (BRAFi) to 37.5 days (oHSV/BRAFi). Kaplan-Meier survival curves were compared using the log-rank (Mantel-Cox) test using Prism Software (GraphPad)=vehicle vs oHSV: ns, vehicle vs BRAFi: *p value 0.0165, vehicle vs oHSV/BRAFi: **p value 0.0011, oHSV vs BRAFi: ns, oHSV vs oHSV/BRAFi: * p value 0.0111, BRAFi vs oHSV/BRAFi: ns. (D) Ex vivo IFNγ memory recall assay. Intracellular IFNγ production, measured by flow cytometry, in CD8+ splenocytes using flow cytometry was used to measure immune cell activation and recall responses towards live TBP-B79 tumor cells. The unrelated murine cell line TC1 was used as control. Statistical analysis: two-way analysis of variance, Tukey’s multiple comparison test was performed using Prism Software (GraphPad). Each experiment was performed twice with similar results. BRAFi, BRAF inhibitor; IFNγ, interferon gamma; ns, not significant; oHSV, oncolytic herpes simplex virus.
Gene expression profiling highlights an important role for activated T cells in oHSV/BRAFi treatment, and suggests the PD-1/PD-L1 axis and CTLA-4 as targets for triple combination therapies
Although we saw effective combination therapy with oHSV/BRAFi in terms of thyroid tumor growth inhibition in vivo, we did not achieve full tumor control and resistance to treatment emerged relatively rapidly. Therefore, gene expression profiles of treated tumors were analyzed using NanoString nCounter technology to more fully characterize the immune microenvironment, covering tumor, stromal and immune components, in an attempt to identify further targets for additional combination strategies. Mechanistically, gene expression data and subsequent cell type clustering of differentially expressed genes suggested that the improved therapy conferred by oHSV/BRAFi combination was mediated through activation of T cells (online supplementary figure 2A and B). Importantly, the gene expression analysis also identified potential targets for further immunotherapy interventions. Specifically, oHSV/BRAFi treatment significantly upregulated both PD-L1 and PD-L2, as well as CTLA-4, providing rationale for the addition of PD-1/PD-L1 or CTLA-4 blockade to oHSV/BRAFi to further improve therapy (online supplementary figure 2B). In contrast, no significant changes were seen in the expression of the other potentially targetable negative immune checkpoint molecules, namely, TIGIT, LAG-3 and TIM-3 (data not shown).
jitc-2020-000698supp003.pdf (58.4KB, pdf)
Both innate and adaptive immunity contribute to the efficacy of oHSV/BRAFi combination treatment through activation of immune effector cells in tumors and in draining lymph nodes
Next, we wanted to explore the ability of combination oHSV/BRAFi treatment phenotypically to activate immune cell subsets. This analysis allowed us to consolidate the flow cytometry data shown for single modality treatment in the orthotopic model shown in online supplementary figure 1, and to test consistency between immune cell flow cytometry characterization, and the transcriptomic data of online supplementary figure 2. Moreover, while direct intratumoral injection of oHSV is currently the only approved oncolytic virotherapy, it remains unclear to what extent the immune consequences of viral administration by this route are restricted to the TME, or whether they are also seen locoregionally, particularly in tdLNs. We, therefore, treated TBP-B79 tumor-bearing mice with oHSV, BRAFi or the combination (figure 4A) and characterized the immune cells within both tumors and tdLNs using flow cytometry. With respect to the innate response, we found an small increase in the percentage of NK cells within CD45+ cells following combination treatment in tumors but not in tdLNs (figure 4B, C). However, NK cells in both tumors and tdLNs were activated by treatment, as measured by their expression of Ki67, GzmB and CD69 (figure 4B, C).
Figure 4.
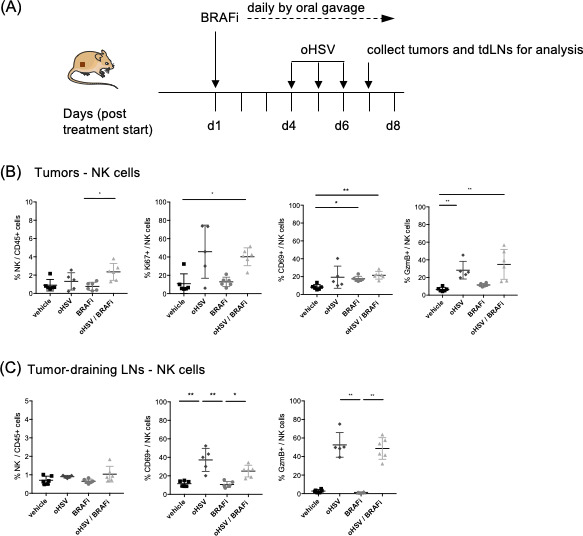
Recruitment and activation of natural killer (NK) cells in tumors. C57Bl/6 mice (n=6/group) were subcutaneously implanted with the murine thyroid cancer cell line TBP-B79 in the right flank (3×106 c/mouse) and mice were treated with BRAFi (40 mg/kg daily by oral gavage) and oHSV (5×105 pfu × 3 injections by intratumoral injection) as single agents or in combination. On treatment day 7, tumors were dissected and tumor-infiltrating lymphocytes (TILs) were analyzed via flow cytometry. (A) Treatment and collection schedule for TBP-B79 tumor model. (B) Percentage of NK cells/CD45+ cells and percentage of Ki67+, CD69+, GzmB+ NK cells in tumors. (C) Percentage of NK cells/CD45+ cells and percentage of CD69+ and GzmB+ NK cells in tumor-draining lymph nodes. Each dot represents an individual mouse. Statistical analysis: non-parametric Kruskal-Wallis test/Dunn’s multiple comparisons were performed using Prism Software (GraphPad). Comparison with p values over 0.05 are deemed not statistically significant. (NK cells: CD45+, CD3-, NK1.1+). The experiment was performed twice with similar results. BRAFi, BRAF inhibitor; oHSV, oncolytic herpes simplex virus; LN, lymph nodes.
With regard to T cells, mice were treated as indicated in figure 5A, and the CD8:Treg ratio in the tumor increased with treatment, as did proliferation (Ki67+), GzmB expression and CXCR3 expression of CD4+ effectors, CD4+ Tregs and CD8+ T cell subsets (figure 5B–E). This was consistent with granzymes and CXCR3 as prominent hits of differentially expressed genes in the tumor transcriptomic data (online supplementary figure 2). PD-1 expression was increased by treatment on tumor CD8+ T cells, but decreased on Treg cells, and was unchanged on CD4+ effectors (figure 5F), while CTLA-4 increased on CD4+ effectors and CD4+ Tregs, but was unchanged on CD8+ cells (figure 5G). In general, the changes in the tumors were mirrored by T cells in the tdLNs; in particular, PD-1 expression increased on CD4+ effectors as well as CD8+ cells, although CTLA-4 increased significantly only on CD4+ effector cells (figure 5H–K). Furthermore, consistent with the gene expression data, we did not detect any changes in TIGIT, LAG-3 or TIM-3 on lymphocytes in tumors (data not shown). Importantly, the CD8:Treg ratio and activation status of T cells in tumors and tdLNs were further validated in a second tumor model using TBP-67 cells (online supplementary figure 3A–I). Taken together, these effects of combination oHSV/BRAFi therapy on T cell activation were consistent with the tumor transcriptomic data and support the targeting of PD-1 and CTLA-4 to further improve therapy.
Figure 5.

Combined treatment with oHSV/BRAFi activates T cells and depletion of CD8+ T cells or natural killer (NK) cells abrogates the effects on tumor growth. C57Bl/6 mice (n=6/group) were subcutaneously implanted with the murine thyroid cancer cell line TBP-B79 in the right flank (3×106 c/mouse) and mice were treated with BRAFi (40 mg/kg daily by oral gavage) and oHSV (5×105 pfu × 3 injections by intratumoral injection) as single agent or in combination. On treatment day 11, tumors and tumor-draining lymph nodes were dissected and lymphocytes analyzed via flow cytometry. (A) Treatment and collection schedule for TBP-B79 tumor model. (B) Ratio of tumor-infiltrating lymphocytes (TILs) (CD8/CD4reg). (C) Percentage of Ki67+ CD4 eff, CD4reg or CD8+ cells. (D) Percentage of GzmB+ CD4 eff, CD4reg or CD8+ cells. (E) Percentage of CXCR3+ CD4 eff, CD4reg or CD8+ cells. (F) Percentage of PD-1+ TILs: CD4eff, CD4reg and CD8+ T cells. (G) Percentage of CTLA-4+ TILs: CD4eff, CD4reg and CD8+ T cells. (H) percentage of Ki67+ CD4 eff, CD4reg or CD8+ cells in tumor-draining lymph nodes. (I) Percentage of CXCR3+ lymphocytes in tumor-draining lymph nodes: CD4eff, CD4reg and CD8+ T cells. (J) Percentage of PD-1+ lymphocytes in tumor-draining lymph nodes: CD4eff, CD4reg and CD8+ T cells. (K) Percentage of CTLA-4+ lymphocytes in tumor-draining lymph nodes: CD4eff, CD4reg and CD8+ T cells. Each dot represents an individual mouse. Statistical analysis: non-parametric Kruskal-Wallis test/Dunn’s multiple comparisons were performed using Prism Software (GraphPad). Comparison with p values over 0.05 is deemed not statistically significant. Teff (CD4+/FoxP3-) and Treg (CD4+/FoxP3+). The experiment was performed four times with similar results. (L) C57Bl/6 mice (n=6/group) were subcutaneously implanted with murine thyroid cancer cell line TBP-B79 in the right flank (3×106 cells/mouse) and mice were treated with single agent BRAFi (40 mg/kg daily by oral gavage), oHSV (5×105 pfu × 3 by intratumoral injection) or both agents in combination. D.Ab: depletion antibodies (isotype, aCD8, aNK1.1, aCD4) were administered every second day 150 μg by intraperitoneal injection for a total of 11 injections. (M) Tumor growth curves showing tumor volume over time. Tumor growth curves were compared using ordinary one-way analysis of variance (ANOVA) on AUC (area under curve) between day 0 and day 41 using Prism Software (GraphPad). oHSV/BRAFi/isotype vs oHSV/BRAFi/aCD4: ns, oHSV/BRAFi/isotype vs oHSV/BRAFi/aCD8: **adj. p value 0.0093, oHSV/BRAFi/isotype vs oHSV/BRAFi/aNK1.1: **adj. p value 0.0057, oHSV/BRAFi/aCD4 vs oHSV/BRAFi/aCD8 ns, oHSV/BRAFi/aCD4 vs oHSV/BRAFi/aNK1.1 ns, oHSV/BRAFi/aCD8 vs oHSV/BRAFi/aNK1.1 ns. BRAFi, BRAF inhibitor; ns, not significant; oHSV, oncolytic herpes simplex virus; LN, lymph node.
jitc-2020-000698supp006.pdf (62.8KB, pdf)
To confirm the contribution of the innate and adaptive arms of the immune response to the in vivo efficacy of oHSV/BRAFi combination therapy, we tested the dependence of therapy on specific immune cell subsets. TBP-B79 tumor-bearing mice were treated with oHSV/BRAFi and depleted of NK cells, CD4+ T cells or CD8+ T cells (figure 5L). This showed that depletion of either NK cells or CD8+ T cells significantly reduced the antitumor efficacy of oHSV/BRAFi treatment compared with isotype control-treated mice (figure 5M). In addition, depletion of CD4+ T cells showed a similar trend, although this did not reach statistical significance (figure 5M).
Blockade of either PD-1 or CTLA-4 further enhances the therapeutic efficacy of oHSV/BRAFi therapy
To investigate if the upregulation of the PD-1/PD-L1 axis and CTLA-4 in tumors and tdLNs (online supplementary figures 2 and 3 and figure 5) could be therapeutically tractable, we set up triple combination experiments including oHSV, BRAFi and anti-PD-1 or anti-CTLA-4 (figure 6A). Since both PD-L1 and PD-L2 were transcriptionally upregulated following combination therapy, we chose to use an anti-PD-1 antibody to target both immune checkpoints in our experiments. Both triple combinations significantly reduced TBP-B79 tumor growth and prolonged survival compared with single or double-agent combinations (figure 6B, C and online supplementary figure 4A‒E). We were not able to address whether the combination of anti-PD1 and anti-CTLA-4 was better than either antibody alone within this triple modality combination, because blockade of even a single negative immune checkpoint resulted in almost complete tumor control in this model. We did not see any weight loss or other toxicity in mice treated with any single, double or triple combination therapies (data not shown). Using the treatment regimen outlined in figure 6A, five out of six animals in each of the triple combination groups were cured of their tumors. Importantly, when these animals were rechallenged with tumor cells on the contralateral flank, previously cured mice did not develop tumors while all naïve control mice did, suggesting that protective immunity had developed (figure 6D). This is consistent with the CD8+ T cell involvement and dependence shown in online supplementary figures 2 and 3 and in figure 5. In addition, mice treated with triple combination therapy had an increase of intratumoral CD45+ cells per gram of tumor compared with other treatment groups (figure 6E). To investigate if the therapeutic benefit of CTLA-4 blockade might be due to Treg depleting properties of the anti-CTLA-4 antibody we analyzed the numbers of Treg cells in the tumors, and found a significant decrease in intratumoral Tregs in triple combination-treated mice compared with oHSV/BRAFi-treated mice (figure 6F).
Figure 6.

Triple combination with oHSV/BRAFi and anti-PD-1 or anti-CTLA-4 further improves therapeutic responses. (A) C57Bl/6 mice (n=6/group) were subcutaneously implanted with murine thyroid cancer cell line TBP-B79 in the right flank (3×106 c/mouse), and mice were treated with BRAFi (40 mg/kg daily by oral gavage) and oHSV (5×105 pfu × 3 by intratumoral injection) and anti-PD-1 (200 µg, intraperitoneal injection) or anti-CTLA-4 (150 µg, intraperitoneal injection) therapeutic antibodies or relevant controls as indicated. (B) Tumor growth curves with data presented as mean±SEM. Tumor growth curves were compared using ordinary one-way analysis of variance (ANOVA) on AUC (area under curve) between day 0 and day 30 using Prism Software (GraphPad). Vehicle vs oHSV/BRAFi: ****adj. p value<0.0001, vehicle vs oHSV/BRAFi/aCTLA-4: ****adj. p value<0.0001, vehicle vs oHSV/BRAFi/aPD-1: ****adj. p value<0.0001, oHSV/BRAF vs oHSV/BRAFi/aCTLA-4: ns, oHSV/BRAFi vs oHSV/BRAFi/aPD-1: ns, oHSV/BRAFi/aCTLA-4 vs oHSV/BRAFi/aPD-1: ns. (C) Kaplan-Meier survival graph displaying the survival of the animals treated with different therapies. Kaplan-Meier survival curves were compared using the log-rank (Mantel-Cox) test using Prism Software (GraphPad). (D) Mice cured of their tumors were rechallenged on the left flank with 3×106 cells and tumor growth monitored. Treatment groups included different numbers of cured mice: oHSV/BRAFi (n=1), oHSV/BRAFi/aPD-1 (n=5) and oHSV/BRAFi/aCTLA-4 (n=5). Six naïve mice served as controls. Tumor growth curves were compared using ordinary one-way ANOVA on AUC (area under curve) between day 0 and day 20 using Prism Software (GraphPad). Naïve vs oHSV/BRAFi/isotype: ***adj. p value 0.0006, naïve vs oHSV/BRAFi/aCTLA-4: ****adj. p value<0.0001 and naïve vs oHSV/BRAFi/aPD-1: ****adj. p value<0.0001. (E) C57Bl/6 mice (n=6/group) were subcutaneously implanted with the murine thyroid cancer cell line TBP-B79 in the right flank (3×106 c/mouse) and mice were treated with BRAFi (40 mg/kg daily by oral gavage) and oHSV (5×105 pfu × 3 injections by intratumoral injection) and anti-CTLA-4 (150 µg) by intraperitoneal injection as single agent or in combination. On treatment day 11, tumors and tumor-draining lymph nodes were dissected and lymphocytes analyzed via flow cytometry. CD45+ cells/ gram tumor is presented. (F) Percentage of FoxP3+CD4 reg/CD45+ cells are presented. Each dot represents an individual mouse. Statistical analysis: non-parametric Kruskal-Wallis test/Dunn’s multiple comparisons were performed using Prism Software (GraphPad). Comparison with p values over 0.05 is deemed not statistically significant. Statistical analysis of flow cytometry data: two-way ANOVA (Tukey’s multiple comparison test), one-way ANOVA or non-parametric Kruskal-Wallis test followed by Dunn’s multiple comparison test, or unpaired non-parametric Mann-Whitney test were performed depending on sample size and numbers of groups analyzed as indicated in the different experiments using Prism Software (GraphPad). Tumor growth curves are presented as mean±SEM and growth curves compared calculating AUC followed by one-way ANOVA. Kaplan-Meier survival curves were compared with the log-rank (Mantel-Cox) test using Prism Software (GraphPad). BRAFi, BRAF inhibitor; ns, not significant; oHSV, oncolytic herpes simplex virus.
jitc-2020-000698supp004.pdf (47.1KB, pdf)
Discussion
PDTC and ATC are very aggressive forms of cancer and among the most rapidly proliferating tumors in humans. Typically, survival for more than 12 months beyond diagnosis is uncommon. In this study, we describe the establishment of murine ATC cell lines that can be used as tools for studies in immunocompetent models to replicate human disease. These mouse cancer cell lines are syngeneic to C57Bl/6 mice, have genetic alterations consistent with human tumors, are transplantable to immunocompetent host mice in orthotopic or subcutaneous models and can be used to study cancer immunology.
Since kinase inhibitors (including BRAFi) are important drugs in the management of aggressive thyroid cancer, we first showed that BRAF mutant ATC is responsive to BRAFi monotherapy, although only transiently. Since BRAFi in the treatment of melanoma has been shown to be potentially immunostimulatory within the TME,35–38 we tested whether BRAFi monotherapy was immunogenic in our ATC model, but found only limited changes in the immune landscape in orthotopic tumors. In contrast, a single intratumoral injection of HSV did impact on the TME in an orthotopic model, increasing both proliferation and activation of CD8+ T cells within targeted tumors.
PDTC and ATC are attractive candidates for treatment with oHSV as they are readily targetable for direct injection. In addition, we found that thyroid cancer cell lines are infectible with, and killed by, oHSV in vitro, although this sensitivity was not affected by the addition of BRAFi. These results contrast somewhat with a study combining oHSV and MEK inhibition in melanoma cells, in which the investigators detected increased cytotoxicity of oHSV, as well as increased in vitro viral replication, if the cells were pretreated with MEKi.39 It is noteworthy that their studies were performed mainly in human cell lines with much greater sensitivity to the virus alone, while all ours were performed in murine systems. Also, there might be differences in models (melanoma vs thyroid cancer) and differences in viral replication in vivo might occur at specific time points that depend on the model system. Importantly, in our study, the combination of oHSV and BRAFi was more effective in vivo, confirming our hypothesis that the immune-stimulatory capacity of oHSV might enhance the cytotoxic effects of BRAFi alone. The immune-mediated effects of oHSV/BRAFi combination treatment were confirmed by their dependence on NK and T cells and the association with priming of specific, adaptive antitumor immunity.
To further explore the consequences of combined oHSV/BRAFi treatment for thyroid cancer, we carried out gene expression analysis, which includes the contribution of non-immune cells within the TME (including tumor cells and fibroblasts) to the tumor transcriptome. Consistent with the immune-mediated activity of the oHSV/BRAFi combination, we found evidence of NK and T cell activation associated with effective therapy. Many of the significantly upregulated genes (covering innate immunity more widely, including interferons, as well as NK and T cell activation), have been previously published as reflecting ‘T-cell inflamed’ tumors, and overlap with genes upregulated in melanoma patients responding to BRAFi/MEKi/anti-PD-1 treatment.11 40 Transcriptomic analysis of tumors can also be used to identify immune-based treatment targets and our data specifically revealed upregulation of CTLA-4, PD-L1 and PD-L2 in tumors, providing a rationale for triple combination strategies including CTLA-4 and PD-1 blockade.41 42 Importantly, inhibitors of CTLA-4 and PD-1/PD-L1 are frequently used in the clinic, have known and manageable toxicity profiles, and are part of standard clinical care in an increasing range of cancers. Addition of either anti-CTLA-4 or anti-PD-1 antibodies significantly further improved oHSV/BRAFi therapy, resulting in almost complete tumor control in our model. Cured mice were then protected from secondary re-challenge, further validating the immune contribution to effective therapy.
The combination MEKi/HSV/aPD-1 led to increased survival in a mouse melanoma model39 and it would, therefore, be tempting to speculate that similar strategies including MEKi would show therapeutic benefit in BRAF wild-type thyroid cancers. Alternative ways to achieve MAPK pathway blockade in combination with oHSV in thyroid cancer could include double inhibition of BRAF and MEK using either BRAFi/MEKi combinations, which is a standard treatment for BRAF mutant melanoma patients, or pan-RAF inhibitors, to overcome or delay resistance to MAPK pathway inhibition and potentially increase therapeutic benefit in combination with oHSV. However, both panRAF and MEK inhibitors are less tumor cell-specific than BRAFV600E inhibitors, and would potentially negatively affect proliferation of immune cells or other non-tumor cells in the TME. Although potentially interesting, these studies incorporating further small molecule drugs are beyond the scope of the current investigation.
It is interesting that a phase III study in melanoma has shown potential clinical benefit and a favorably altered TME from combining oHSV (TVEC) with anti-PD-1 (pembrolizumab)43 and other clinical trials involving combinations of T-VEC with anti-PD-1 therapy in sarcoma, lung cancer studies, breast and melanoma are ongoing (clinicaltrials.gov). Of interest is a recent paper by Zhu and colleagues who used a modified oHSV2 virus encoding an antibody against PD-1. oHSV2-aPD-1 treatment induced durable antitumor response and had superior therapeutic efficacy over unmodified oHSV2 or aPD-1 blockade alone.44
Analysis focused on innate (NK) and adaptive (CD8+ effectors, CD4+ effectors and CD4+ Treg) tumor-infiltrating immune cells at the protein level by flow cytometry supported the tumor gene expression data. Of note is the fact that increased numbers and activity of NK cells were detected when we analyzed tumors at an early time point. Tumor NK cells were activated by treatment, effects which were shown to be due primarily to oHSV rather than BRAFi, when monotherapy and combination treatment groups were compared. Interestingly, human ATC cells are able to attract CXCR3-positive NK cells and are sensitive to NK cell-mediated lysis.45 The same pattern was seen on activation of T cells within tumors by virus. With regard to tumor T cell checkpoint protein expression, CTLA-4 was increased on CD4+ T effectors and Treg though this change was not clearly associated with oHSV rather than BRAFi treatment. Given the potential role for Treg depletion by anti-CTLA-4 antibodies in immunotherapy,46 we specifically investigated the numbers of Tregs following triple combination treatment (oHSV/BRAFi/anti-CTLA-4) and found fewer Tregs in this treatment context. This suggests that anti-CTLA-4-mediated Treg depletion may play a role in this triple combination treatment regimen, although the various mechanisms by which immune checkpoint blockade may act remain to be fully elucidated in this and other contexts. For example, studies of human samples have not detected depletion of FoxP3+ Tregs by anti-CTLA-4 treatment, although these differences might depend on factors such as the time points available post-treatment for analysis.47 In our study, PD-1 expression showed a virus-mediated increase on tumor CD8+ T cells and reduction on Tregs, despite the lack of significant upregulation in PD-1 expression as assessed by NanoString in whole tumors (log2 fold change 0.38, p value: 0.357). However, consistent with this, we have previously found that PD-1 transcription is not necessarily mirrored by protein expression levels.48 We have not specifically investigated levels of PD-1 on NK or other cells, and we cannot exclude that the therapeutic benefit seen with PD-1 blockade could be due to its effect on cells other than T cells.
Analysis of tdLNs showed very similar changes in NK and T cell activation as those seen in tumors on treatment, suggesting that even local intervention by oHSV injection can modify the locoregional, as well as the tumor, immune microenvironment.49 However, the relative contribution to therapy of tumor versus locoregional immune activation remains unclear. To investigate if local administration of oHSV activates immune effector cells and molecules both in the TME and in the systemic immune system, spleens in addition to tdLNs could be analyzed. Interestingly, oHSV2-aPD-1 treatment has been shown to induce a similar trend of immune activation of splenocytes compared with tumors using NanoString Technology,44 suggesting that localized oHSV application induces activation of immune effectors both in the TME and systemically. This could provide an explanation for the induced antitumor memory detected in our rechallenge experiment. tdLNs are key regulators in antitumor immune responses and analysis of specific memory T cell subpopulations within tdLNs by flow cytometry using markers such as CD44 and CD62L for effector and central memory, would provide further insight into treatment strategies and potential virus-induced therapeutic tumor vaccination effects. Tumor antigens drain primarily to tdLNs through transportation by CD103+ dendritic cells (DCs) which leads to priming of T cells, and these lymph nodes have been shown to control the magnitude of therapeutic efficacy to aPD-1 checkpoint therapy in mouse models.50 Overall, further analysis of the functional role of APC populations in tdLNs (and beyond in the spleen) would be worthwhile, in particular to inform the clinical uncertainty around whether treatment of tdLNs, by surgery and/or radiotherapy, is likely to enhance or restrict priming of effective antitumor immunity. Of note is our finding that, although the NanoString data did not suggest any major contribution of DCs to immune changes in the tumor, flow cytometry analysis of conventional DCs (CD45+/CD11c+/MHCII+) revealed low numbers that decreased in the tumors with treatment at 10 days, while significant upregulation was seen in tdLNs (data not shown). This illustrates how changes in numbers and activation status of various immune cells in both tumor and tdLNs are likely time-point-specific and might vary between models, as well as being influenced by levels of virus affecting the balance between priming of antiviral and antitumoral responses.
Conclusions
Our data support the potential for successful immunotherapy in aggressive, BRAF-mutant thyroid cancer. BRAFi alone instigated limited changes only in the immune TME, but these were significantly boosted, and associated with effective therapy, on addition of immunostimulatory intratumoral oncolytic HSV injection. Gene expression analysis and flow cytometry profiling of tumors and draining lymph nodes confirmed activation of NK cell and T cell immunity by oHSV/BRAFi treatment, which was predominantly virus-driven, and directed successful incorporation of anti-CTLA-4 or anti-PD-1 into triple combination treatment regimens. Combining the cytotoxic and/or immunogenic effects of BRAFi and oHSV to generate a favorable immune response in the context of drug-mediated tumor cell kill, which can be further boosted by immune-checkpoint blockade, is worthy of further testing in the clinic.
Acknowledgments
This work was supported by Oracle Cancer Trust, Get Ahead Charitable Trust, Anthony Long Charitable Trust and Cancer Research UK (C16708/A21855). We thank Barbara Langford and her family for the generous support of our research. We also thank Breast Cancer Now for funding this work as part of Programme Funding to the Breast Cancer Now Tony Robins Research Centre. We thank Suzanne Thomas for providing the viruses.
Footnotes
Contributors: EC-R performed the majority of the in vitro and ex vivo experiments including cell and tissue culture, establishment of murine cell lines, virus infections, proliferation assays, plaque assays, processing of murine tumor samples, western blot analysis, colony formation assays, acquired flow cytometry data and analyzed flow cytometry data. KB performed the orthotopic thyroid tumor model in vivo as well as performed the flow cytometry experiments and analysis of the orthotopic model. She also performed subcutaneous thyroid tumor in vivo experiments, drug treatments and acquired flow cytometry data and analyzed flow cytometry data. HW, GB, and SF provided technical assistance for the in vivo experiments. EP provided technical assistance with the flow cytometry experiments and expertise with the design of flow cytometry multicolor panels and analysis of flow cytometry data. KT performed the histologic examination of the murine thyroid tumors. RB ran the NanoString assay. SH and GM analyzed the NanoString data. KN and RV contributed to the design of the work and the interpretation of the data. RC provided the oncolytic herpes simplex virus, contributed to the design of the work and the interpretation of the data. DK provided funding for the project, contributed to the design of the work and the interpretation of the data. MML contributed to the design of the work, technical assistance as well as contributed with analysis and interpretation of the data. AM designed the study, analyzed and interpreted the data and wrote the manuscript, as well as provided funding for the project. KH designed the study, analyzed and interpreted the data and wrote the manuscript, as well as provided funding for the project. MP designed the study and the experiments, performed in vivo experiments including generation of the transgenic model and establishment of murine thyroid cancer cell lines as well as performed subcutaneous tumor model experiments in vivo. She also supervised the project, analyzed and interpreted the data, wrote the manuscript, as well as provided funding for the project. All authors read and approved the final manuscript. AAM, KJH and MP are joint senior authors.
Funding: This work was supported by Oracle Cancer Trust, Get Ahead Charitable Trust, Anthony Long Charitable Trust and Cancer Research UK (C16708/A21855).
Competing interests: RC: employment and share ownership with Replimune. RV: research grant funding Oncolytics Biotech. AM: research grant funding from AstraZeneca/Medimmune, Bristol Myers Squibb and Oncolytics Biotech; advisory board membership, honoraria and/or speakers’ bureaus from Amgen, AstraZeneca, Bristol-Myers-Squibb, Merck-Serono, Turnstone Biologics. KH: research grant funding from AstraZeneca/Medimmune, Boehringer-Ingelheim, Merck Sharp Dohme and Replimune; advisory board membership, honoraria and/or speakers’ bureaus from Amgen, AstraZeneca, Bristol-Myers-Squibb, Boehringer-Ingelheim, Merck-Serono, Merck Sharp Dohme, Oncolys, Pfizer and Replimune.
Patient consent for publication: Not required.
Ethics approval: All procedures involving animals were approved by the Animal Ethics Committee at the Institute of Cancer Research in accordance with National Home Office Regulations under the Animals (Scientific Procedures) Act 1986.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are either included in this published article and its supplementary information files or available from the corresponding author on reasonable request.
References
- 1.Jin L, Chen E, Dong S, et al. Braf and TERT promoter mutations in the aggressiveness of papillary thyroid carcinoma: a study of 653 patients. Oncotarget 2016;7:18346–55. 10.18632/oncotarget.7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xing M, Alzahrani AS, Carson KA, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 2013;309:1493–501. 10.1001/jama.2013.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Xu X, Peng C, et al. BRAFV600E-induced KRT19 expression in thyroid cancer promotes lymph node metastasis via EMT. Oncol Lett 2019;18:927–35. 10.3892/ol.2019.10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim S-jin, Lee KE, Myong JP, et al. Braf V600E mutation is associated with tumor aggressiveness in papillary thyroid cancer. World J Surg 2012;36:310–7. 10.1007/s00268-011-1383-1 [DOI] [PubMed] [Google Scholar]
- 5.Rosove MH, Peddi PF, Glaspy JA. Braf V600E inhibition in anaplastic thyroid cancer. N Engl J Med 2013;368:684–5. 10.1056/NEJMc1215697 [DOI] [PubMed] [Google Scholar]
- 6.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36. 10.1056/NEJMoa1502309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prager GW, Koperek O, Mayerhoefer ME, et al. Sustained Response to Vemurafenib in a BRAFV600E-Mutated Anaplastic Thyroid Carcinoma Patient. Thyroid 2016;26:1515–6. 10.1089/thy.2015.0575 [DOI] [PubMed] [Google Scholar]
- 8.Lim AM, Taylor GR, Fellowes A, et al. Braf inhibition in BRAFV600E-Positive anaplastic thyroid carcinoma. J Natl Compr Canc Netw 2016;14:249–54. 10.6004/jnccn.2016.0030 [DOI] [PubMed] [Google Scholar]
- 9.McFadden DG, Vernon A, Santiago PM, et al. P53 constrains progression to anaplastic thyroid carcinoma in a BRAF-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A 2014;111:E1600–9. 10.1073/pnas.1404357111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subbiah V, Kreitman RJ, Wainberg ZA, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol 2018;36:7–13. 10.1200/JCO.2017.73.6785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ribas A, Lawrence D, Atkinson V, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25:936–40. 10.1038/s41591-019-0476-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ascierto PA, Dummer R. Immunological effects of BRAF+MEK inhibition. Oncoimmunology 2018;7:e1468955. 10.1080/2162402X.2018.1468955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ascierto PA, Ferrucci PF, Fisher R, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019;25:941–6. 10.1038/s41591-019-0448-9 [DOI] [PubMed] [Google Scholar]
- 14.Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19:1480–92. 10.1016/S1470-2045(18)30700-9 [DOI] [PubMed] [Google Scholar]
- 15.Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345–56. 10.1056/NEJMoa1709684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunda V, Gigliotti B, Ndishabandi D, et al. Combinations of BRAF inhibitor and anti-PD-1/PD-L1 antibody improve survival and tumour immunity in an immunocompetent model of orthotopic murine anaplastic thyroid cancer. Br J Cancer 2018;119:1223–32. 10.1038/s41416-018-0296-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brauner E, Gunda V, Vanden Borre P, et al. Combining BRAF inhibitor and anti PD-L1 antibody dramatically improves tumor regression and anti tumor immunity in an immunocompetent murine model of anaplastic thyroid cancer. Oncotarget 2016;7:17194–211. 10.18632/oncotarget.7839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunda V, Gigliotti B, Ashry T, et al. Anti-Pd-1/Pd-L1 therapy augments lenvatinib's efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int J Cancer 2019;144:2266–78. 10.1002/ijc.32041 [DOI] [PubMed] [Google Scholar]
- 19.Ulisse S, Tuccilli C, Sorrenti S, et al. Pd-1 ligand expression in epithelial thyroid cancers: potential clinical implications. Int J Mol Sci 2019;20. 10.3390/ijms20061405. [Epub ahead of print: 20 Mar 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kollipara R, Schneider B, Radovich M, et al. Exceptional response with immunotherapy in a patient with anaplastic thyroid cancer. Oncologist 2017;22:1149–51. 10.1634/theoncologist.2017-0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyer PC, Dadu R, Gule-Monroe M, et al. Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma. J Immunother Cancer 2018;6:68. 10.1186/s40425-018-0378-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andtbacka RHI, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015;33:2780–8. 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 23.Lin S-F, Gao SP, Price DL, et al. Synergy of a herpes oncolytic virus and paclitaxel for anaplastic thyroid cancer. Clin Cancer Res 2008;14:1519–28. 10.1158/1078-0432.CCR-07-4628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan M, Romano G, Coroniti R, et al. Progress in oncolytic virotherapy for the treatment of thyroid malignant neoplasm. J Exp Clin Cancer Res 2014;33:91. 10.1186/s13046-014-0091-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J-N, Xu L-H, Zeng W-G, et al. Treatment of human thyroid carcinoma cells with the g47delta oncolytic herpes simplex virus. Asian Pac J Cancer Prev 2015;16:1241–5. 10.7314/APJCP.2015.16.3.1241 [DOI] [PubMed] [Google Scholar]
- 26.Bressy C, Benihoud K. Association of oncolytic adenoviruses with chemotherapies: an overview and future directions. Biochem Pharmacol 2014;90:97–106. 10.1016/j.bcp.2014.05.003 [DOI] [PubMed] [Google Scholar]
- 27.Lavie M, Struyf S, Stroh-Dege A, et al. Capacity of wild-type and chemokine-armed parvovirus H-1PV for inhibiting neo-angiogenesis. Virology 2013;447:221–32. 10.1016/j.virol.2013.09.019 [DOI] [PubMed] [Google Scholar]
- 28.Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic BRAF induces melanocyte senescence and melanoma in mice. Cancer Cell 2009;15:294–303. 10.1016/j.ccr.2009.02.022 [DOI] [PubMed] [Google Scholar]
- 29.Suzuki A, Yamaguchi MT, Ohteki T, et al. T cell-specific loss of PTEN leads to defects in central and peripheral tolerance. Immunity 2001;14:523–34. 10.1016/S1074-7613(01)00134-0 [DOI] [PubMed] [Google Scholar]
- 30.Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004;119:847–60. 10.1016/j.cell.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 31.Kusakabe T, Kawaguchi A, Kawaguchi R, et al. Thyrocyte-specific expression of Cre recombinase in transgenic mice. Genesis 2004;39:212–6. 10.1002/gene.20043 [DOI] [PubMed] [Google Scholar]
- 32.Girotti MR, Lopes F, Preece N, et al. Paradox-breaking Raf inhibitors that also target Src are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015;27:85–96. 10.1016/j.ccell.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waggott D, Chu K, Yin S, et al. NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics 2012;28:1546–8. 10.1093/bioinformatics/bts188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donia M, Fagone P, Nicoletti F, et al. Braf inhibition improves tumor recognition by the immune system: potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology 2012;1:1476–83. 10.4161/onci.21940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18:1386–94. 10.1158/1078-0432.CCR-11-2479 [DOI] [PubMed] [Google Scholar]
- 37.Liu C, Peng W, Xu C, et al. Braf inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res 2013;19:393–403. 10.1158/1078-0432.CCR-12-1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frederick DT, Piris A, Cogdill AP, et al. Braf inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225–31. 10.1158/1078-0432.CCR-12-1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bommareddy PK, Aspromonte S, Zloza A, et al. Mek inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci Transl Med 2018;10. 10.1126/scitranslmed.aau0417. [Epub ahead of print: 12 Dec 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. 10.1172/JCI91190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang H, Liang Y, Anders RA, et al. Pd-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest 2018;128:580–8. 10.1172/JCI96061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin H, Wei S, Hurt EM, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 2018;128:1708. 10.1172/JCI120803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017;170:e10:p. 1109–1119. 10.1016/j.cell.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu Y, Hu X, Feng L, et al. Enhanced therapeutic efficacy of a novel oncolytic herpes simplex virus type 2 encoding an antibody against programmed cell death 1. Mol Ther Oncolytics 2019;15: :201–13. 10.1016/j.omto.2019.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wennerberg E, Pfefferle A, Ekblad L, et al. Human anaplastic thyroid carcinoma cells are sensitive to NK cell-mediated lysis via ULBP2/5/6 and chemoattract NK cells. Clin Cancer Res 2014;20:5733–44. 10.1158/1078-0432.CCR-14-0291 [DOI] [PubMed] [Google Scholar]
- 46.Arce Vargas F, Furness AJS, Litchfield K, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 2018;33:649–63. 10.1016/j.ccell.2018.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma A, Subudhi SK, Blando J, et al. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer Res 2019;25:1233–8. 10.1158/1078-0432.CCR-18-0762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samson A, Bentham MJ, Scott K, et al. Oncolytic reovirus as a combined antiviral and anti-tumour agent for the treatment of liver cancer. Gut 2018;67:562–73. 10.1136/gutjnl-2016-312009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res 2010;16:4005–15. 10.1158/1078-0432.CCR-10-0196 [DOI] [PubMed] [Google Scholar]
- 50.Fransen MF, Schoonderwoerd M, Knopf P, et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 2018;3. 10.1172/jci.insight.124507. [Epub ahead of print: 06 Dec 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-000698supp001.pdf (69.8KB, pdf)
jitc-2020-000698supp002.pdf (116.6KB, pdf)
jitc-2020-000698supp005.pdf (46.6KB, pdf)
jitc-2020-000698supp003.pdf (58.4KB, pdf)
jitc-2020-000698supp006.pdf (62.8KB, pdf)
jitc-2020-000698supp004.pdf (47.1KB, pdf)