

Abstract
Histone deacetylases (HDACs) have been shown to alleviate renal fibrosis, however, the role of individual HDAC isoforms in this process is poorly understood. In this study, we examined the role of HDAC8 in the development of renal fibrosis and partial epithelial-mesenchymal transitions (EMT). In a murine model of renal fibrosis induced by unilateral ureteral obstruction (UUO), HDAC8 was primarily expressed in renal tubular epithelial cells and time-dependently upregulated. This occurred in parallel with the deacetylation of contactin, a nonhistone of HDAC8, and increased expression of three fibrotic markers: α-smooth muscle actin, collagen 1 and fibronectin. Administration of PCI34051, a highly selective inhibitor of HDAC8, restored acetylation of contactin and reduced expression of those proteins. PCI34051 treatment also reduced the number of renal tubular epithelial cells arrested at the G2/M phase of the cell cycle and suppressed phosphorylation of Smad3, STAT3, β-catenin and expression of Snail after ureteral obstruction. In contrast, HDAC8 inhibition reversed UUO-induced downregulation of BMP7 and Klotho, two renoprotective proteins. In cultured murine proximal tubular cells, treatment with PCI34051 or specific HDAC8 siRNA was also effective in inhibiting transforming growth factor β1-induced deacetylation of contactin, EMT, phosphorylation of Smad3, STAT3 and β-catenin, upregulation of Snail, and downregulation of BMP7 and Klotho. Collectively, these results suggest that HDAC8 activation is required for the EMT and renal fibrogenesis by activation of multiple profibrotic signaling and transcription factors, and suppression of antifibrotic proteins. Therefore, targeting HDAC8 may be novel therapeutic approach for treatment of renal fibrosis.
Keywords: Histone deacetylase 8, epithelial-mesenchymal transition, transforming growth factor β1, β-catenin, unilateral ureteral obstruction, renal fibrosis
Introduction
Chronic kidney disease (CKD), defined as a progressive reduction of glomerular filtration rate, has become a major public health problem due to its rising epidemic and mortality rate (1, 2). It can be caused by primary kidney injury and injuries secondary to other chronic diseases such as diabetes and hypertension (2). So far, the underlying mechanism remains incompletely clear, and there are no available therapeutic treatments to halt the progression of renal fibrosis. Thus, it is necessary to identify novel therapeutic targets in order to develop effective treatments for this disease process.
A large body of evidence points to renal fibrosis as a direct consequence of maladaptive repair after renal injury no matter what the underlying cause of the injury is (3). Fibrosis involves activation of renal interstitial fibroblasts and an excess accumulation of extracellular matrix. Partial epithelial-mesenchymal transition (EMT) of tubular epithelial cells also contributes to this process (4). Unlike complete EMT, partial EMT in kidneys is characterized by loss of epithelial cell polarity and acquisition by epithelial cells of mesenchymal features and a motile phenotype without conversion into fibroblasts (4). These epithelial cells co-express epithelial and mesenchymal markers, but still reside within the basement membrane (4). However, they are arrested at the G2/M phase of cell cycle, acquiring an ability to produce numerous profibrotic cytokines and growth factors, including transforming growth factor β1 (TGFβ1) (5, 6), a potent cytokine that can induce renal interstitial fibroblast activation and renal fibrogenesis (7). Interaction of TGF-β1 with its receptors induces Smad3 phosphorylation; phosphorylated Smad3 together with Smad4 is then translocated to the nucleus where it transcriptionally drives expression of numerous profibotic genes, including collagen 1 and connective tissue growth factor (CTGF) (8). Activation of TGFβ receptors and many other growth factors can also induce activation of STAT3 and β-catenin signaling pathways, which are involved in the EMT and crosstalk with TGF-β1/Smad3 signaling during renal fibrosis (9). Snail and twist are two major transcription factors that drive the EMT process of renal tubular cells in various fibrotic diseases (10, 11).
The profibrotic machinery is counteracted by renoprotective proteins, many of which have been identified. Of them, Klotho and bone morphogenetic protein 7 (BMP-7) exert strong antifibrotic effects in various renal diseases (12, 13). BMP-7 is one of the members of the TGFβ superfamily that can negatively regulate the TGFβ/Smad signaling pathway (14). Klotho is a membrane-bound protein widely expressed in kidney tissue that can also bind to the type II TGFβ receptor and inhibit its activation (15). However, expression of BMP-7 and Klotho in most renal fibrotic diseases is downregulated, leading to reduction or loss of their renoprotective effects (12, 13). As such, elucidating the molecular machinery that leads to suppression of their expression and developing a novel approach to restore their expression will aid prevention and treatment of renal fibrosis.
Protein acetylation plays a critical role in controlling gene expression and is implicated in the regulation of many diseases, including tissue fibrosis (16). This modification occurs in histone and many nonhistone proteins and is controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs can remove acetyl groups from hyperacetylated proteins and suppress general gene transcription. There are 11 HDAC members composed of four classes: class I HDACs (HDAC1, 2, 3, and 8); class II HDACs (HDAC4, 5, 6, 7, 9, and 10); class III HDACs (SIRT1–7); and class IV (HDAC11) (17). HDAC8 is a ubiquitously expressed protein that can localize to either the nucleus or the cytoplasm. Although many of its substrates, in particular, those expressed in the cytosol, have not been fully identified, cotactin, a cortical actin-binding protein, has been found to be a unique substrate of HDAC8 (18). HDAC8 participates in numerous signaling networks and links with a wide range of diseases, including Cornelia de Lange syndrome (19), childhood neuroblastoma and T cell lymphoma (20), and parasitic and viral infections (21). Our previous studies and those by other groups showed that inhibition of class I and class II HDACs could attenuate renal interstitial fibrosis (22, 23). However, it remains unclear whether selective inhibition of HDAC8 would offer an antifibrotic effect in chronic kidney injury.
In this study, we investigated the impact of HDAC8 inhibition on the development of renal fibrosis in a murine model of unilateral ureteral legation (UUO), as well as EMT of renal tubular epithelial cells and mechanism involved in vivo and in vitro. Our data suggested that HDAC8 is a novel therapeutic target for the treatment of renal fibrosis.
Materials and Method
Reagents and Antibodies
PCI34051 was purchased from Selleckchem (Radnor, PA). Antibodies to α-SMA, α-Tubulin were purchased from MilliporeSigma (Burlington, MA). Antibodies to collagen 1, HDAC8, GAPDH, Cortactin, CTGF and Small interfering RNA (siRNA) specific for HDAC8 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to p-Smad3, Smad3, p-Stat3, Stat3, p-β-Catenin, β-Catenin and Snail were purchased from Cell Signaling Technology (Danvers, MA). Antibodies to Fibronectin, BMP7, Klotho were purchased from Abcam (Cambridge, MA). Antibodies to p-Histone H3 (Ser10) and acetyl-Cortactin were purchased from EMD Millipore (Temecula, CA). TGF-β1 was purchased from R&D Systems (Minneapolis, MN). All other reagents and chemicals were purchased from Millipore Sigma (St. Louis, MO).
Cell culture and treatments
Murine renal proximal tubular cells (TKPT) were kindly provided by Dr. E. Bello-Reuss (University of Texas Medical Branch, Galveston, USA) and cultured and in DMEM-F12 with 5% fetal bovine serum and 0.5% penicillin (Thermo Fisher Scientific, Waltham, MA) in an atmosphere of 5% CO2 at 37°C. For TGF-β1 treatment, TKPT were washed and starved for 24 hours in DMEM-F12 containing 0% FBS prior to any stimulation. PCI34051 (6.25 μM) was added to the culture and then incubated for 1 hour before TGF-β1 exposure (R&D Systems, Minneapolis, MN).
Western blot analysis
Western blot analysis was performed as described in our previous article (24). Tissues or cells were processed in lysis buffer (Cell Signaling Technology, Danvers, MA,) containing protease inhibitors cocktail (Roche Life Science, Indianapolis, IN) and then centrifuged. The supernatants were collected for densitometry analysis. Bands were detected using the Pierce ECL Western Blotting Substrate from Thermo Fisher Scientific (Waltham, MA). The intensity of immunoblot results was determined by ImageJ Software (version 1.52a, NIH).
Small interfering RNA (siRNA) transfection
TKPT cells were plated to 50–60% confluence in 6-well plate with antibiotic-free medium. The HDAC8 siRNA (90 pmol) were transfected into cells using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. As a control, 90 pmol control siRNA was co-incubated with TKPT cells. After transfection for 8 hours, cells were cultured in DMEM-F12 with 5% FBS for further treatment.
Unilateral ureteral obstruction mice model and PCI34051 treatment
The UUO model was created in male C57BL/6 mice (The Jackson Laboratory, BarHarbor, ME, USA) as described previously (24). All animal experiments were approved by the Institutional Animal Care and Use Committee at RIH (Providence, RI) and were performed in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animals. After UUO injury, PCI34051 at 20 mg/kg was intraperitoneally administered immediately and then given at the same dosage every 12 hours for 7 days before mice were euthanized. DMSO treated mice were used as controls. 5–6 mice were used in each group.
Masson staining and immunofluorescent Staining
Histological experiment was performed according to our previous studies (24). For evaluation of renal fibrosis quantitatively, the tissues were fixed in 10% formalin and sliced in paraffin sections at 5 μm before Masson trichrome staining. The collagen tissue area was measured by ImageJ Software (version 1.52a, NIH). For immunofluorescent, α-SMA antibody and HDAC8 antibody used as primary antibodies. The signal intensity was quantified using CellSens software (Olympus Life Science, Tokyo).
Statistical analysis
All in vitro experiments were performed at least three times. Statistical analysis was performed by SPSS (version 20.0, IBM Corporation, Armonk, NY, USA). Results are shown as the mean±SD. T test was used for comparisons between two groups. One-way ANOVA was used for multiple group comparisons. P<0.05 was considered as a statistically significant difference.
Results
UUO induces HDAC8 expression, which parallels to the development of interstitial fibrosis in the kidney
To understand the role of HDAC8 in the activation of renal interstitial fibroblasts, we first examined HDAC8 expression, and its relation to renal interstitial fibrosis at different time after unilateral ureteral obstruction in the murine model by immunoblot analysis. Renal fibrosis, as evidenced by expression of α-SMA, collagen 1, fibronectin, developed after injury in a time dependent manner (Figure 1, A–D). The expression of HDAC8 and its unique substrate, cortactin, was also increased overtime in the obstructed kidneys, which was clearly detected at day 7 and further increased at day 14 (Figure 1 A,E–G). Of note, both HDAC8 and cortactin were also slightly expressed in the sham-operated kidney. In contrast, renal expression of acetylcortactin declined over time after UUO injury and seemingly disappeared at day 14. Immunofluorescence microscopy demonstrated that HDAC8 was primarily distributed in the cytosol of renal epithelial cells and not co-stained with α-SMA, a hallmark of myofibroblasts, in interstitial cells, indicating that HDAC8 is not expressed in myofibroblasts. In addition, HDAC8 was not expressed in small blood vessels in either control or UUO injured kidneys as well (Figure 1, H). These results illustrate that HDAC8 expression and activation are correlated with expression of renal fibrotic markers and limited to proximal tubular cells.
Figure 1. UUO induces HDAC8 expression and development of interstitial fibrosis over time.
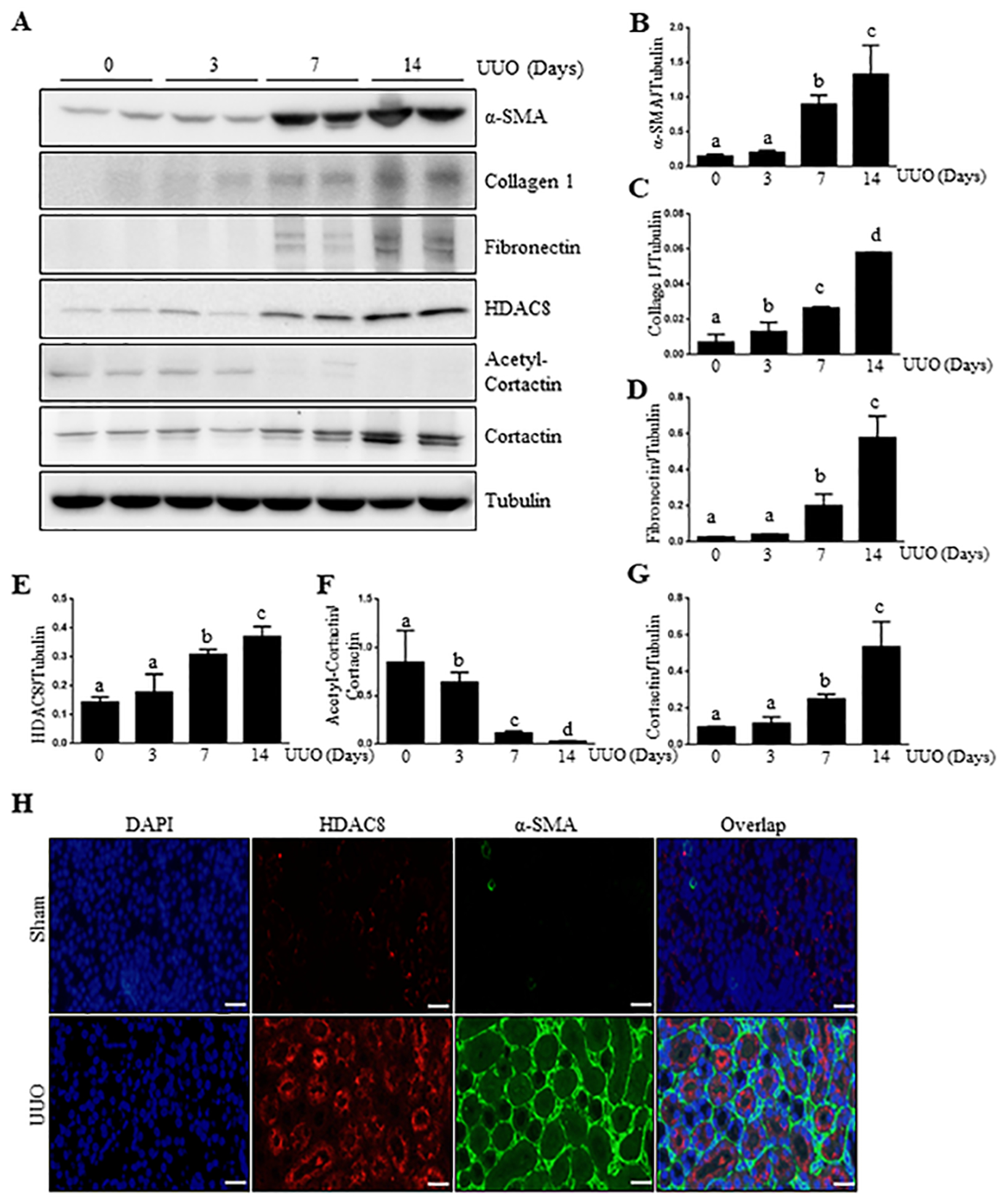
(A) The kidneys were harvested at 0, 3, 7, 14 days after surgery with left ureteral ligation and then homogenized. Kidney tissue lysates were subjected to immunoblot analysis with antibodies against α-SMA, Collagen1, Fibronectin, HDAC8 and tubulin, respectively. Expression levels of α-SMA, Collagen1, Fibronectin, HDAC8 and Tubulin were performed were quantified by densitometry and the levels of α-SMA (B), Collagen1 (C), Fibronectin (D), HDAC8 (E) and Cortactin (G) were normalized with Tubulin. Acetyl-Cortactin (F) was normalized with Cortactin. Values are the means ± S.D (n = 6). Means with different superscript letters (a-d) are significantly different from one another (P<0.05). (H) Photomicrographs illustrating HDAC8 (red), α-SMA (green) and nuclei (blue) immunofluorescent staining of kidney tissue after UUO treatments (200X). Scale bar: 50 μM.
HDAC8 inhibition with PCI34051 attenuates renal interstitial fibrosis after UUO injury
To elucidate the role of HDAC8 in renal fibrosis, we examined the effect of PCI34051, a high selective HDAC8 inhibitor, on the pathological change and expression of fibrosis markers in the kidney after UUO injury. Masson trichrome staining of renal cortex illustrated that a large amount of collagen fibers existed in the renal interstitium of UUO kidney compared with the sham group. By contrast, the renal interstitial collagen in UUO group treated with PCI34051 was significantly reduced compared to those without this treatment at day 7(Figure 2, A and B).
Figure 2. Administration of PCI34051 (PCI) attenuates development of renal fibrosis in obstructed kidneys.

Kidneys were harvested at 7 days after various treatments as indicated. Photomicrographs illustrating Masson trichrome staining of kidney tissue (200X) (A). The Masson trichrome-positive tubulointerstitial area (blue) relative to the whole area from 6 random cortical fields was analyzed (B). Kidney tissue lysates were subjected to immunoblot analysis with antibodies against α-SMA, Collagen1, Fibronectin, and HDAC8, acetyl-Cortactin and Cortacin and tubulin. Expression levels of all those proteins were quantified by densitometry and α-SMA (D), Collagen1 (E), Fibronectin (F), HDAC8 (G) and Cortacin (I) were normalized with Tubulin, respectively. Expression levels of acetyl-Cortactin were normalized with Cortacin (H). Values are the means ± S.D (n = 6). Means with different superscript letters (a-d) are significantly different from one another (P<0.05). Scale bar: 50 μM.
Chronic renal fibrosis is characterized by activation of renal interstitial fibroblasts and overproduction of ECM proteins (5, 6). To confirm the role of HDAC8 in regulating renal fibrosis, we further examined the effect of PCI34051 on the expression of α-SMA and deposition of fibronectin and collagen type 1, three fibrotic markers, in obstructed kidneys by immunoblot analysis. As indicated in Figure 2, C–F, treatment with PCI34051 (20 mg/kg) significantly reduced the levels of α-SMA, fibronectin, and collagen. This dose of PCI34051 was sufficient to restore UUO-induced downregulation of cortatin acetylation, indicative of its effectiveness (Figure 2, C, G–I). In addition, PCI34051 treatment also slightly reduced HDAC8 expression. These data suggest that HDAC8 is a critical mediator of renal fibrosis.
PCI34051 inhibits the EMT of renal epithelial cells in culture
Since HDAC8 was mainly expressed in renal tubular cells, we investigated the possible role of HDAC8 in mediating EMT in cultured mouse proximal tubular cells (TKPT). Immunostaining indicates that TGFβ1 exposure induced increased expression of HDAC8, which was mainly distributed in the cytosol of TKPTs (Figure 3, A). TGFβ1 treatment also increased expression of α-SMA, collagen 1 and fibronectin in this cell type; presentation of PCI34051 largely inhibited α-SMA expression and reduced collagen 1 and fibronectin expression to the basal levels. At this concentration, PCI3405 also reduced HDAC8 expression and restored cortactin acetylation (Figure 3, B, F–H). This suggests that HDAC8 plays an essential role in mediating the conversion of renal epithelial cells to a dedifferentiated phenotype, also called EMT. However, we did not observe the inhibitory effect of PCI34051 on the down-regulation of E-cadherin, an epithelial marker (data not shown). These results agree with a previous observation that restoration of TGFβ1-induced E-cadherin down-regulation was not seen in renal epithelial cells treated with PCI34051 (25).
Figure 3. PCI34051(PCI) suppresses TGFβ1-induced EMT of cultured renal epithelial cells.
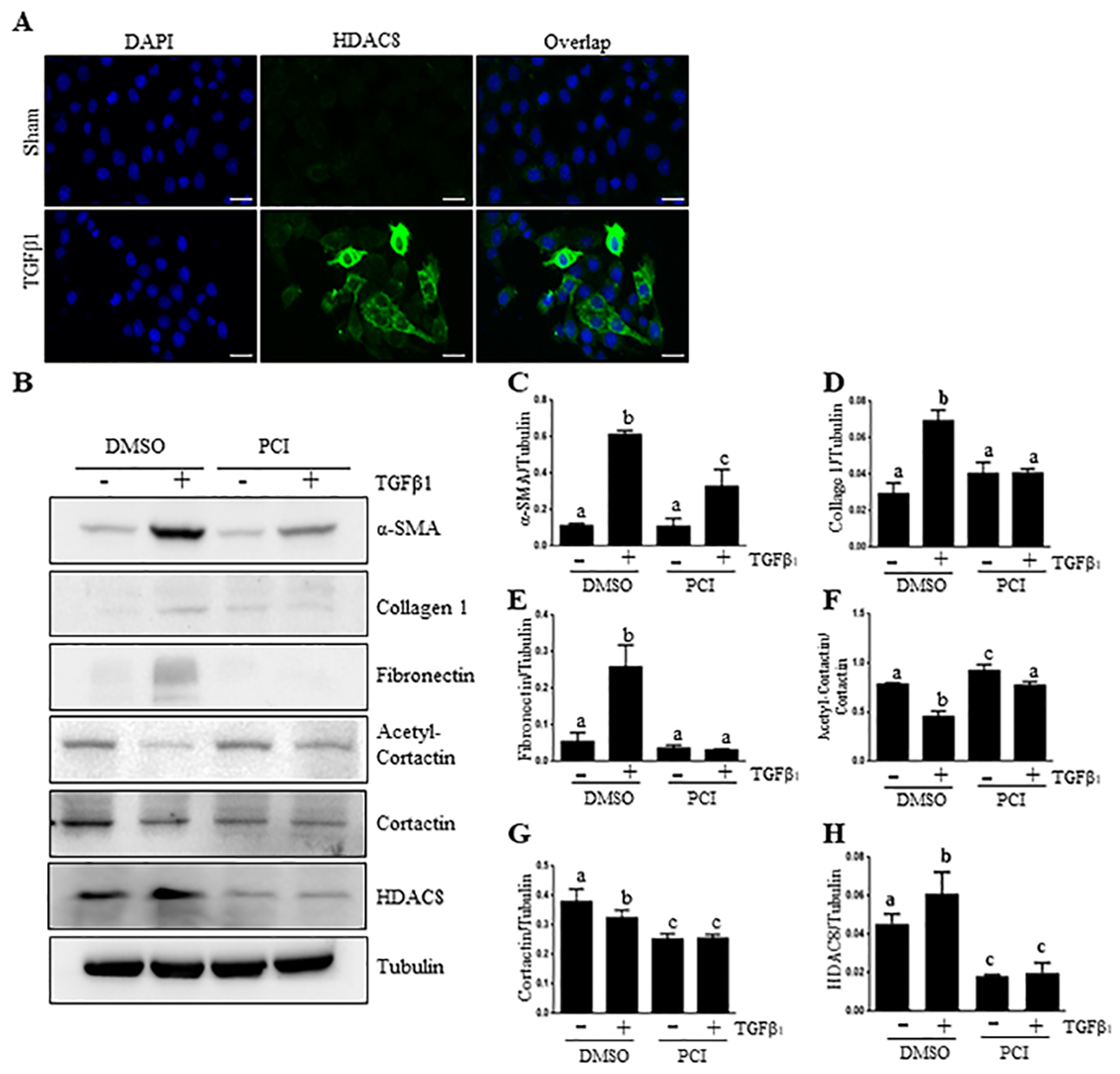
TKPT cells were serum starved for 24 hours and then treated with PCI (6.25 μM) for 1 hour and then incubated for an additional 24 hours in the absence or presence of TGFβ1 (5 ng/ml). Cells were stained with using an antibody against HDAC8 (green) and DAPI (blue) (X 200) (A). Cell lysates were prepared for immunoblot analysis with antibodies against α-SMA, Collagen1, Fibronectin, acetyl-Cortactin, Cortacin, HDAC8 and Tubulin (B). All those proteins were quantified by densitometry, and were normalized to Tubulin, respectively (C, D, E, F, G, H). Acetyl-Cortactin was normalized to Cortacin. The values are the means ± S.D. (n = 6). Bars with different letters (a–c) are significantly different from one another (P < 0.05). Scale bar: 50 μM.
Knockdown of HDAC8 inhibits EMT of renal epithelial cells in culture
We further confirmed the role of HDAC8 in mediating the process of EMT by siRNA-mediated silencing of HDAC8. Figure 4 demonstrated that transfection of small interfering RNA (siRNA) specifically targeting HDAC8 blocked expression of α-SMA, collagen 1 and fibronectin in TKPT cells (Figure 4, A–D). Of note, HDAC8 was knocked down by approximately 70% in HDAC8 siRNA transfected cells compared to those transfected with control siRNA, in line with what we observed using PCI34051. SiRNA-mediated silencing of HDAC8 also restored the expression of acetyl-cortacin to the control level (Figure 4, E–H). These data support our conclusions that HDAC8 is involved in the transformation of renal epithelial cells to a dedifferentiated phenotype.
Figure 4. HDAC8 siRNA suppresses TGFβ1-induced EMT of proximal tubular epithelial cells.

TKPT cells were serum starved for 24 hours and transfected with HDAC8 siRNA or scrambled siRNA for 24 hours followed by exposure of cells to TGFβ1 (5 ng/ml) for an additional 24 hours. Cell lysates were prepared for immunoblot analysis with antibodies against α-SMA, Collagen1, Fibronectin and Tubulin (A) or acetyl-cortactin, cortactin, HDAC8, or Tubulin (E). All those proteins were quantified by densitometry, and were normalized to Tubulin (B-D, F-H). The values are the means ± S.D. (n = 6). Bars with different letters (a–b) are significantly different from one another (P < 0.05).
Blocking HDAC8 inhibits arrest of renal epithelial cells at the G2/M phase in the kidney after UUO injury
During renal fibrosis, partial EMT induces a cell-cycle arrest at G2/M check point and produces profibrotic growth factors/cytokines, including CTGF (26). This process is derived by Snail and Twist, two transcriptional factors (27, 28). To address whether HDAC8 mediates this process, we investigated the effect of HDAC8 inhibition on the expressions of phospho-histoneH3 at serine 10, a marker of cell cycle arrest in the G2/M phase, in the kidney after UUO injury. The results from our immunostaining demonstrated an increase in the number of renal tubular cells with expression of p-H3 at serine 10 in the kidney after UUO injury; PCI34051 treatment reduced its expression (Figure 5, A–B). Immunoblot analysis indicated that injury to the kidney resulted in increased expression of p-histoneH3 (Ser10) as well as that of CTGF, Snail, (Figure 5, C–F) and Twist (Supplemental figure 1, A,B). PCI34051 was also effective in blocking their expression.
Figure 5. Pharmacological blocking of HDAC8 PCI34051(PCI) inhibits renal epithelial cells arrested at G2/M phase of cell cycles in the kidney after UUO injury.
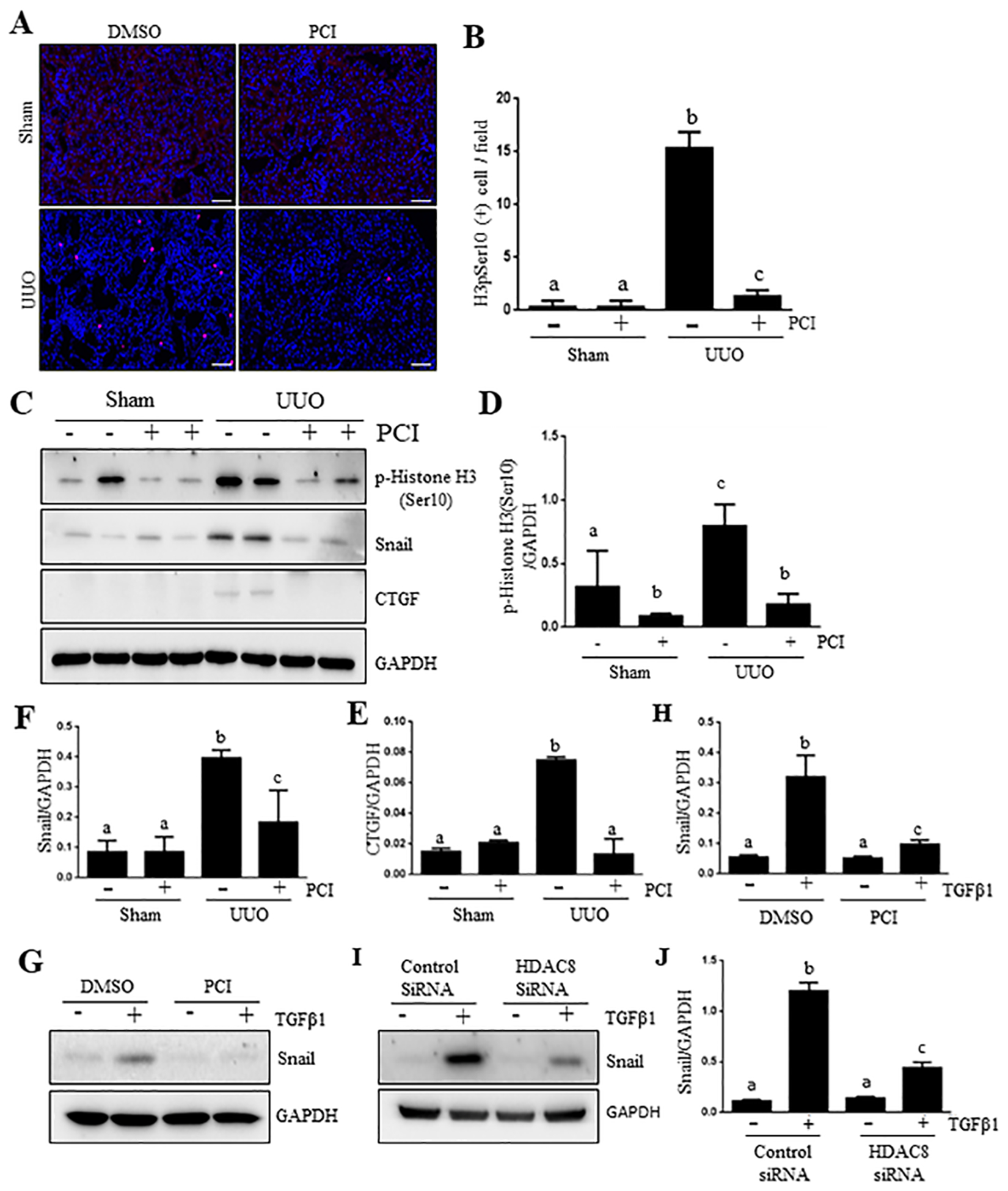
Kidneys were collected at 7 days after various treatments as indicated. Photomicrographs (x200) illustrate p-histone H3 (Serine 10) staining of the kidney tissues (A). Epithelial cells with positive staining for phistone H3 were counted in 10 high-power fields and expressed as means ± S.D.(B). Lysates of kidney tissues collected at day 7 after sham and UUO treatment were subjected to immunoblot analysis with specific antibodies against p-histone H3 (ser 10), Snail, CTGF and Tubulin (C). The expression levels of p-histone H3 (ser10) (D), Snail (E) or CTGF (F) were quantified by densitometry and normalized with Tubulin, respectively. Data are means ± SEM (n =6). Means with different symbols are significantly different from one another. P < 0.05. Scale bar: 50 μM.
Given the importance of Snail in regulating partial EMT of renal tubular cells and G2/M phase arrest, we examined the effect of HDAC8 inhibition on the expression of Snail in cultured TKPT. As shown in Figure 5, G–J, treatment with either PCI34051 or HDAC8 siRNA inhibited TGFβ1-induced Snail expression.
Taken together, our data suggest that HDAC8 plays an important role in regulating partial EMT and epithelial cell arrest at the G2/M phase of cell cycle.
HDAC8 is required for the activation of TGFβ1/Smad3 pathway in vivo and in vitro
TGFβ1/Samd signaling is primarily involved in the development of renal fibrosis in various models (29). To elucidate the underlying mechanism by which HDAC8 inhibition attenuates renal fibrosis in UUO model, we also analyzed the impact of HDAC8 inhibition on the expression of TGFβ1 and phosphorylation of Smad3 (p-Smad3), a key downstream factor of TGF-β signaling. Both TGFβ1 and p-Smad3 were barely detected in the sham-operated kidney, but their expression levels were increased after UUO injury. Administration of PCI34051 completely blocked expression of TGFβ1 and p-Smad3 in the injured kidney. Total Smad3 expression levels remained the same in kidneys with and without UUO injury and was not affected by PCI34051 treatment (Figure 6 A–C). To validate the effect of PCI34051 on the activation of Smad3 signaling, we also examined the effect of HDAC8 inhibition on Smad3 phosphorylation in vitro. As expected, TGF-β1 addition to the culture of TKPT increased expression levels of p-Smad3. Treatment with either PCI34051 or HDAC8 siRNA inhibited its expression to the basal level (Figure 4 D–G). Therefore, these data suggest that HDAC8 is involved in the activation of TGF-β1/Smad3 signaling during renal fibrogenesis and renal epithelial cell transformation.
Figure 6. HDAC8 inhibition suppresses activation of TGFβ/Smad3 signaling in the kidney after UUO injury and cultured TKPT exposed to TGF-β1.

Kidneys were collected at 7 days after UUO treatments with or without PCI34051(PCI) (A). TKPT were serum starved for 24 hours and then treated with PCI34051 at 6.25 μM for 1 hour (D) or transfected with HDAC8 siRNA or scrambled siRNA for 24 hours (F) followed by exposure of cells to TGFβ1 (5ng/ml) for an additional 24 hours. Kidney and cell lysates were subjected to immunoblot analysis with antibodies against TGFF-β1 (A), phosphorylated Smad3 (p-Smad3), Smad3 and GAPDH (A, D, F). Expression levels of all those proteins were quantified by densitometry and TGF-β1 was normalized with GAPDH (B), and p-Smad3 was normalized with Smad3 (C, E, G). Values are the means ± S.D (n = 6). Bars with different letters (a–c) are significantly different from one another (P < 0.05).
Blocking HDAC8 reduces phosphorylation of Stat3 and β-catenin in UUO injured kidney and TGFβ1-treated renal tubular cells.
Studies by our and others have shown that activation of STAT3 and β-catenin contributes to renal fibrogenesis (30–32). We hypothesized that HDAC8 may also regulate activation of these two signaling pathways. Figure 7A–C demonstrates that p-Stat3 and p-β-catenin were not detectable in the control kidney; but their expression levels were increased in UUO kidneys, and reduced by HDAC8 inhibition. However, both UUO injury and HDAC8 inhibition did not affect expression levels of total STAT3 and β-catenin. Similar to those observations, HDAC8 inhibition blocked TGFβ1-indued phosphorylation of STAT3 and β-catenin in cultured TKPT exposed to PCI34051 or transfected with HDAC8 specific siRNA (Figure 7, D–I). Therefore, HDAC8 is also required for the activation of STAT3 and β-catenin signaling pathways during renal fibrosis and renal epithelial cell transformation.
Figure 7. HDAC8 inhibition suppresses activation of STAT3 and β-catenin signaling in the kidney after UUO injury and cultured TKPT exposed to TGF-β1.
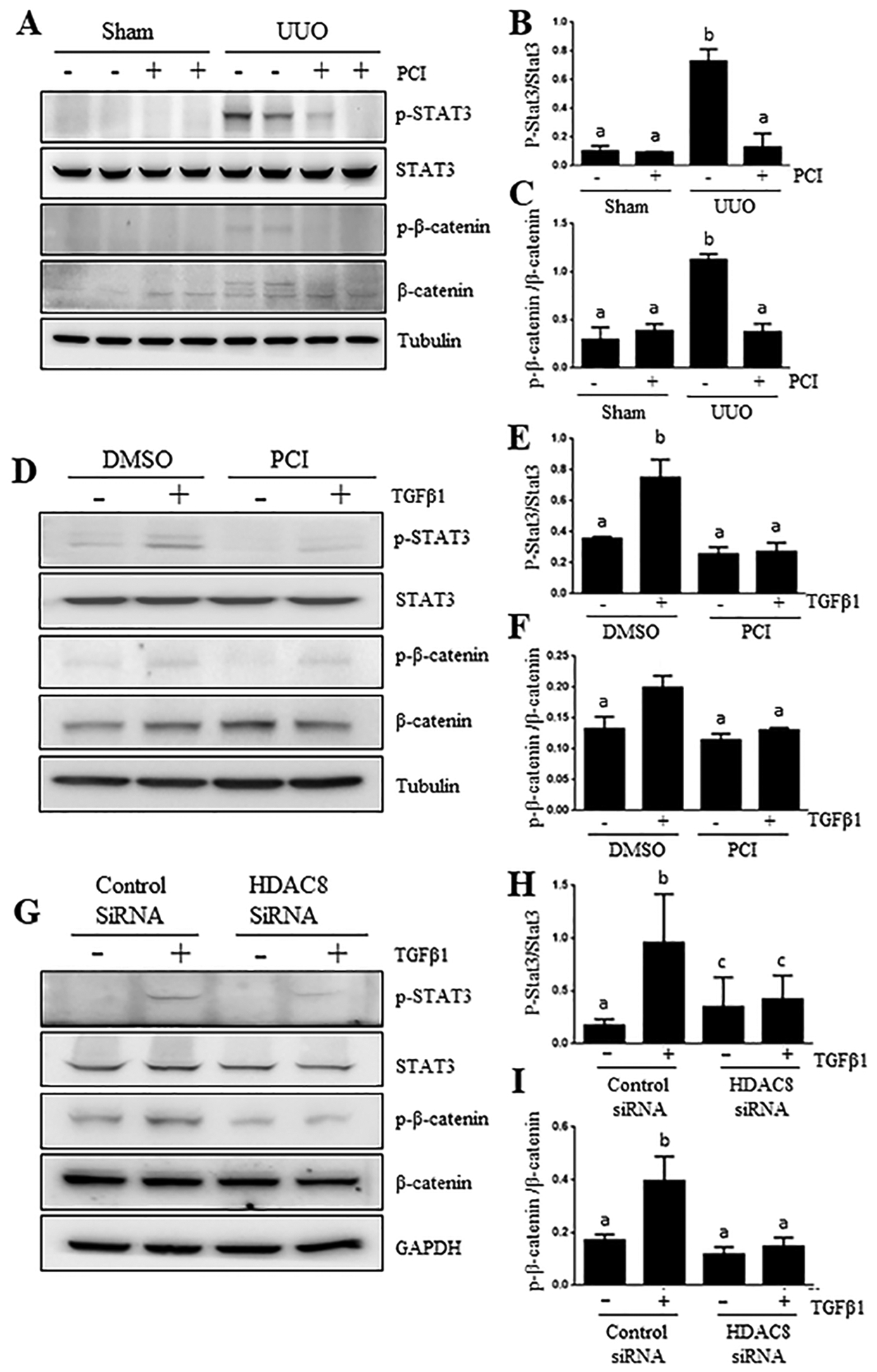
Kidneys were collected at 7 days after various treatments (A). TKPT were serum starved for 24 hours and then treated with PCI34051 (PCI) at 6.26 μM for 1 hour (D) or transfected with HDAC8 siRNA or scrambled siRNA for 24 hours (G) followed by exposure of cells to TGFβ1 (5 ng/ml) for an additional 24 hours. Kidney (A-C) and cell lysates (D-F, G-I) were subjected to immunoblot analysis with antibodies against phosphorylated STAT3 (p-STAT3), STAT3, β-catenin, p-β-catenin, Tubulin and GAPDH (A, D, G). Expression levels of all those proteins were quantified by densitometry and normalized with Tubulin (B-C, E-F), or GAPDH (H, I). Values are the means ± S.D (n = 6). Bars with different letters (a–c) are significantly different from one another (P < 0.05).
Effects of HDAC8 inhibition on the expression of BMP-7, Klotho, matrix metalloproteinase 2/9 in UUO injured kidney and/or TGFβ1-treated renal tubular cells.
BMP-7 is essential for the formation of renal tubular epithelium through promotion of mesenchymal to epithelial transition (MET) and exerts an anti-fibrotic effect in animal models of chronic renal injury, such as UUO (33). Klotho is a kidney-derived endogenous anti-fibrotic factor and can protect against renal fibrosis (15). Therefore, we further investigated whether HDAC8 regulated expression of BMP7 and Klotho in vivo and in vitro. Immunoblot analysis showed that UUO injury reduced BMP7 and Klotho expression in the kidney, which was resumed in part by HDAC8 inhibition with PCI34051 (Figure 8A–C). Similar to those observations, TKPT cells exposed to TGFβ1 showed a significant decline in BMP7 and Klotho but that was reversed by either treatment with either PCI34051 or HDAC8 SiRNA (Figure 7 D–I). In addition, we found that PCI34051 was effective in reducing expression of MMP2 and MMP9, two matrix metalloproteinases that are involved in renal fibrosis, in the UUO injured kidney (Supplemental figure 2, A–C). Thus, preservation of BMP-7 and Klotho expression and suppression of MMP2/9 may also contribute to protection against renal fibrosis elicited by HDAC8 inhibitors.
Figure 8. HDAC8 inhibition preserves expression of BMP-7 and klotho in the kidney after UUO injury and cultured TKPT exposed to TGF-β1.
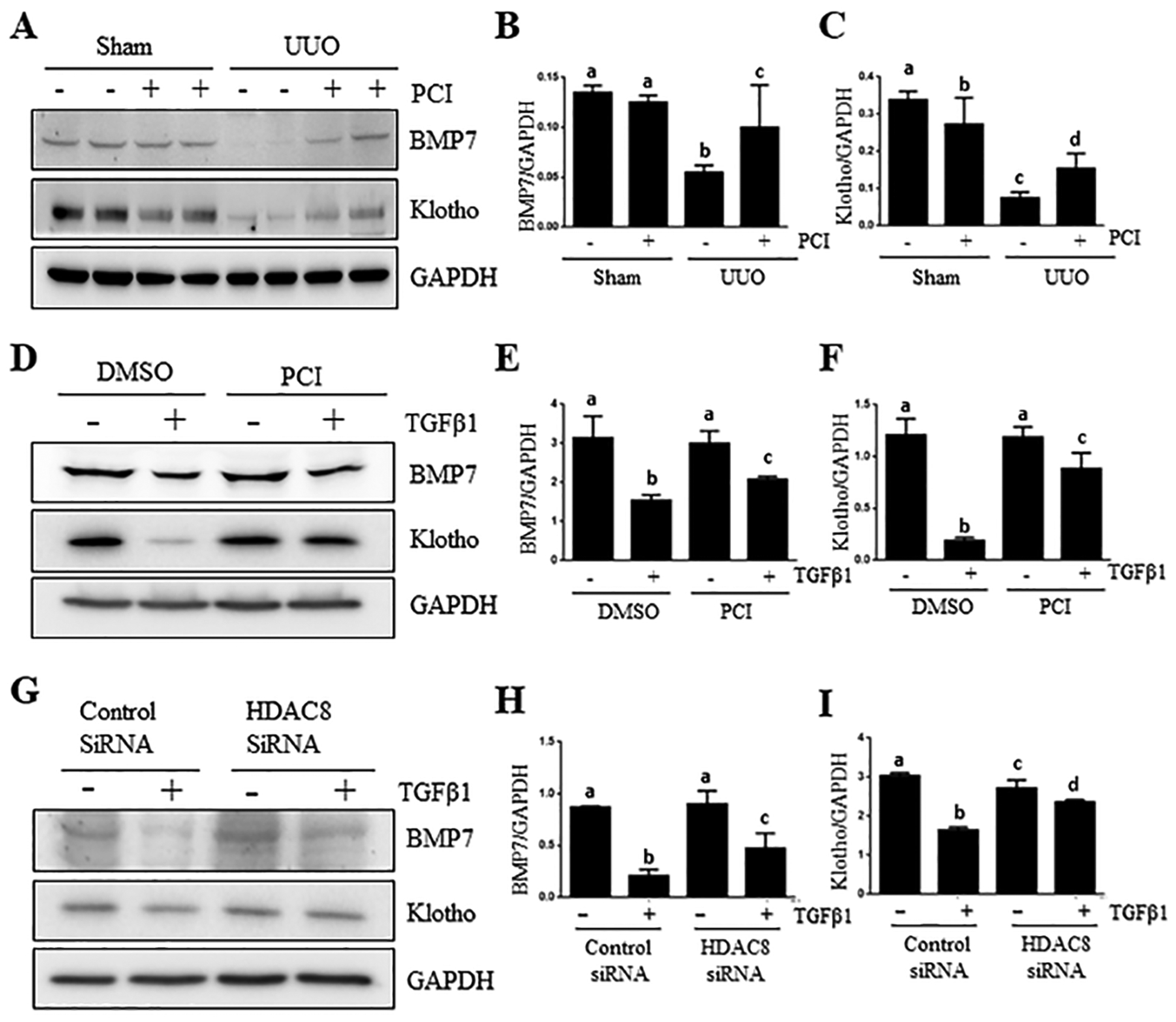
Kidneys were collected at 7 days after UUO treatments with or without PCI34051 (PCI) (A). TKPT were serum starved for 24 hours and then treated with PCI34051 (PCI) at 6.26 μM for 1 hour (D) or transfected with HDAC8 siRNA or scrambled siRNA for 24 hours (G) followed by exposure of cells to TGFβ1 (5 ng/ml) for an additional 24 hours. Kidney (A-C) and cell lysates (D-F, G-I) were subjected to immunoblot analysis with antibodies against BMP-7, Klotho and GAPDH (A, D, G). Expression levels of all those proteins were quantified by densitometry and normalized with GAPDH (B-C, E-F. H-I). Values are the means ± S.D (n = 6). Bars with different letters (a–c) are significantly different from one another (P < 0.05).
Discussion
Our and other studies have demonstrated that blocking class I HDACs inhibits development of renal fibrosis (22, 34), but the role of individual HDAC isoforms in this pathological process remains less explored. In this study, we investigated the role of HDAC8, one of class I HDACs, in partial epithelial-mesenchymal transformation of renal tubular cells and fibrogenesis. We demonstrated four novel findings: First, HDAC8 was abundantly expressed and located in renal epithelial cells of UUO kidneys, and inhibition of HDAC8 with PCI34051, a HDAC8 selective inhibitor, attenuated renal fibrosis. Second, either pharmacological blockade of HDAC8 or siRNA-mediated silencing of it inhibited TGF-β1 induced EMT. Third, HDAC8 inhibition resulted in dephosphorylation of several key profibrotic signaling molecules (Smad3, STAT3, β-catenin), reduction of the number of renal epithelial cells arrested at G2/M phase of cell cycle, and suppression of transcription factor Snail. Fourth and finally, HDAC8 inhibition reversed UUO and TGF-β1-induced downregulation of BMP-7 and Klotho (Figure 9). These results suggest that HDAC8 is a critical mediator of EMT and renal fibrogenesis and it regulates these processes by adjusting the balance of profibrotic and antiprofirotic signaling in the kidney.
Figure 9. The proposed mechanism of HDAC8-mediated renal fibrosis.
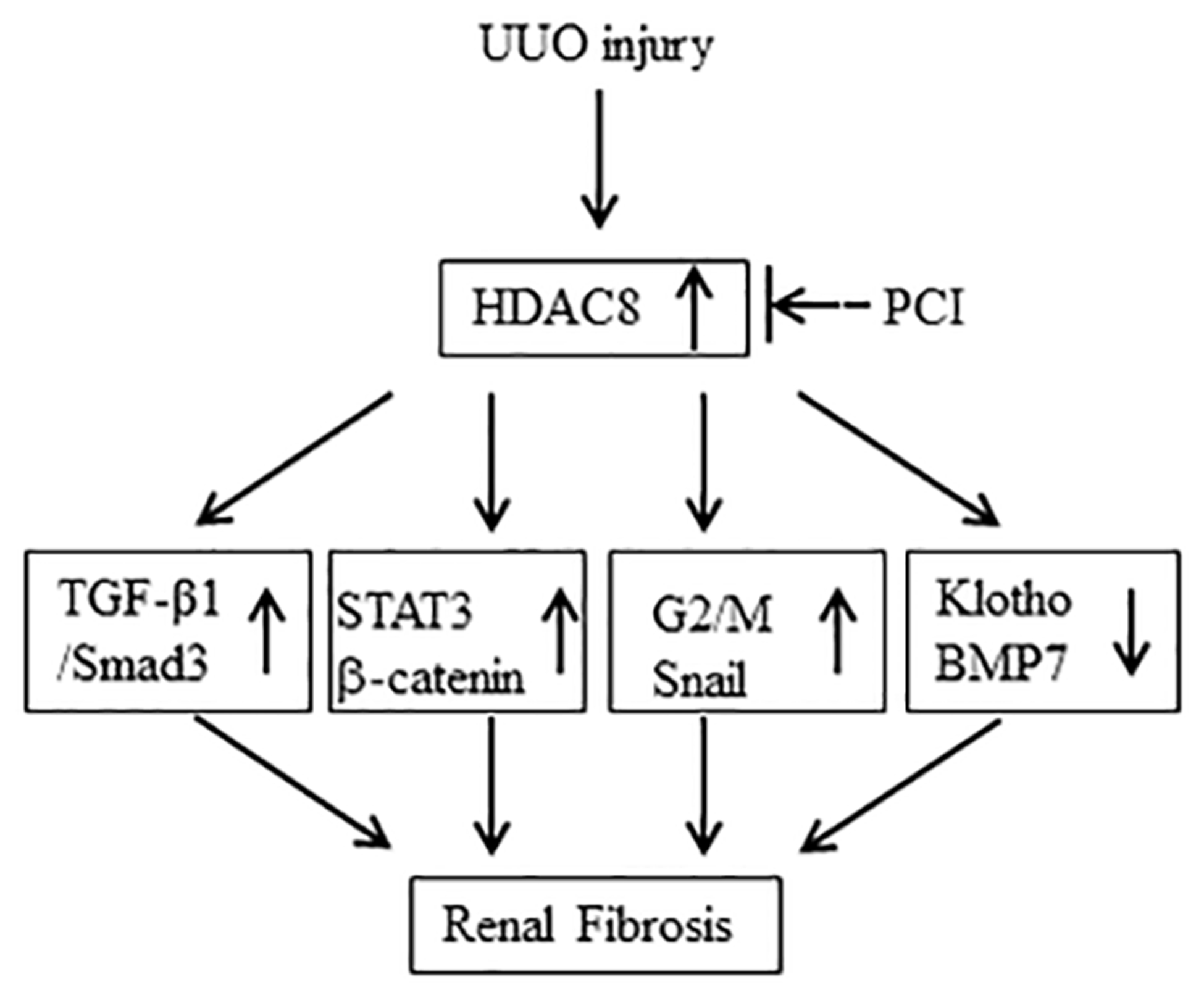
Injury to the kidney results in increased expression of HDAC8 in renal tubular cells. Increased HDAC8 activity leads to the activation of TGFβ/Smad3, STAT3, β-catenin signaling, induction of epithelial cells arrested at G2/M phase of cell cycle, upregulation of Snail and downregulation of klotho and BMP-7. Blocking class HDAC8 inhibits all these profibrotic responses.
HDAC8 is a unique member of class I HDAC family: Unlike other isoforms (HDAC1–3) in this class, HDAC8 is located in either the nucleus or cytoplasm and does not contain a C-terminal protein-binding domain (18). Although HDAC 8 has been reported to induce deacetylation of histone H3 and H4, several studies failed to demonstrate that histones are its substrate (18). In agreement with those observations, we observe that pharmacological HDAC 8 inhibition did not alter acetylation levels of histone H3 in the kidney following UUO injury and in cultured renal epithelial cells in response to TGF-β1. A possible explanation is that global acetylation modification of histone H3 may mask the specific acetylation by HDAC8; another explanation is that HDAC 8 may be translocated from the nucleus to the cytoplasm in response to those insults. In support of the latter hypothesis, we found that 1) HDAC8 was located in the cytosol of renal epithelial cells following UUO injury and cultured renal epithelial cells exposed to TGFβ1, and 2) HDAC8 inhibition restored acetylation of cortactin, a HDAC8 substrate expressed in the cytosol of renal epithelial cells, after UUO injury in vivo and TGF β1 exposure in vitro. Given that cortactin is only expressed in the cytoplasm of cells, our results also imply that HDAC8 may regulate renal fibrotic process through a mechanism primarily involved in the regulation of intracellular proteins in the cytosol.
Indeed, we observed that HDAC8 inhibition led to inactivation of several profibrotic intracellular signaling molecules including Smad3, STAT3 and β-catenin in vivo and in vitro. Smad3 is a key mediator in TGF-β profibrotic signaling and is phosphorylated upon TGFβ1 stimulation in the cytosol and then translocated to the nucleus where drives expression of multiple genes associated with tissue fibrosis, including collagen 1 and fibronectin (29). In support of the role of HDAC8 in regulating Smad3 and renal fibrosis, we demonstrated that either pharmacological blockade of HDAC8 or siRNA-mediated downregulation of it reduced Smad3 phosphorylation and expression of α-SMA, collagen1, and fibronectin in the UUO kidney and culture renal epithelial cells. This suggests that HDAC8 is coupled to the TGFβ/Smad3 signaling pathway. Although it remains poorly understood how HDAC8 regulates this signaling pathway, there is the possibility that it acts on any components of this signaling pathway from the membrane to the nucleus, theoretically. In this study, we found that HDAC8 inhibition suppressed TGF-β1 expression in the kidney after injury, suggesting that HDAC8 may directly or indirectly regulate expression and release of TGF-β1 from the injured kidney and thereby promote Smad3 phosphorylation. Of course, we cannot rule out the possibility that HDAC8 also promotes Smad3 phosphorylation by reducing expression of the intracellular proteins against Smad-3 phosphorylation. This hypothesis is supported by one of our observations that inhibition of class II HDAC upregulates Smad7, an inhibitory protein of the TGF-β1/Smad3 signaling pathway (23). Further studies are needed to examine the effect of HDAC8 inhibition on the expression of this or other proteins that have anti-fibrotic effects in our model system.
STAT3 and β-catenin are two intracellular signaling molecules. Like Smad3, they are also phosphorylated in the cytosol and then translocated to the nucleus to regulate expression of genes associated with renal fibrosis (32, 35). For example, Bienaimé et al found that injury to the epithelium leads to release of platelet-derived growth factor-BB, matrix metalloproteinase1, and lipocalin 2 in a STAT3-dependent manner (31). These factors contribute to fibroblast survival, proliferation, and activation (36). Similarly, activation of β-catenin is required for the expression of a wide variety of mediators implicated in kidney fibrosis, such as fibronectin, Snail1, matrix metalloproteinase-7, and various components of the renin-angiotensin system (37). Here, we found that inhibition of HDAC8 completely blocked phosphorylation of STAT3 and β-catenin in the kidney after UUO injury and renal epithelial cells with exposure to TGF-β1, suggesting the importance of HDAC8 in mediating activation of these two molecules. Currently, the mechanisms underlying HDAC8 inhibition-elicited inactivation of these two pathways remain unclear. This may occur indirectly through suppression of the membrane receptors and intracellular signaling molecules such as cytokines that lead or STAT3 and β-catenin phosphorylation or directly through an acetylation switch dependent mechanism. In support of these possibilities, we have recently demonstrated that treatment with MS-275, a class I family of HDACs, blocked UUO-induced phosphorylation and expression of EGFR, a critical upstream activator of STAT3 (34), and that administration of Trichostatin A (TSA), a class I/II HDAC inhibitor, resulted in STAT3 acetylation at Lys 608 and dephosphorylation at Tyr 375 in cultured renal fibroblasts (38). Similarly, other groups have reported that selective inhibition of HDAC6, another cytosolic HDAC isoform, led to increased acetylation of Lys49 on β-catenin in neural progenitor cells, which was accompanied by a decreased ubiquitination of β-catenin, and an increased localization of β-catenin at the plasma membrane (39). Given that an increase of β-catenin in membrane location would reduce its translocation to the nucleus (40), it is assumed that this will reduce its transcription activity. This hypothesis is worthy of investigations in the future. However, whatever mechanisms are involved in the regulation of STAT3 and β-catenin by HDAC8, these observations, together with HDAC8 inhibition elicited-suppression of renal fibrosis and the TGFβ/Smad3 signaling, suggest that these two pathways may be in concert with Smad3, transducing HDAC8 activation of renal fibrogenesis.
HDAC8 has been linked to EMT by deregulated expression or interaction with transcription factors critical to tumorigenesis (18). Since injury-induced reactivation of Snail is key to driving partial EMT and renal fibrosis, we examined the possible impact of HDAC8 inhibition on EMT in vivo and in vitro. Our studies suggest that HDAC8 is a critical epigenetic player in regulating partial EMT of renal epithelial cells. This is evidenced by following observations: 1) HDAC8 expression is limited to renal tubular cells and increased in the kidney overtime after UUO injury, 2) pharmacological inhibition of HDAC8 largely blocked UUO or TGF-β1-induced upregulation of renal α-SMA, collagen I and fibronectin, and 3) HDAC8 inhibition suppressed expression of Snail in both the injured kidney and renal epithelial cells Moreover, since arrest of epithelial cells at the G2/M phase of cell cycle is a prerequisite for EMT, our finding that HDAC8 inhibition resulted in the blockage of G2/M phase of cell cycle, also favors the hypothesis that HDAC8 epigenetically regulates EMT of renal epithelial cells.
HDAC8 inhibition mediated attenuation of renal fibrosis may also be associated with preservation of Klotho and BMP-7. Klotho is a transmembrane protein that protects against acute and chronic injury (41); BMP7 is a protein of the TGF-β super family and regarded as a counteracting molecule against TGF-β signaling. Numerous studies have shown that expression levels of Klotho and BMP-7 are reduced in the kidneys in several animal models of CKD and their downregulation promotes renal fibrosis (41). Recent studies indicated that treatment with the pan-HDAC inhibitor trichostatin A can restore Klotho and BMP-7 expression and their renoprotective effects (42), showing the importance for renal function of HDAC activation in mediating expression of these two proteins. Along with these observations, we found that selective inhibition of HDAC8 with PCI largely restored UUO-induced repression of Kloth and BMP-7 in the kidney and TGFβ1-induced repression in cultured TKPT. Therefore, HDAC8 may be a critical isoform of HDACs in mediating regulation of Klotho and BMP-7 expression in the kidney. The detailed mechanism underlying HDAC8 regulation of Klotho and BMP-7 expression has not yet been described. It has been reported that Klotho preservation by trichostatin A (TSA)-inhibition of HDACs occurred at the transcription level and is mediated by acetylation of peroxisome Proliferation-activated receptor γ (PPARγ), a major transcription factor (42). However, since HDAC8 is mainly distributed in the cytosol of renal tubular cells as indicated in this study, it is less likely that this is a major regulatory mechanism for reduced expression of Klotho in UUO kidney. As with BMP-7, it has been reported that TSA-mediated HDAC inhibition was accompanied by augmented renal expression of BMP-7 in the injured kidney or human renal epithelial cells undergoing EMT, but there are so far no mechanistic studies (43, 44). Further investigations are thus needed to examine the cellular basis for HDAC8-regulated expression of both klotho and BMP-7.
HDAC8 has emerged as an attractive target for isotype-selective drug development in a variety of diseases, such as cancer, parasitic and viral infections, as well as neurodegenerative diseases (18). Despite selective inhibition of HDAC8 with NCC170 has been shown to offer a beneficial effect to pulmonary fibrosis in an animal model of idiopathic pulmonary fibrosis (45), the role of this enzyme and efficacy of HDAC8 selective inhibitors in other models of diseases associated with tissue fibrosis remain largely unknown. In this work, we demonstrated a remarkable inhibitory effect of PCI34051 on HDAC8, EMT and renal fibrosis. This suggests that HDAC8 could be a novel therapeutic target in fibrotic diseases and that selective inhibition of HDAC8 would be a promising therapeutic treatment for protecting against this pathological condition. Additional investigation is required to define the role of HDAC8 and determine the effectiveness and side effect of its selective inhibitors in other models of renal fibrosis.
In this study, we demonstrated for the first time that selective HDAC8 inhibition attenuates renal fibrogenesis through suppression of partial EMT and multiple profibrotic signaling pathways. In particular, we found that HDAC8 is coupled to the regulation of expression of Snail, a key transcription factor that drive EMT. Although the exact mechanism by which HDAC8 regulate Snail and other signaling pathways remains unclear, we provide evidence that HDAC8-mediated epigenetic regulation is critically involved in trigging the machinery of EMT and fibrosis in the injured kidney. Thus, selective inhibition of HDAC8 inhibitors should be a potential therapeutic approach for the treatment of renal fibrotic diseases.
Supplementary Material
Supplemental Figure 1. Pharmacological inhibition of HDAC8 with PCI34051(PCI) abolishes expression of twist in the kidney after unilateral ureteral obstruction (UUO). Kidneys were collected at 7 days after various treatments as indicated. Lysates of kidney tissues after sham and UUO treatment were subjected to immunoblot analysis with specific antibodies against twist or GAPDH (A). The expression levels of Twist (B) were quantified by densitometry and normalized with GAPDH. Data are means ± SEM (n =6). Means with different letters are significantly different from one another. P < 0.05.
Supplemental 2. Pharmacological inhibition of HDAC8 with PCI34051(PCI) reduces expression of MMP-9 and MMP-2 and in the kidney after unilateral ureteral obstruction (UUO). Kidneys were collected at 7 days after various treatments as indicated. Lysates of kidney tissues after sham and UUO treatment were subjected to immunoblot analysis with specific antibodies against MMP-9, MMP-2 or GAPDH (A). The expression levels of MMP-2 (C) and MMP-9 (B) were quantified by densitometry and normalized with GAPDH. Data are means ± SEM (n =6). Means with different letters are significantly different from one another. P < 0.05.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81670623 and 81830021 to S.Z.), the Branch Grant of National Key Grants of the Ministry of Science and Technology (2018YFA0108802 to SZ) and the US National Institutes of Health (1R01DK113256-01A1 to S.Z).
Abbreviations:
- HDACs
Histone deacetylases
- EMT
epithelial-mesenchymal transitions
- UUO
unilateral ureteral obstruction
- TGFβ1
including transforming growth factor β1
- CTGF
connective tissue growth factor
- BMP-7
bone morphogenetic protein 7
- HATs
histone acetyltransferases
- TKPT
murine renal proximal tubular cells
- MET
mesenchymal to epithelial transition
- PPARγ
peroxisome Proliferation-activated receptor γ
- TSA
trichostatin A
Footnotes
Conflict of Interest Statement
No conflict of interest
References
- 1.Chen W, and Bushinsky DA (2017) Chronic kidney disease: KDIGO CKD-MBD guideline update: evolution in the face of uncertainty. Nature reviews. Nephrology 13, 600–602 [DOI] [PubMed] [Google Scholar]
- 2.Ng JK, and Li PK (2018) Chronic kidney disease epidemic: How do we deal with it? Nephrology (Carlton, Vic.) 23 Suppl 4, 116–120 [DOI] [PubMed] [Google Scholar]
- 3.Docherty MH, O’Sullivan ED, Bonventre JV, and Ferenbach DA (2019) Cellular Senescence in the Kidney. Journal of the American Society of Nephrology : JASN 30, 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lovisa S, Zeisberg M, and Kalluri R (2016) Partial Epithelial-to-Mesenchymal Transition and Other New Mechanisms of Kidney Fibrosis. Trends in endocrinology and metabolism: TEM 27, 681–695 [DOI] [PubMed] [Google Scholar]
- 5.Basile C (2008) The long-term prognosis of acute kidney injury: acute renal failure as a cause of chronic kidney disease. Journal of nephrology 21, 657–662 [PubMed] [Google Scholar]
- 6.Liu Y (2010) New insights into epithelial-mesenchymal transition in kidney fibrosis. Journal of the American Society of Nephrology : JASN 21, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI, and Tomic-Canic M (2016) Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell and tissue research 365, 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loboda A, Sobczak M, Jozkowicz A, and Dulak J (2016) TGF-beta1/Smads and miR-21 in Renal Fibrosis and Inflammation. Mediators Inflamm 2016, 8319283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan J, Shi M, Li L, Liu J, Guo F, Feng Y, Ma L, and Fu P (2019) Pterostilbene, a bioactive component of blueberries, alleviates renal fibrosis in a severe mouse model of hyperuricemic nephropathy. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 109, 1802–1808 [DOI] [PubMed] [Google Scholar]
- 10.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, and Kalluri R (2015) Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nature medicine 21, 998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, and Wysocka J (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, and Hu MC (2017) Klotho/FGF23 Axis in Chronic Kidney Disease and Cardiovascular Disease. Kidney diseases (Basel, Switzerland) 3, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li RX, Yiu WH, and Tang SC (2015) Role of bone morphogenetic protein-7 in renal fibrosis. Frontiers in physiology 6, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji F, Wang K, Zhang Y, Mao XL, Huang Q, Wang J, Ye L, and Li Y (2019) MiR-542–3p controls hepatic stellate cell activation and fibrosis via targeting BMP-7. Journal of cellular biochemistry 120, 4573–4581 [DOI] [PubMed] [Google Scholar]
- 15.Mencke R, Olauson H, and Hillebrands JL (2017) Effects of Klotho on fibrosis and cancer: A renal focus on mechanisms and therapeutic strategies. Adv Drug Deliv Rev 121, 85–100 [DOI] [PubMed] [Google Scholar]
- 16.Tang J, and Zhuang S (2015) Epigenetics in acute kidney injury. Current opinion in nephrology and hypertension 24, 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhuang S (2018) Epigenetic targeting for acute kidney injury. Nephrology (Carlton, Vic.) 23 Suppl 4, 21–25 [DOI] [PubMed] [Google Scholar]
- 18.Chakrabarti A, Oehme I, Witt O, Oliveira G, Sippl W, Romier C, Pierce RJ, and Jung M (2015) HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci 36, 481–492 [DOI] [PubMed] [Google Scholar]
- 19.Kaiser FJ, Ansari M, Braunholz D, Concepcion Gil-Rodriguez M, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, Di Donato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillen-Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, McKee S, Mehta SG, Micule I, Mohammed S, Moran E, Mortier GR, Moser JA, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Perez-Aytes A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheuerle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Wilson LC, Hakonarson H, Quintero-Rivera F, Wierzba J, Musio A, Gillessen-Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pie J, Christianson DW, Krantz ID, Fitzpatrick DR, and Deardorff MA (2014) Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Human molecular genetics 23, 2888–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, and Buggy JJ (2008) A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 22, 1026–1034 [DOI] [PubMed] [Google Scholar]
- 21.Yamauchi Y, Boukari H, Banerjee I, Sbalzarini IF, Horvath P, and Helenius A (2011) Histone deacetylase 8 is required for centrosome cohesion and influenza A virus entry. PLoS pathogens 7, e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang M, Chen G, Zhang X, Guo Y, Yu Y, Tian L, Chang S, and Chen ZK (2019) Inhibition of class I HDACs attenuates renal interstitial fibrosis in a murine model. Pharmacol Res 142, 192–204 [DOI] [PubMed] [Google Scholar]
- 23.Xiong C, Guan Y, Zhou X, Liu L, Zhuang MA, Zhang W, Zhang Y, Masucci MV, Bayliss G, Zhao TC, and Zhuang S (2019) Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. Faseb j 33, 8249–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan Y, Ma L, Zhou X, Ponnusamy M, Tang J, Zhuang MA, Tolbert E, Bayliss G, Bai J, and Zhuang S (2016) Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney international 89, 68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SY, Kee HJ, Kurz T, Hansen FK, Ryu Y, Kim GR, Lin MQ, Jin L, Piao ZH, and Jeong MH (2016) Class I HDACs specifically regulate E-cadherin expression in human renal epithelial cells. J Cell Mol Med 20, 2289–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canaud G, and Bonventre JV (2015) Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol Dial Transplant 30, 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon-Tillaux N, and Hertig A (2017) Snail and kidney fibrosis. Nephrol Dial Transplant 32, 224–233 [DOI] [PubMed] [Google Scholar]
- 28.Ning X, Zhang K, Wu Q, Liu M, and Sun S (2018) Emerging role of Twist1 in fibrotic diseases. Journal of cellular and molecular medicine 22, 1383–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng XM, Nikolic-Paterson DJ, and Lan HY (2016) TGF-beta: the master regulator of fibrosis. Nature reviews. Nephrology 12, 325–338 [DOI] [PubMed] [Google Scholar]
- 30.Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, Chin YE, Yan H, Dworkin LD, and Zhuang S (2010) A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney international 78, 257–268 [DOI] [PubMed] [Google Scholar]
- 31.Bienaime F, Muorah M, Yammine L, Burtin M, Nguyen C, Baron W, Garbay S, Viau A, Broueilh M, Blanc T, Peters D, Poli V, Anglicheau D, Friedlander G, Pontoglio M, Gallazzini M, and Terzi F (2016) Stat3 Controls Tubulointerstitial Communication during CKD. Journal of the American Society of Nephrology : JASN 27, 3690–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuo Y, and Liu Y (2018) New insights into the role and mechanism of Wnt/beta-catenin signalling in kidney fibrosis. Nephrology (Carlton, Vic.) 23 Suppl 4, 38–43 [DOI] [PubMed] [Google Scholar]
- 33.Higgins DF, Ewart LM, Masterson E, Tennant S, Grebnev G, Prunotto M, Pomposiello S, Conde-Knape K, Martin FM, and Godson C (2017) BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim Biophys Acta Mol Basis Dis 1863, 3095–3104 [DOI] [PubMed] [Google Scholar]
- 34.Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, Bayliss G, Zhao TC, Yan H, and Zhuang S (2013) Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PloS one 8, e54001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsui F, and Meldrum KK (2012) The role of the Janus kinase family/signal transducer and activator of transcription signaling pathway in fibrotic renal disease. The Journal of surgical research 178, 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duffield JS (2016) Beyond EMT: Epithelial STAT3 as a Central Regulator of Fibrogenesis. Journal of the American Society of Nephrology : JASN 27, 3502–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hao S, He W, Li Y, Ding H, Hou Y, Nie J, Hou FF, Kahn M, and Liu Y (2011) Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. Journal of the American Society of Nephrology : JASN 22, 1642–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, and Zhuang S (2009) Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. American journal of physiology. Renal physiology 297, F996–f1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iaconelli J, Huang JH, Berkovitch SS, Chattopadhyay S, Mazitschek R, Schreiber SL, Haggarty SJ, and Karmacharya R (2015) HDAC6 inhibitors modulate Lys49 acetylation and membrane localization of beta-catenin in human iPSC-derived neuronal cells. ACS chemical biology 10, 883–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das L, Kokate SB, Dixit P, Rath S, Rout N, Singh SP, Crowe SE, and Bhattacharyya A (2017) Membrane-bound beta-catenin degradation is enhanced by ETS2-mediated Siah1 induction in Helicobacter pylori-infected gastric cancer cells. Oncogenesis 6, e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuro OM (2019) The Klotho proteins in health and disease. Nature reviews. Nephrology 15, 27–44 [DOI] [PubMed] [Google Scholar]
- 42.Lin W, Li Y, Chen F, Yin S, Liu Z, and Cao W (2017) Klotho preservation via histone deacetylase inhibition attenuates chronic kidney disease-associated bone injury in mice. Scientific reports 7, 46195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imai N, Hishikawa K, Marumo T, Hirahashi J, Inowa T, Matsuzaki Y, Okano H, Kitamura T, Salant D, and Fujita T (2007) Inhibition of histone deacetylase activates side population cells in kidney and partially reverses chronic renal injury. Stem cells (Dayton, Ohio) 25, 2469–2475 [DOI] [PubMed] [Google Scholar]
- 44.Marumo T, Hishikawa K, Yoshikawa M, and Fujita T (2008) Epigenetic regulation of BMP7 in the regenerative response to ischemia. Journal of the American Society of Nephrology : JASN 19, 1311–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito S, Zhuang Y, Suzuki T, Ota Y, Bateman ME, Alkhatib AL, Morris GF, and Lasky JA (2019) HDAC8 inhibition ameliorates pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 316, L175–L186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Pharmacological inhibition of HDAC8 with PCI34051(PCI) abolishes expression of twist in the kidney after unilateral ureteral obstruction (UUO). Kidneys were collected at 7 days after various treatments as indicated. Lysates of kidney tissues after sham and UUO treatment were subjected to immunoblot analysis with specific antibodies against twist or GAPDH (A). The expression levels of Twist (B) were quantified by densitometry and normalized with GAPDH. Data are means ± SEM (n =6). Means with different letters are significantly different from one another. P < 0.05.
Supplemental 2. Pharmacological inhibition of HDAC8 with PCI34051(PCI) reduces expression of MMP-9 and MMP-2 and in the kidney after unilateral ureteral obstruction (UUO). Kidneys were collected at 7 days after various treatments as indicated. Lysates of kidney tissues after sham and UUO treatment were subjected to immunoblot analysis with specific antibodies against MMP-9, MMP-2 or GAPDH (A). The expression levels of MMP-2 (C) and MMP-9 (B) were quantified by densitometry and normalized with GAPDH. Data are means ± SEM (n =6). Means with different letters are significantly different from one another. P < 0.05.