

Abstract
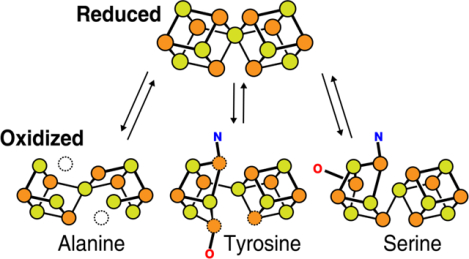
Molybdenum nitrogenase catalyzes the reduction of dinitrogen into ammonia, which requires the coordinated transfer of eight electrons to the active site cofactor (FeMoco) through the intermediacy of an [8Fe-7S] cluster (P-cluster), both housed in the molybdenum-iron protein (MoFeP). Previous studies on MoFeP from two different organisms, Azotobacter vinelandii (Av) and Gluconacetobacter diazotrophicus (Gd), have established that the P-cluster is conformationally flexible and can undergo substantial structural changes upon two-electron oxidation to the POX state, whereby a backbone amidate and an oxygenic residue (Ser or Tyr) ligate to two of the cluster’s Fe centers. This redox-dependent change in coordination has been implicated in the conformationally gated electron transfer in nitrogenase. Here, we have investigated the role of the oxygenic ligand in Av MoFeP, which natively contains a Ser ligand (βSer188) to the P-cluster. Three variants were generated in which 1) the oxygenic ligand was eliminated (βSer188Ala), 2) the P-cluster environment was converted to the one in Gd MoFeP (βPhe99Tyr/βSer188Ala), and 3) two oxygenic ligands were simultaneously included (βPhe99Tyr). Our studies have revealed that the P-cluster can become compositionally labile upon oxidation and re-versibly lose one or two Fe centers in the absence of the oxygenic ligand, while still retaining wild-type like dinitrogen reduction activity. Our findings also suggest that Av and Gd MoFeP’s evolved with specific preferences for Ser and Tyr ligands, respectively, and that the structural control of these ligands must extend beyond the primary and secondary coordination spheres of the P-cluster. The P-cluster adds to the increasing number of examples of inherently labile Fe-S clusters whose compositional instability may be an obligatory feature to enable redox-linked conformational changes to facilitate multi-electron redox reactions.
Graphical Abstract
Introduction
Nitrogenase is the only known enzyme capable of catalyzing the reduction of dinitrogen to ammonia:1–3
The most commonly studied form of this enzyme, the molybdenum-containing nitrogenase, is composed of two proteins: the homodimeric (γ2) iron-protein (FeP) and the heterotetrameric (α2β2) molybdenum-iron protein (MoFeP) (Figure 1). FeP is an ATPase containing a [4Fe-4S]-cluster that reduces the catalytic MoFeP through transient proteinprotein interactions controlled by ATP binding and hydrol-ysis.4 In line with its unique chemical reactivity, MoFeP houses two exclusive metalloclusters. The iron-molybdenum cofactor (FeMoco, a [7Fe-1Mo-9S-1C] cluster) is the catalytic site for N2 reduction,5–7 whereas the P-cluster (an [8Fe-7S] cluster)8 acts as the electron relay site between the [4Fe-4S] cluster of FeP and FeMoco. Justifiably, research efforts to understand nitrogenase function and its mechanistic details have primarily focused on FeMoco.9–12 Detailed structural, spectroscopic and biochemical investigations in the last two decades have firmly established the composition of this cluster,5,7 narrowed down the possible sites of substrate binding and activation,13 and have begun to put forth plausible mechanistic scenarios for the complex 8 e−/8 H+ catalytic process.3,12,14 Excitingly, recent crystallo-graphic findings indicate that FeMoco can even undergo changes in composition whereby an integral bridging sulfide in the cluster may be labilized during the catalytic cycle to enable substrate binding,13 and that FeVco contains a labile sulfide that can be replaced by a carbonate bridging ligand.15,16 We report here that the P-cluster is similarly redox-metastable and it may, under certain circumstances, also undergo alterations in its composition, reflective of its proposed role as a conformationally active electron relay site.
Figure 1.
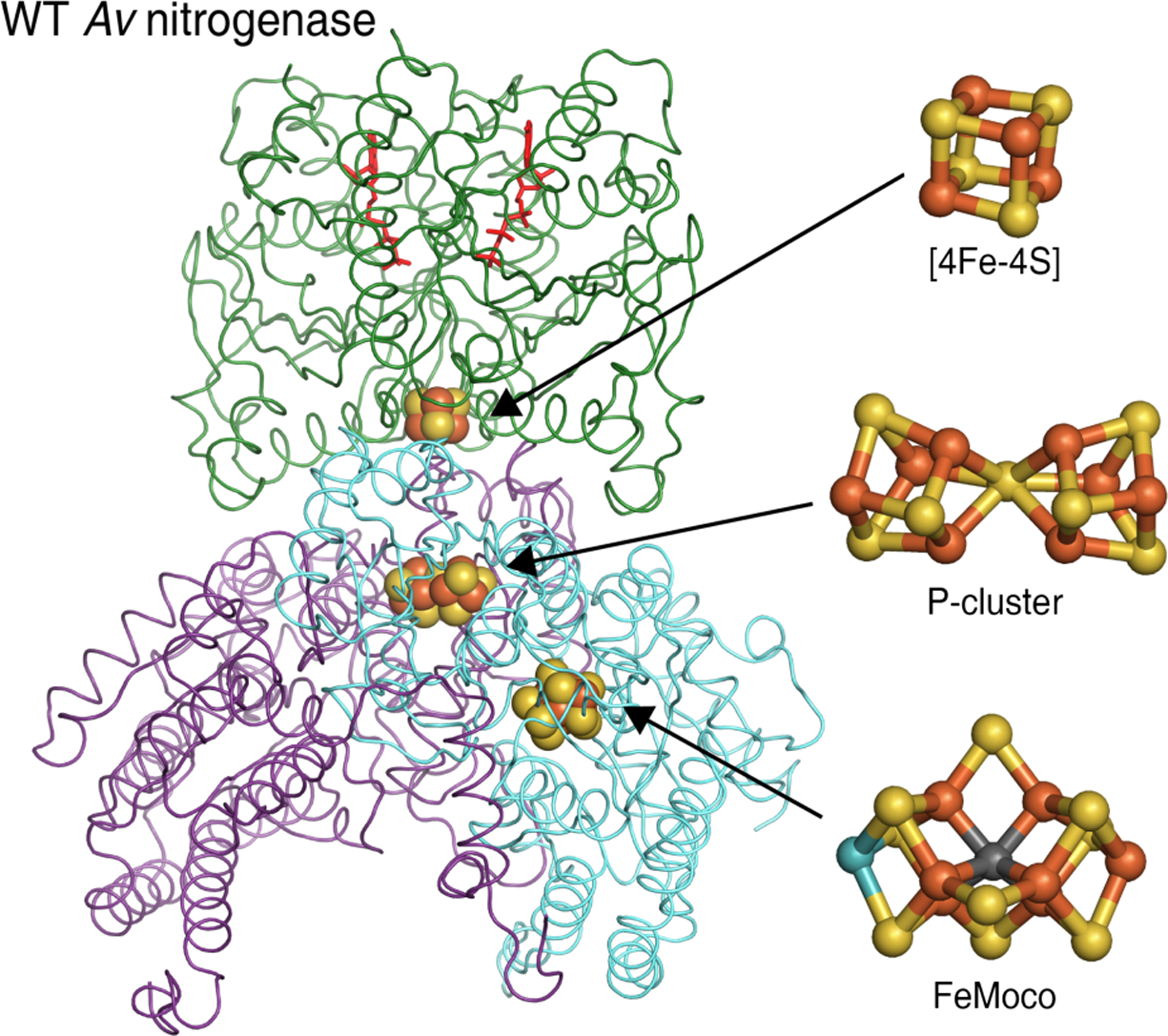
One of the two equivalent halves of the Av nitrogenase FeP-MoFeP complex. FeP (green) is an ATPase (nucleotides in red) containing a [4Fe-4S] cluster. In MoFeP, the P-cluster bridges the α (blue) and β (purple) subunits, and FeMoco is housed in the a subunit. (PDB ID: 4WZB)
There are several lines of evidence-though circumstantial-suggesting that the unique composition of the P-cluster is critical for nitrogen fixation and may have specifically evolved for its role as a special electron donor to FeMoco. First, nitrogenase homologs which carry out different and arguably less complex multi-electron catalytic reactions such as the dark-operative protochlorophyllide oxidoreductase17 and the methanogenic oxidoreductase CfbD18,19 contain a more typical [4Fe-4S] cluster in the equivalent position of the P-cluster. Second, the only known reductant capable of activating MoFeP for nitrogen reduction is ATP-bound FeP1,2 despite its modest reduction potential (−420 mV).20 It is hypothesized that FeP binding must induce conformational changes within MoFeP that permit an initial electron transfer (ET) from the all-ferrous P-cluster (PN) to FeMoco,21 followed by the reduction of the one or two-electron oxidized P-cluster (P+1 or POX) by the [4Fe-4S] cluster of FeP22. Such a gated ET mechanism would require the P-cluster to be dynamic, whereby the FeP-induced conformational changes are transmitted to the P-cluster to change its redox equilibrium, enabling it to reduce FeMoco. This possibility is supported by recent electronic structure calculations which have revealed that the P-cluster has many accessible, low-energy electronic states that may help accommodate the different electron demands (i.e., redox potentials) of the many intermediate states of FeMoco populated during catalytic turnover.23 Importantly, it has been crystal-lographically observed that PN in Azotobacter vinelandii (Av) can undergo a large conformational change upon oxidation to the POX state.8 This transformation involves the opening of one of the two [4Fe-4S] cubane halves, where Fe5 and Fe6 dissociate from the central bridging sulfide and ligate to the negatively charged backbone amidate N of αC88 and the βS188 serinate, respectively, to stabilize two Fe3+ centers (Figure 2a).8,24 Interestingly, βS188 is not strictly conserved and is frequently replaced by alanine in different nitrogen-fixing organisms25,26, which has called into question its functional role. Yet, through investigations of one such MoFeP homolog from Gluconacetobacter diazotrophicus (Gd), we recently established that an equally unusual FeS-cluster ligand, the phenolate group from βY99 (Av numbering), could stabilize the POX state through coordination to Fe8 (Figure 2b).25,26 Notably, our sequence alignment of 95 nitrogenase sequences (including MoFeP’s and the alternate VFeP’s and FeFeP’s)revealed that β188 and β99 positions were covariant in 92 instances, such that one of the positions is an oxygenic ligand (Tyr or Ser) and the other a non-coordinating ligand (Table S1).
Figure 2.
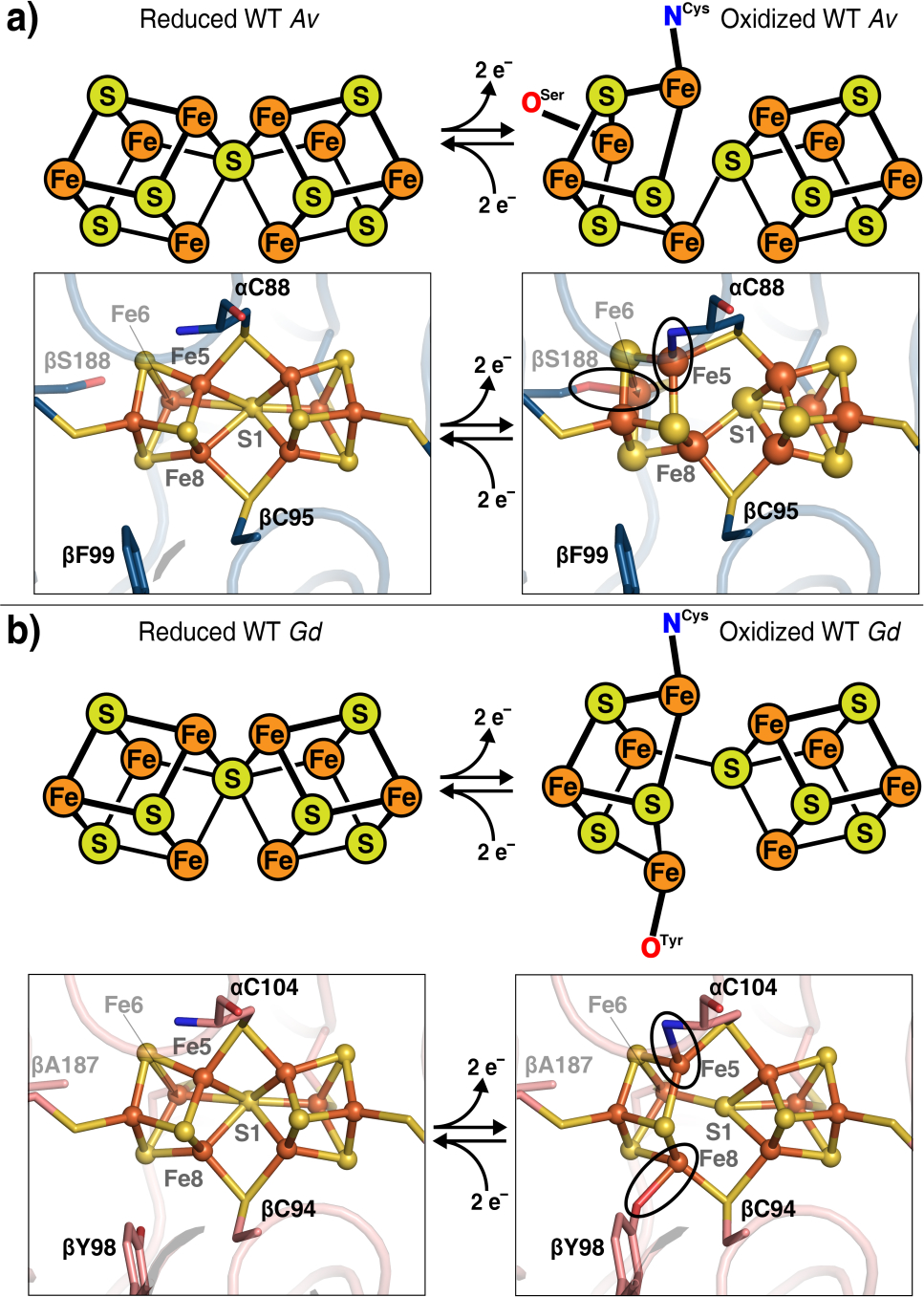
Wild-type DT-reduced (left) and IDS-oxidized (right) P-clusters. (a) In Av, two-electron oxidation of the P-cluster results in ligation of βS188 to Fe6 of the backbone amide of αC88 to Fe5 (circled). (PDB IDs: 3MIN and 2MIN) (b) In Gd, ligation of βY98 (βY99, Av numbering) to Fe8 and of the backbone amide of αC104 (αC88, Av numbering) to Fe5 occurs upon two-electron oxidation (circled). (PDB IDs 5KOH and 5KOJ)
Given that this covariance strongly implies that the Ser and Tyr residues must be functionally important, we set out to further investigate the role of these residues on the redox-mediated reorganization of the P-cluster and whether they can be interchanged. To this end, we have generated, purified to homogeneity, and characterized three Av MoFeP mutants in which there is (1) no oxygenic ligand (βS188A), (2) only tyrosine (βF99Y/βS188A) (replicating the coordination environment in Gd MoFeP), and (3) both serine and tyrosine (βF99Y) (Figures S1 and S2).
Results and Discussion
We first determined the crystal structures of βS188A-MoFeP in dithionite (DT)-reduced and indigo disulfonate (IDS)-oxidized states (both at 2.3 Å resolution, Table S2) to probe what, if any, redox-dependent reorganization of the P-cluster occurs in the absence of the native oxygenic ligand, serine. In the DT-reduced state, the P-cluster displays the expected PN conformation that is superimposable on the wild-type cluster (Figures 3a-left and S3a-left). Surprisingly, the two-electron oxidation of the P-cluster leads to complete loss of two Fe centers, Fe1 and Fe5, yielding a [6Fe-7S] P-cluster (Figures 3a-center, S3a-center, S4, and Table S3) - this new configuration can be viewed as two equivalent [3Fe-4S] clusters that share a bridging sulfide (S1). This finding indicates that a negatively charged, oxygenic ligand is not only necessary to stabilize the oxidized states (POX or the P+1) of the P-cluster, but also to keep its composition intact. It is important to note that the loss of both Fe centers occurs from the central, “over-coordinated” sulfide, rather than the peripheral position (Fe6) that normally coordinates βS188 upon oxidation to Fe3+. In the process, the two bridging Cys sidechains (αC88 and βC95) are also converted into terminal ligands. These changes give the most stable cluster configuration with 4-coordinate Fe centers and non-bridging Cys ligands, with the typical coordination metrics of canonical Fe-S clusters (Table 1). Inspection of the anomalous electron density maps calculated near the Fe K-edge did not reveal any Fe atoms associated with MoFeP except the P-cluster, FeMoco, and the “16th Fe-site”27 suggesting that the lost Fe ions either diffused completely out of MoFeP or are loosely bound to the protein. Interestingly, the [6Fe-7S] P-cluster could be fully converted back to the native-like [8Fe-7S] P-cluster upon re-reduction with DT (Figures S3a-right, and S5a). This behavior parallels the compositional lability of FeMoco as well as the metalloclusters in aconitase28 biotin synthase,29 lipoyl synthase,30 and CODH.31 Although the source of the incoming Fe atoms is unclear as there was no added Fe in the reconstitution solutions, the possibility exists that some MoFeP is cannibalized for its iron content, analogous to the reconstitution process of aconitase.32 Regardless, the efficient Fe2+ capture and structural recovery highlights the inherent stability of the [8Fe-7S] P-cluster in the reduced state, and conversely its inherent instability upon oxidation. In both structures of βS188A MoFeP, FeMoco is observed in its native (MN) conformation (Figure S6). Since the insertion of FeMoco into MoFeP only occurs after assembly of the intact and active P-cluster,33–35 it follows that the P-cluster in βS188A must have been properly assembled in vivo.
Figure 3.
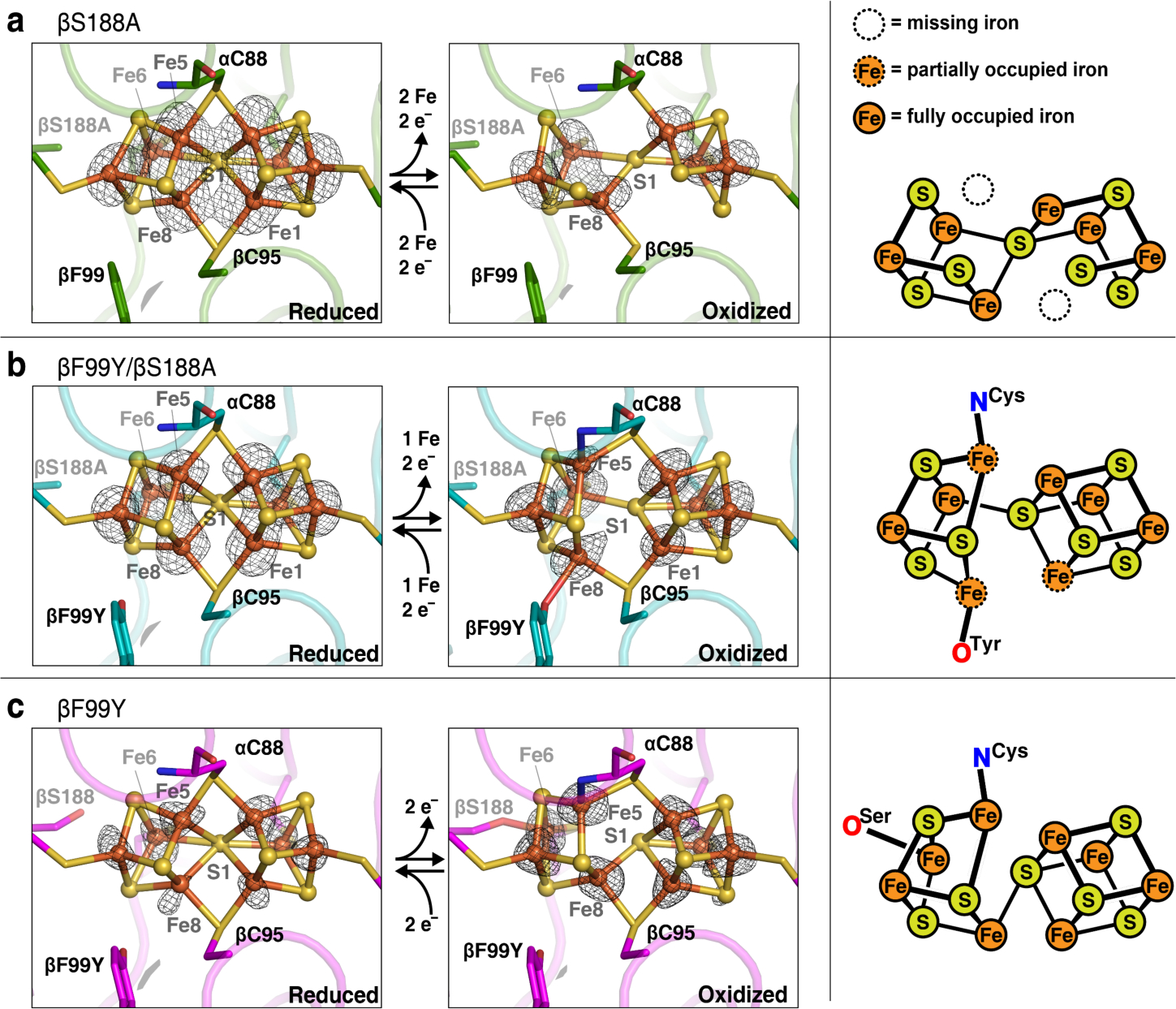
X-ray crystal structures of the P-clusters of Av MoFeP variants. Anomalous electron density difference maps determined using X-ray diffraction data collected near the Fe K-edge are shown in black mesh. (a) Av βS188A P-cluster in DT-reduced (left) and IDS-oxidized (middle) states (7.13 keV, Fe f” = 3.93 and 12.0 keV, Fe f’ = 1.64). Maps are contoured at 8.0 and 3.5 σ, respectively. Fe1 and Fe5 are both redox-labile (right). (b) Av βF99Y/βS188A P-cluster in DT-reduced (left) and IDS-oxidized (middle) states (7.13 keV, Fe f” = 3.93 and 8.0 keV, Fe f” = 3.18). Maps are contoured at 8.0 and 6.0 σ, respectively. Fe1, Fe5, and Fe8 are partially occupied (right). (c) Av βF99Y P-cluster in DT-reduced (left) and IDS-oxidized (middle) states (7.13 keV, Fe f’ = 3.93 and 13.0 keV, Fe f’ = 1.43). Maps are contoured at 3.5 σ. This mutant contains no redox-labile iron centers (right).
Table 1.
Crystallographically determined changes in Fe-S distances upon IDS-mediated oxidation of the PN state in MoFeP variants.
| Variant | Sl-Fe6 (Å) | S 1-Fe8 (Å) | Oser-Fe6 (Å) | Otyr-Fe8 (Å) | Ncys-Fe5 (Å) |
|---|---|---|---|---|---|
| βS188A | 2.7 → 2.5 | 2.4 → 2.3 | n/a | n/a | n/a |
| βF99Y/PS188A | 3.0 → 2.5 | 2.3 → 3.5 | n/a | 3.5 → 2.3 | 3.0 → 2.2 |
| βF99Y ox | 2.7 → 3.8 | 2.4 → 2.4 | 3.3 → 2.0 | 3.5 → 3.9 | 3.3 → 2.2 |
| wild-type Av | 2.5 → 4.0 | 2.4 → 2.5 | 3.4 → 1.9 | n/a | 3.4 → 2.1 |
| wild-type Gd | 2.5 → 2.4 | 3.6 → 2.1 | n/a | 3.6 → 2.1 | 3.4 → 2.6 |
It is well established that native Av P-cluster is EPR-silent in the PN state, whereas POX exhibits an integer-spin (S ≥ 3) with a feature at g ≈ 12 in the parall el collection mod e (Figures 4).20,25,36,37 in contrast, we found that the parallel-mode EPR spectrum of IDS-oxidized βS188A MoFeP is devoid of any spectral features (Figure 4c-green), suggesting that the majority of the P-clusters present in the sample are of a [6Fe-7S] stoichiometry in an all ferrous state, and that the two labile Fe atoms have dissociated from the cluster as Fe3+ (although we cannot definitively rule out the loss of two Fe2+ ions to yield an EPR-silent cluster containing four Fe2+ and two Fe3+). We note that the perpendicular-mode EPR spectrum of DT-reduced βS188A MoFeP is identical to that of native MoFeP, dominated by the S = 3/2 signature of FeMoco (Figure 4-solid green and dark blue).36,37 Oxidized βS188A MoFeP, on the other hand, displays weak absorption features near g ≈ 2 region that are not seen in the POX state of the native enzyme (Figure 4b-green). We postulate that these signals may arise from a minor population of P-clusters that have lost a single Fe3+ ion, yielding a non-integerspin signature reminiscent of the P+1 state previously observed in a previously characterized Av βS188C MoFeP38 and during potentiometric titrations of the wild-type Av P-cluster.36
Figure 4.
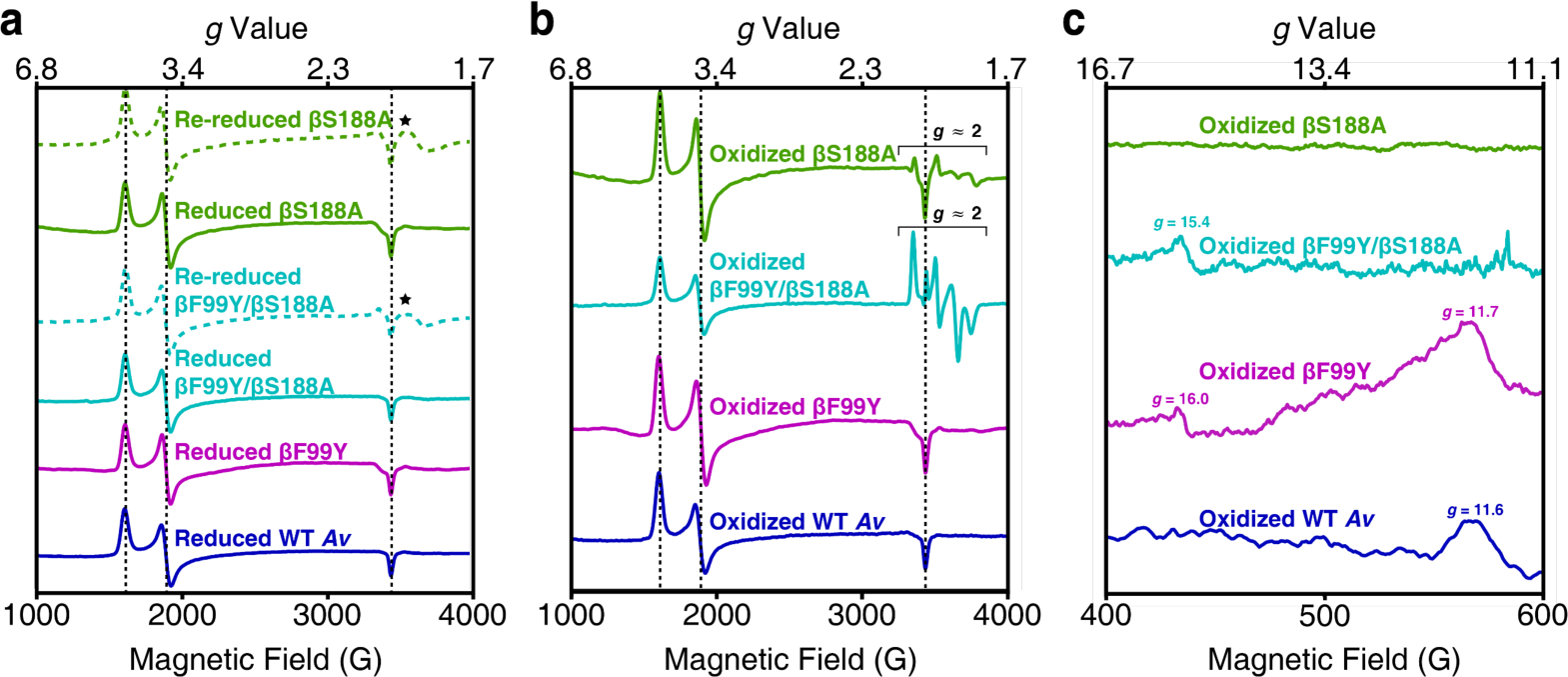
X-band EPR spectra of the Av MoFeP variants. All spectra were collected at 5–10 K. Dashed black lines mark the features that arise from the S = 3/2 signal associated with FeMoco. (a) Perpendicular-mode EPR spectra of DT-reduced and DT-re-reduced MoFeP. Reduced mutants have the same FeMoco-associated features present in wild-type Av. The spectra of re-reduced βS188A and βF99Y/βS188A MoFeP display new features in the g ≈ 1.9 region (⋆) that may arise from some amount of degraded protein. The data (from top to bottom) were collected at 8 K, 6 K, 9 K, 5 K, 6 K, and 8 K. (b) Perpendicular-mode EPR spectra of IDS-oxidized MoFeP. The spectra of βS188A and βF99Y/βS188A MoFeP have features in the g ≈ 2.0 region attributed to a population of MoFeP containing only one Fe3+ per P-cluster. Data (from top to bottom) were collected at 5 K, 9 K, 6 K, and 8 K. (c) Parallel-mode spectra of the oxidized Av MoFeP demonstrate that ligation of the oxygenic ligand to the P-cluster results in an integer spin. Tyrosine ligation (βF99Y/βS188A and βF99Y) results in features at g ≈ 16, and serine ligation (βF99Y and wild-type) at g ≈ 12.
Using our βF99Y/βS188A Av MoFeP mutant, we next investigated if a tyrosine residue in the equivalent position as native Gd MoFeP would provide enough stability to maintain the stoichiometry of the [8Fe-7S] P-cluster upon oxidation. The structure of the DT-reduced P-cluster of βF99Y/βS188A (2.3 Å resolution) is superimposable onto that of its wild-type counterpart (Figure 3b-left and S3b-left), whereas the structure of the IDS-oxidized cluster (1.8 Å resolution) reveals an unusual mixture of multiple conformational states (Figures 3b-center and 5). We observe that βF99Y indeed ligates Fe8 as planned, and the backbone amide of αC88 coordinates Fe5. Yet, Fe1, Fe5, and Fe8 (all coordinated to the central sulfide in the PN state) are only partially occupied with a summed average of 2.0 Fe atoms (Figures 3b-right and Table S3) indicating that each P-cluster has on average one labile Fe and possesses a [7Fe-7S] stoichiometry. The electron densities are well-described by a structural model involving the equilibrium of three conformational states in which any one of Fe1, Fe5 or Fe8 is missing (Figure 5). Such an equilibrium is consistent with a flat free energy landscape governing the redox-dependent conformational changes of the P-cluster and implies that Fe3+ ligation by the oxygenic ligand (tyrosinate or serinate) and the αC88 backbone amidate are nearly isoenergetic. This, in turn, accords with the observation that the chemical oxidation of the P-cluster is a two-electron process at pH 8 (PN→POX),39 and that it is difficult to populate the singly oxidized form of the P-cluster (P+1) due to the approximately equal midpoint potentials of both redox couples.20,39 The unusual dynamic structural properties of the [7Fe-7S] cluster are also reflected in the EPR spectra of βF99Y/βS188A-MoFeP (Figure 4-light blue). Strong features are observed near g ≈ 2 in the perpendicular mode, suggesting the presence of an electronic state with non-integer spin (likely S = ½),36,37 whereas the signal at g ≈ 15.4 in the parallel mode indicates the simultaneous population of an integer-spin state (S ≥ 3) that is characteristic of tyrosinate coordination in POX (Figures 4b and 4c-light blue).25,37 As in the case of βS188A, the re-reduction of the iron-deficient cluster in βF99Y/βS188A, leads to the reconstitution of the intact [8Fe-7S] cluster (Figures S3b-right and S5b).
Figure 5.

Equilibrium conformational states of the oxidized [7Fe-7S] P-cluster of Av βF99Y/βS188A MoFeP. The non-native tyrosine ligates the cluster in two of the equilibrium states, and Fe1, Fe5, and Fe8 are redox-labile.
It is interesting to consider that the βF99Y/βS188A double mutation converts the Av P-cluster coordination environment to the one in Gd. In fact, a structural superposition of oxidized Av βF99Y/βS188A MoFeP protein and native Gd MoFeP shows that P-cluster environments in the two proteins are essentially identical (RMSD = 0.165 Å), including all side chain orientations within 7–8 Å of the cluster (Figure S7). Yet, the oxidation of the mutant Av βF99Y/βS188A MoFeP leads to an Fe-deficient [7Fe-7S] cluster whereas wild-type Gd MoFeP retains its native [8Fe-7S] composition25. This distinction suggests that the Av and Gd MoFePs have evolved with specific preferences for Ser and Tyr ligands, respectively, and that the structural control of coordination by these ligands must extend beyond the primary and secondary coordination spheres of the P-cluster. Indeed, the structural analysis of the IDS-oxidized Av βF99Y variant (1.4 Å resolution), which contains both βS188 and βY99 residues, reveals that the POX state in Av MoFeP is governed by serinate coordination (Figures 3c-center and S3c-center). The parallel-mode EPR spectrum also shows a predominant, albeit broad, signal near g ≈ 12 associated with serinate coordination, with only a small signal near g ≈ 16 associated with tyrosinate coordination (Figure 4c -pink).25,37 Taken together, these observations are consistent with a structural equilibrium between βS188- and βY99-co-ordinated states with a preference for the former, at least within the Av MoFeP scaffold and at the low temperatures (5 – 10 K and 100 K) used in the EPR and X-ray crystallography experiments.
Lastly, to probe the functional effects of the P-cluster mutants, we examined their substrate reduction activities. All three Av variants expressing these mutants have doubling times similar to wild-type under diazotrophic growth conditions, with βS188A and βF99Y/βS188A demonstrating a slightly longer lag phase. (Figure S8). The 2-e− acetylene (C2H2) reduction activities of the isolated MoFeP mutants are reduced by ~20 to 40% compared to the native enzyme at pH 8.0 (Table 2 and Figure S9). Unexpectedly, the 6-e− N2 reduction activities are indistinguishable from wild-type values (Table 2 and Figure S9). There are several possible explanations: 1) the kinetics of redox-dependent metal substitution processes at the P-cluster during enzymatic turnover may be faster than the rate-limiting step(s) of substrate binding/reduction processes at FeMoco, which themselves may involve changes in cluster composition - thus, any possible perturbations in electron flux from the P-cluster to FeMoco would not drastically alter catalytic substrate reduction; 2) under diazotrophic growth or in vitro assay conditions (containing 13 mM DT) which are highly reducing, the kinetics of reduction of P+1 and POX states by exogenous electron donors may be faster than that of Fe dissociation from the P-cluster; or 3) the structures of the chemically induced oxidized states of the P-cluster in isolated MoFeP may be different from those populated during enzymatic turnover. The physiological relevance of the βS188A and βF99Y mutants is supported by the fact that MoFeP of at least one diazotrophic organism (Methanococcus aeolicus Nankai-3) contains neither a Tyr nor a Ser ligand to the P-cluster, and MoFeP in two organisms (Geobacter sulfurreducens PCA and Pelobacter carbinolicus DSM2380) contains both (Table S1). Finally, it must be noted that the effect of P-cluster mutations on relative C2H2 and N2 reduction activities is somewhat counterintuitive, as any perturbation in the electron flux from the P-cluster to FeMoco could be expected to have greater influence on a 6-e− catalytic process rather than a 2-e− one. This points to the possibility that different rate-limiting steps or even catalytic sites may be operative in the catalytic reduction of different substrates by MoFeP. Detailed studies with additional variants (e.g., those lacking FeMoco) and substrates are currently underway to explore such possibilities.
Table 2.
Substrate reduction activities of βS188A, βF99Y/ βS188A, and βF99Y Av MoFeP variants relative to the wild-type enzyme at pH 8.0 (see Figure S9 for activity assays).
| βS188A | βF99Y/ βS188A | βF99Y | |
|---|---|---|---|
| C2H2 reduction | 79 ± 3% | 82 ± 4% | 60 ± 7 %o |
| N2 reduction | 87 ± 19 %o | 96 ± 8 %o | 95 ± 14% |
Conclusions
Our results establish that the P-cluster is an intrinsically labile Fe-S cluster that exists on the brink of instability, and that an oxygenic ligand (Ser or Tyr) is critical for controlling its redox-metastability. The P-cluster thus adds to the growing number of systems such as the C-cluster in carbon monoxide dehydrogenase,31 the [4Fe-3S] cluster in oxygen tolerant hydrogenases,40,41 FeVco,15,16 and FeMoco,13 which also appear to involve reversible changes in their composition as an integral part of their function. The P-cluster possesses many energetically close-lying, redox-dependent structural and electronic states,23 which are controlled not only by the immediate coordination environment of the cluster but also by structural and dynamic elements in MoFeP that extend beyond its primary and secondary coordination spheres. The nature of those factors, which relate to conformational ET gating in nitrogenase, remain to be established. The unique dynamic properties of the P-cluster are undoubtedly an evolutionary requirement for an electron relay center that supports a catalytic center, i.e., FeMoco, which must accommodate an abundance of N2 reduction intermediates at different redox potentials while also avoiding unnecessary electron loss to the more favorable H+ reduction. Thus, a full mechanistic elucidation of biological nitrogen fixation reaction will require a deeper understanding of how the P-cluster actively controls electron flux to FeMoco.
Materials and Methods
Reagents.
All reagents were obtained from Fischer Scientific, Sigma Aldrich, VWR International or Praxair, unless otherwise noted and used without further purification.
Growth media.
All Azotobacter vinelandii (Av) nitrogenase cultures were grown in Burke’s media (BM+) containing 2.0% sucrose, 0.9 mM CaCl2, 1.67 mM MgSO4, 0.035 mM FeSO4, 0.002 mM Na2Mo2O4, 181 mM C12H22O11, 10 mM K3PO4 pH 7.5, 10 mM NH4Cl. Burke’s media lacking a fixed source of nitrogen (BM−) did not contain NH4Cl. Solid Burke’s medium contained 23 g/L agar. All liquid cultures were shaken at 30°C and 200 rpm unless otherwise noted.
Construction of nifK plasmids.
A pGEM-T Easy plasmid (Promega) containing the C-terminal region of the nifD gene, the complete nifK gene, and the N-terminal region of the nifT gene had the βF99Y, βS188A, and βF99Y/βS188A mutations introduced via PCR-based site-directed mutagenesis using the primers listed below (Integrated DNA technologies, mutated codon is underlined). Plasmid DNA was amplified using E. coli XL1-Blue cells.
βS188A forward primer: 5’-ATACCCCGGCCTTCGTGGGC-3’
βS188A reverse primer: 5’-GCCCACGAAGGCCGGGGTAT-3’
βF99Y forward primer: 5’-TGCGTCGCCTACTACCGCTCCTACTTCAAC-3’
βF99Y reverse primer: 5’-GTTGAAGTAGGAGCGGTAGTAGGCGACGCA-3’
Mutagenesis of A. vinelandii genome.
All nitrogenase mutants were generated in the native organism, Av, using a two-step process as previously described.42–44 First, a nifK deletion strain deficient in nitrogen fixation was iron-starved in modified liquid Burke’s media (no FeSO4, 0.09 mM CaCl2) and shaken at 150 rpm. The deletion strain, Av DJ 200, contained a kanamycin resistance cassette disrupting nifK and was thus deficient in its ability to fix nitrogen (ΔnifK :: KanR Av generously provided by V. Cash and D. Dean). Cells turned fluorescent green due to siderophores secreted under iron-starvation conditions. At OD600 0.5, 50 μL of the iron-starved cells were transformed in 50 μL buffer (20 mM MOPS, pH 7.5, 20 mM MgSO4) with 1 – 5 μg of the purified rescue plasmid containing the mutation of interest, restoring nitrogen fixation ability. Plasmid DNA incorporated into the genome by double homologous recombination, replacing the kanamycin resistance cassette. Transformants were screened for both restoration of nitrogen fixation ability on BM− agar and for loss of kanamycin resistance. Multiple passes on BM− were required to obtain a consensus sequence, which was verified with sequencing of colony PCR products (Figure S1).
A. vinelandii culture growth.
Each A. vinelandii strain was grown using a 100-mL BM− starter culture in a 500- mL Erlenmeyer flask until cells reached an OD600 of 1.5 – 2.0, followed by inoculation of 1.0 L BM− in a 2.8-L Erlenmeyer flask with 10 – 25 mL of the starter culture. At an OD600 > 1.5, 400 mL of the starter culture were used to inoculate 50 L of BM+ (only 3 mM NH4Cl) in a 60-L fermenter (New Brunswick Scientific). Cells were harvested 4 h after the start of derepression, as indicated by a spike in dissolved oxygen. Cells were concentrated to ~5 L using a Pellicon 2 Tangential flow membrane (Eppendorf), followed by centrifugation at 5,000 rpm and 4°C. Cell pellets ranged from 120 – 210 g and were stored at −80°C until protein purification.
Protein purification.
Cell lysis and purification were carried out on a Schlenk line under ultra-high purity Ar or in a Coy anaerobic chamber under 90% Ar/10% H2. All columns and buffers used were prepared under anaerobic conditions. The bacterial cell pellets were thawed and resuspended in ~200 mL buffer Eq (50 mM TRIS, pH 7.75, 200 mM NaCl, 5 mM sodium dithionite (DT), 0.1 mg/mL DNase I), then lysed using a microfluidizer at 16,000 psi N2 (Microfluidics). The dark brown lysate was spun anaerobically at 12,000 rpm for 75 min. The supernatant was loaded onto a DEAE Sepharose column and washed overnight with 1.5 L buffer Eq. The protein was eluted using a linear NaCl gradient (200 to 500 mM NaCl at 2.5 mL/min for 1.0 L). MoFeP and FeP eluted at conductivities of ~25 and ~30 mS/cm, respectively. Fractions containing MoFeP and FeP were identified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Fractions were pooled and diluted two-fold with salt-free buffer (50 mM TRIS, pH 7.75). Protein was concentrated on a small DEAE Sepharose column in a high salt buffer (50 mM TRIS, pH 7.75, 500 mM NaCl, 5 mM DT). Proteins were further purified via gel filtration with a Sepharose 200 column (GE Healthcare) with high salt buffer (50 mM TRIS, pH 8.0, 500 mM NaCl 5 mM DT). Fractions containing pure nitrogenase component proteins were determined with SDS-PAGE, combined, and concentrated in a 10-kDa cutoff Amicon concentrator (EMD/Millipore) at ~15 psi 90% Ar/10% H2 (Figure S2). Concentrated proteins were stored in ~500 μL aliquots in cryovials in liquid nitrogen. Each cell pellet yielded 30 – 160 mg FeP and 120 – 280 mg MoFeP. FeP and MoFeP protein concentrations were determined using the Bradford assay as well an Fe-chelation assay with 6.2 M guanidine-HCl and 2 mM 2,2-bipyridine in 10% glacial acetic acid at 522 nm using an extinction coefficient of 8650 M−1 cm−1.
Crystallographic data collection and refinement.
All crystals were prepared using the sitting drop vapor diffusion method at room temperature in a Coy anaerobic chamber under 90% Ar/10% H2 with 500 μL precipitation solution in the reservoir, and 2 μL + 2 μL drops of protein solution (125 μM) and precipitation solution in the well. Crystals took 3 days − 2 weeks to reach full size, ranging from ~50 μm – 200 μm along the longest dimension. DT-reduced crystals were dark brown in color, and IDS-oxidized crystals were dark blue. Cryoprotection of the crystals using perfluoropolyether and their subsequent freezing in liquid N2 were carried out in an anaerobic chamber. Data were collected with multi-wavelength synchrotron radiation at SSRL beamlines 9–2 and 12–2 and ALS beamline 5.0.2.
X-ray data collected from ALS beamlines were indexed, integrated and scaled using XDS,45,46 and all other data sets were integrated and scaled using iMosflm and aimless.47 Molecular replacement was carried out using Phaser-MR of the PHENIX suite with the search model PDB 2MIN, then refined with phenix.refine and Coot.48 Structures were refined with riding hydrogens that were added with phenix.reduce.48 For data collection and refinement parameters and statistics, see Table S2. All crystal structure figures were produced with PyMOL (www.pymol.org).
Electron paramagnetic resonance spectroscopy of MoFeP.
Sample preparation was carried out in a Coy anaerobic chamber under 90% Ar/10% H2. DT was removed from the solution by buffer exchanging MoFeP into 50 mM TRIS, pH 8.0, 500 mM NaCl using 10-DG desalting columns (BioRad). MoFeP was concentrated to 50 μM using 10-kDa cutoff Microcon filters (EMD/Millipore). Concentrated protein samples were either reduced with 10 mM DT or oxidized with 5 mM IDS. Mutants containing labile iron-centers also had a re-reduced sample prepared in which the IDS-oxidized protein was desalted over another 10-DG column, concentrated, then re-reduced with 10 mM DT. All data were collected on an X-band Bruker EMX spectrometer with a liquid helium cryostat at 5 – 10 K. Spectra were recorded with a modulation frequency of 100.0 kHz and modulation amplitude of 9.8 G. Perpendicular and parallel mode spectra were collected at microwave frequencies of ~9.62 GHz and ~9.39 GHz, and microwave power of 6 mW and 127 mW, respectively. All perpendicular-mode spectra shown in figures were background subtracted.
Nitrogenase MoFeP NH3 and H2 assays.
Substrate reduction assays were completed at least in triplicate. All assay steps involving protein were carried out on a Schlenk line under ultra-high purity N2 or in a Coy anaerobic chamber under 90% Ar/10% H2. NH3 and H2 were measured from the same 10.0 mL reaction vial. Reactions contained 1.00 mL ATP regeneration solution (50 mM HEPES, pH 8.0, 10 mM MgCl2, 0.125 mg/mL creatine kinase, 5 mM Na2ATP, 30 mM phosphocreatine). All reaction vials were degassed under ultra-high purity N2 to remove oxygen, and 1 M DT stock solution (pH 8.0) was added for a final concentration of 13 mM. FeP and MoFeP were buffer exchanged from TRIS to HEPES (50 mM HEPES pH 8.0, 500 mM NaCl) using 10-DG desalting columns. Protein concentrations were 0.4 μM MoFeP and 0 – 16 μM FeP. Reactions were carried out at 30.0°C for 20.0 min and then quenched with 400 μL of 4 M NaCl. H2 production was measured using gas chromatography (SRI 8610C) with a 5A 80/100 molecular sieve column (Alltech) and a TCD detector. An H2 calibration curve was prepared daily with each experiment using injections of pure H2 diluted in ultra-high purity N2.
After measuring H2 production, NH3 production was quantified using a modified o-phthalaldehyde (OPA) fluorescence detection method (HORIBA Jobin Yvon fluorimeter).49 All sample preparation with OPA was completed in the dark. Proteins were removed from the assay reaction by filtering 500 μL of the solution with a 10-kDa cutoff Microcon filtration device. The flow-through solution was diluted 10-fold with water. 200 μL of the diluted flow-through solution was added to 1.8 mL of another solution containing 50 mM PO43−, 5 mM OPA, 3 mM Na2SO3, 12.5% MeOH, and allowed to react for 2 h.* Fluorescence was measured at an excitation wavelength of 365 nm and an emission wavelength of 422 nm. Calibration curves were prepared daily using NH4Cl standards (0–875 μM NH4Cl) prepared in parallel to the samples. H2 and NH3 specific activity curves were plotted with standard deviation represented by error bars (Figure S9a–c).
*Fluorescence solutions were prepared fresh daily by combining equal parts OPA stock solution (10 mM OPA in 25% MeOH) and sulfite stock solution (6 mM Na2SO3, 100 mM PO43−, pH 11.0).
Nitrogenase MoFeP C2H4 activity assay.
All assays were completed at least in triplicate and were carried out on a Schlenk line under ultra-high purity Ar or in a Coy anaerobic chamber under 90% Ar/10% H2. Reactions in 10.0 mL reaction vials contained 1.00 mL ATP regeneration solution (50 mM TRIS, pH 8.0, 10 mM MgCl2, 0.125 mg/mL creatine kinase, 5 mM Na2ATP, 30 mM phosphocreatine and 13 mM DT. All reaction vials were degassed with ultra-high purity Ar. 1.0 mL C2H2 (room temperature, atmospheric pressure) was added to the headspace of each vial. Protein concentrations were 0.2 μM MoFeP and 0 – 8 μM FeP. Reactions were carried out at 30°C for 15.0 min and quenched with 400 μL of 4 M NaCl. C2H4 production was measured using gas chromatography (SRI 8610C) with a HayeSep N packed column (SRI instruments) and an FID detector. A C2H4 calibration curve was prepared with each experiment using injections of pure C2H4 diluted in ultra-high purity Ar. C2H4 specific activity curves were plotted with standard deviation represented by error bars (Figure S9d).
Supplementary Material
ACKNOWLEDGMENT
We thank M. Green (UC Irvine) for the use of EPR instrumentation, R. Subramanian and R. Alberstein for helpful discussions, C. Morrison and J. Bailey for help with anaerobic crystallography and structure refinement, and V. Cash and D. Dean (Va. Tech) for generously providing the Av nifK knockout strain DJ200. This work was supported by the National Institutes of Health (Grant GM099813 to F.A.T.). H.L.R. was additionally supported by the Molecular Biophysics Training Grant (NIH Grant T32 GM008326).
Footnotes
Supporting Figures 1 – 10 and Supporting Tables 1 – 3. This material is available free of charge via the Internet at http://pubs.acs.org. Coordinate and structure factor files have been deposited into RCSB databank under the following PDB IDs:6O7L, 6O7M, 6O7N, 6O7O, 6O7P, 6O7Q, 6O7R, 6O7S.
The authors declare no competing financial interests.
REFERENCES
- 1.Burgess BK; Lowe DJ Mechanism of molybdenum nitrogenase. Chem. Rev 1996, 96, 2983. [DOI] [PubMed] [Google Scholar]
- 2.Howard JB; Rees DC Structural basis of biological nitrogen fixation. Chem. Rev 1996, 96, 2965. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman BM; Lukoyanov D; Yang ZY; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tezcan FA; Kaiser JT; Mustafi D; Walton MY; Howard JB; Rees DC Nitrogenase complexes: Multiple docking sites for a nucleotide switch protein. Science 2005, 309, 1377. [DOI] [PubMed] [Google Scholar]
- 5.Spatzal T; Aksoyoglu M; Zhang LM; Andrade SLA; Schleicher E; Weber S; Rees DC; Einsle O Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Einsle O; Tezcan FA; Andrade SLA; Schmid B; Yoshida M; Howard JB; Rees DC Nitrogenase MoFe-protein at 1.16 angstrom resolution: A central ligand in the FeMo-cofactor. Science 2002, 297, 1696. [DOI] [PubMed] [Google Scholar]
- 7.Lancaster KM; Roemelt M; Ettenhuber P; Hu YL; Ribbe MW; Neese F; Bergmann U; DeBeer S X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters JW; Stowell MHB; Soltis SM; Finnegan MG; Johnson MK; Rees DC Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 1997, 36, 1181. [DOI] [PubMed] [Google Scholar]
- 9.Thorneley RN; Lowe DJ Molybdenum Enzymes; Wiley: New York, 1985. [Google Scholar]
- 10.Rohde M; Sippel D; Trncik C; Andrade SLA; Einsle O The Critical E4 State of Nitrogenase Catalysis. Biochemistry 2018, 57, 5497. [DOI] [PubMed] [Google Scholar]
- 11.Montgomery JM; Mazziotti DA Strong Electron Correlation in Nitrogenase Cofactor, FeMoco. J. Phys. Chem. A 2018, 122, 4988. [DOI] [PubMed] [Google Scholar]
- 12.Harris DF; Yang ZY; Dean DR; Seefeldt LC; Hoffman BM Kinetic Understanding of N2 Reduction versus H2 Evolution at the E4(4H) Janus State in the Three Nitrogenases. Biochemistry 2018, 57, 5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spatzal T; Perez KA; Howard JB; Rees DC Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. Elife 2015, 4, e11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raugei S; Seefeldt LC; Hoffman BM Critical computational analysis illuminates the reductive-elimination mechanism that activates nitrogenase for N2 reduction. Proc. Natl. Acad. Sci. USA 2018, 115, E10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sippel D; Einsle O The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol 2017, 13, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484. [DOI] [PubMed] [Google Scholar]
- 17.Muraki N; Nomata J; Ebata K; Mizoguchi T; Shiba T; Tamiaki H; Kurisu G; Fujita Y X-ray crystal structure of the light-independent protochlorophyllide reductase. Nature 2010, 465, 110. [DOI] [PubMed] [Google Scholar]
- 18.Zheng KY; Ngo PD; Owens VL; Yang XP; Mansoorabadi SO The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 2016, 354, 339. [DOI] [PubMed] [Google Scholar]
- 19.Hu YL; Ribbe MW Nitrogenase and homologs. J. Biol. Inorg. Chem 2015, 20, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanzilotta WN; Seefeldt LC Changes in the midpoint potentials of the nitrogenase metal centers as a result of iron protein molybdenum-iron protein complex formation. Biochemistry 1997, 36, 12976. [DOI] [PubMed] [Google Scholar]
- 21.Danyal K; Mayweather D; Dean DR; Seefeldt LC; Hoffman BM Conformational Gating of Electron Transfer from the Nitrogenase Fe Protein to MoFe Protein. J. Am. Chem. Soc 2010, 132, 6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danyal K; Dean DR; Hoffman BM; Seefeldt LC Electron Transfer within Nitrogenase: Evidence for a Deficit-Spending Mechanism. Biochemistry 2011, 50, 9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhendong L; Sheng G; Qiming S; Garnet KL C. The electronic landscape of the P-cluster of nitrogenase. arXiv:1810.10196 2018. [Google Scholar]
- 24.Keable SM; Zadvornyy OA; Johnson LE; Ginovska B; Rasmussen AJ; Danyal K; Eilers BJ; Prussia GA; LeVan AX; Raugei S; Seefeldt LC; Peters JW Structural characterization of the P(1+) intermediate state of the P-cluster of nitrogenase. J. Biol. Chem 2018, 293, 9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owens CP; Katz FEH; Carter CH; Oswald VF; Tezcan FA Tyrosine-Coordinated P-Cluster in G. diazotrophicus Nitrogenase: Evidence for the Importance of O-Based Ligands in Conformationally Gated Electron Transfer. J. Am. Chem. Soc 2016, 138, 10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howard JB; Kechris KJ; Rees DC; Glazer AN Multiple amino acid sequence alignment nitrogenase component 1 : insights into phylogenetics and structure-function relationships. PLoS One 2013, 8, e72751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang LM; Kaiser JT; Meloni G; Yang KY; Spatzal T; Andrade SLA; Einsle O; Howard JB; Rees DC The 16th Fe in the Nitrogenase MoFe-Protein. Angew. Chem. Int. Ed. Engl 2013, 52, 10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robbins AH; Stout CD Structure of Activated Aconitase - Formation of the [4Fe-4S] Cluster in the Crystal. Proc. Natl. Acad. Sci. USA 1989, 86, 3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berkovitch F; Nicolet Y; Wan JT; Jarrett JT; Drennan CL Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 2004, 303, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLaughlin MI; Lanz ND; Goldman PJ; Lee KH; Booker SJ; Drennan CL Crystallographic snapshots of sulfur insertion by lipoyl synthase. Proc. Natl. Acad. Sci. USA 2016, 113, 9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittenborn EC; Merrouch M; Ueda C; Fradale L; Leger C; Fourmond V; Pandelia ME; Dementin S; Drennan CL Redox-dependent rearrangements of the NiFeS cluster of carbon monoxide dehydrogenase. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beinert H; Kennedy MC; Stout CD Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev 1996, 96, 2335. [DOI] [PubMed] [Google Scholar]
- 33.Hu Y; Fay AW; Lee CC; Ribbe MW P-cluster maturation on nitrogenase MoFe protein. Proc. Natl. Acad. Sci. USA 2007, 104, 10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu YL; Fay AW; Lee CC; Yoshizawa J; Ribbe MW Assembly of nitrogenase MoFe protein. Biochemistry 2008, 47, 3973. [DOI] [PubMed] [Google Scholar]
- 35.Ribbe MW; Hu YL; Hodgson KO; Hedman B Biosynthesis of Nitrogenase Metalloclusters. Chem. Rev 2014, 114, 4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tittsworth RC; Hales BJ Detection of Epr Signals Assigned to the 1-Equiv-Oxidized P-Clusters of the Nitrogenase Mofe-Protein from Azotobacter vinelandii. J. Am. Chem. Soc 1993, 115, 9763. [Google Scholar]
- 37.Surerus KK; Hendrich MP; Christie PD; Rottgardt D; Orme-Johnson WH; Münck E Mossbauer and Integer-Spin EPR of the Oxidized P-Clusters of Nitrogenase - P(ox) Is a NonKramers System with a Nearly Degenerate Ground Doublet. J. Am. Chem. Soc 1992, 114, 8579. [Google Scholar]
- 38.Chan JM; Christiansen J; Dean DR; Seefeldt LC Spectroscopic evidence for changes in the redox state of the nitrogenase P-cluster during turnover. Biochemistry 1999, 38, 5779. [DOI] [PubMed] [Google Scholar]
- 39.Lanzilotta WN; Christiansen J; Dean DR; Seefeldt LC Evidence for coupled electron and proton transfer in the [8Fe-7S] cluster of nitrogenase. Biochemistry 1998, 37, 11376. [DOI] [PubMed] [Google Scholar]
- 40.Fritsch J; Scheerer P; Frielingsdorf S; Kroschinsky S; Friedrich B; Lenz O; Spahn CMT The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature 2011, 479, 249. [DOI] [PubMed] [Google Scholar]
- 41.Noor NDM; Matsuura H; Nishikawa K; Tai HL; Hirota S; Kim J; Kang JY; Tateno M; Yoon KS; Ogo S; Kubota S; Shomura Y; Higuchi Y Redox-dependent conformational changes of a proximal [4Fe-4S] cluster in Hyb-type [NiFe] hydrogenase to protect the active site from O2. Chem. Commun 2018, 54, 12385. [DOI] [PubMed] [Google Scholar]
- 42.Glick BR; Brooks HE; Pasternak JJ Transformation of Azotobacter vinelandii with plasmid DNA. J. Bacter 1985, 162, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Page WJ; Von Tigerstrom M Optimal Conditions for Transformation of Azotobacter vinelandii. J. Bacter 1979, 139, 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth LE; Tezcan FA ATP-Uncoupled, Six-Electron Photoreduction of Hydrogen Cyanide to Methane by the Molybdenum-Iron Protein. J. Am. Chem. Soc 2012, 134, 8416. [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch W Xds. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr 1994, 50, 760. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genfa Z; Dasgupta PK Fluorometric Measurement of Aqueous Ammonium Ion in a Flow Injection System. Anal. Chem 1989, 61, 408. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.