

Abstract
Amyloid diseases are global epidemics with profound health, social and economic implications and yet remain without a cure. This dire situation calls for research into the origin and pathological manifestations of amyloidosis to stimulate continued development of new therapeutics. In basic science and engineering, the cross-β architecture has been a constant thread underlying the structural characteristics of pathological and functional amyloids, and realizing that amyloid structures can be both pathological and functional in nature has fuelled innovations in artificial amyloids, whose use today ranges from water purification to 3D printing. At the conclusion of a half century since Eanes and Glenner’s seminal study of amyloids in humans, this review commemorates the occasion by documenting the major milestones in amyloid research to date, from the perspectives of structural biology, biophysics, medicine, microbiology, engineering and nanotechnology. We also discuss new challenges and opportunities to drive this interdisciplinary field moving forward.
Keywords: structure, amyloid disease, pathological amyloid, functional amyloid, artificial amyloid, gut microbiota
Graphical Abstract
1. Introduction
The X-ray diffraction pattern of filamentous amyloid from human liver and spleen, as described by Eanes and Glenner in 1968, contained “a sharp, intense ring at 4.75Å overlaying a diffuse halo at 4.3Å and a broad and less intense ring at 9.8Å”.1 This followed the first report by Astbury et al. in 1934 on the presence of the cross-β motifs in chicken egg proteins.2 Much progress has been made since then. Within the realm of structural biology, the cross-β structure of human amyloid first revealed in that seminal study has underpinned our understanding (or the lack thereof) of the aggregation of amyloid proteins as well as their associated pathologies, from cerebral amyloid angiopathy (CAA), tauopathies and synucleinopathies to amyloid light-chain (AL) amyloidosis, transthyretin (TTR) amyloidosis,3, 4 amyotrophic lateral sclerosis (ALS) and rheumatoid arthritis (RA), and from the endogenous aggregation of insulin and human islet amyloid polypeptide (hIAPP) to the cross talk5 between amyloid proteins of different physiological origins and the transmission of prion diseases6 across animal species. Recent discovery of amyloid-like assemblies of metabolites and their associated toxicities7 has shed new light on the molecular mechanisms of human diseases, and blurred the boundary between functional and pathological amyloid. As bacterial multidrug resistance (MDR) has become a global health crisis, understanding and exploiting the structural and pathological roles of bacterial amyloid may offer new solutions for the development of novel therapeutics. The recent finding of a gut-brain neural circuit for nutrient sensory transduction8 points to a connection between the gut microbiome and neurological disorders,9 each of which is associated with amyloid architectures. Major discoveries have been made in recent years and months, implicating the gut microbiome as causative for obesity, type 2 diabetes (T2D), neurological disorders, cancer, depression and social behaviour.10–14
Amyloidosis refers to the accumulation and deposition of amyloid fibrils,15 whose aggregation kinetics contains contributions of both primary and secondary nucleation,16 giving rise to toxic intermediates of oligomers17, 18 and protofibrils en route. The amyloid state is characterized by steric zippers of the amyloid cross-β spine,19 and the state is proposed to be accessible by virtually all proteins under physiological or artificial conditions. The crystalline form of amyloid proteins has recently been identified as the absolute free-energy ground state20, 21 over the native or amyloid state of proteins. On a mesoscopic scale, amyloid fibrils possess polymorphism, displaying a prevalent yet nonexclusive left-handedness likely originated from the biased chirality of amino acids.22 In engineering, amyloids of whey proteins have found new applications in iron fortification, water purification and in vivo sequestration of pathological amyloid proteins,23–25 while functional amyloid-nanocomposites yield new mechanical, thermal and electronic properties appealing to nanoelectronics, biotechnology and environmental engineering.26
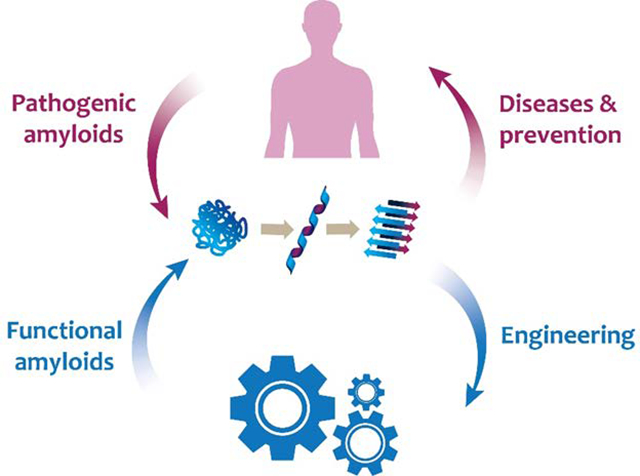
To commemorate five decades of research since Eanes and Glenner’s landmark study of human amyloid,1 here we reflect on major milestones in the field of amyloid to date, shared among the three major classes of amyloids: the pathological, functional and artificial amyloids, and we discuss emerging opportunities and grant challenges of the amyloid science moving forward, from the perspectives of basic science, medicine and engineering (Fig. 1).
Fig. 1.

Amyloidosis is a biophysical phenomenon of protein self-assembly under natural or artificial conditions, underpinned by a ubiquitous cross-β architecture (middle, in cyan). For over a half century, or arguably much longer, investigations into the structures of pathological and functional amyloids within the human anatomy (left, in blue), the microbiota (left, in green) and beyond (right, in dark blue) have revealed their inner workings as well as their entangled implications for biology, medicine and engineering.
2. Amyloidosis, a prevalent yet peculiar form of protein misfolding
Protein folding is one of the most perplexing problems in molecular biology, despite many decades of extensive research.27, 28 In short, protein folding is a complex process through which a protein molecule acquires the unique native structure for carrying out its specific biological functions. However, under certain pathological conditions, proteins can misfold, resulting in structures that expose the hydrophobic residues at the core of the folded protein to the solvent. These misfolded proteins can self-assemble into a variety of aggregate structures, including large, insoluble fibrillar entities known as the amyloids.28 As mentioned above, a number of diseases, including Alzheimer’s disease (AD) and T2D, are associated with the presence of amyloid. Although proteins involved in amyloid diseases are dissimilar in both sequences and folds, the end-products of their aggregation bear striking structural similarities including the fibrillar structure and cross-β backbone as revealed by X-ray diffraction.1, 29 Since many proteins that are not associated with diseases also form amyloid fibrils, it has been suggested that under certain conditions, any protein is capable of forming an amyloid,30 indicating amyloidosis might be a prevalent yet peculiar form of protein misfolding (i.e., amyloid formation might represent a special type of evolving protein folding free energy landscape, more below). In addition to protein misfolding, it has also been recognized that some proteins have no single well-defined tertiary structure. These proteins are termed intrinsically disordered proteins (IDPs) which are often involved in cellular signaling and regulation.31, 32 Given the very large number of degrees of freedom in an unfolded polypeptide chain, the protein molecule has an astronomical number of possible conformations. From one estimation, for a ~100 residue protein, it would take ~1011 years to fold if the protein needs to explore all the possible conformation states, while in reality it takes merely milliseconds to seconds for a typical protein to fold in vivo. This is called the Levinthal paradox,33 proposed by Cyrus Levinthal five decades ago in 1969. To overcome this paradox,33 several folding models, from classical nucleation-propagation model to the folding funnel model, have been proposed to complement experiments towards better understanding of this complex folding process.
The widely adopted protein folding funnel model has evolved from both experiment and theory through the use of simplified mechanical models developed by Wolynes, Onuchic, Dill and colleagues35–37 and more recently by Mezzenga and coworkers.20, 21 Fig. 2 illustrates the folding funnel which is a simplified 2D representation of the very high-dimensional conformational space accessible to a protein during its folding.34 The broad top of the funnel represents a vast number of conformations present in the fully unfolded or stretched state, while the narrow bottom of the funnel depicts the unique native structure of the protein.35 The separation between the top and bottom of the funnel represents other energies (solute enthalpy, solvent entropy and enthalpy) contributing to each protein conformation. However, the chaperone effect (chaperonin-mediated folding) or multi-protein co-folding effect (folding upon binding, etc.) is not included in this picture due to its simplicity.35, 36, 38 Starting from the ensemble of unfolded conformations, the folding funnel allows many different pathways to proceed rapidly to the global free energy minimum occupied by amyloids, recently refined into a series of closely-positioned local minima, occupied by different amyloid polymorphs, with the amyloid crystals alone occupying the absolute minimum (for an extended discussion on the energetic levels of different amyloid polymorphs see section 4, “Mesoscopic structures of amyloids and the energy landscape”).20, 21 As the chain folds to lower energy conformations, and before reaching the metastable local minima of the various amyloid polymorphs, intermediate states along the sides of the funnel are also populated. During this process, the kinetic traps might hinder or promote formation of native structures depending on their depths and the barriers between the traps and next energy minima. According to statistical mechanics, the number and depth of local kinetic traps on the funnel landscape correspond to the degree of frustration of the protein sequence.35 Following the concept of the folding funnel diagrams, an off-pathway aggregation can be incorporated as second “aggregation funnel”.39 Like intramolecular folding, the association of two or more non-native protein molecules can form an “amyloidosis formation funnel” through intermolecular contacts (Fig. 2). The process is largely driven by hydrophobic forces and primarily results in the formation of amorphous structures (“amorphous aggregates”; Fig. 2).34 Subsequently, aggregation can lead to the formation of amyloid fibrils. These simplistic folding funnel models provide a conceptual framework for understanding the complex process of amyloid formation.20, 21, 34
Fig. 2. Energy landscape of protein folding and aggregation.

The purple surface shows the multitude of conformations ‘funneling’ to the native state via intramolecular contacts and the pink area shows the conformations moving toward amorphous aggregates or amyloid fibrils via intermolecular contacts. Both parts of the energy surface overlap. Aggregate formation can occur from intermediates populated during de novo folding or by destabilization of the native state into partially folded states and is normally prevented by molecular chaperones. Toxic oligomers may occur as off-pathway intermediates of amyloid fibril formation. Reproduced with permission from ref. 34, copyright 2009 Nature Publishing Group.34
Meanwhile, recent advances in experimental techniques that probe amyloid formation at different stages have shed light on the nature of both the kinetics and thermodynamics of this complex process (more in the following sections). However, many of the underlying molecular mechanisms and interactions involved in amyloid protein/peptide misfolding and aggregation pathways remain elusive. Computer simulations performed at various levels of complexity ranging from simple lattice models, models with continuum solvent, to all atom models with explicit solvent have been used to offer complementary and valuable insights that cannot be obtained by experimental methods alone.40 In particular, the important role of water molecules in promoting the formation of protofilaments, the basic building blocks of amyloid fibrils, has been investigated using fully atomic molecular dynamics (MD) simulations.41
Although the hydrophobic effect is known to have a significant impact on protein self-assembly in water, the precise mechanism of how it operates as well as the exact role of water in facilitating this assembly remains controversial. In a recent study,41 a model protofilament comprised of two parallel β-sheets of Alzheimer Aβ16–22 peptides (Ac-K16-L17-V18-F19-F20-A21-E22-NH2) was employed to study amyloid formation and the role of water molecules during the process using MD simulation. Each β-sheet presented a distinct hydrophobic face and a hydrophilic face, which together self-assembled into a stable protofilament with a core consisting of purely hydrophobic residues (L17, F19, A21), with the two charged residues (K16, E22) pointing to the solvent (Fig. 3A). The simulation results revealed a subtle interplay between a water mediated assembly and one driven by favorable energetic interactions between specific residues forming the interior of the protofilament. Overall, the role of water during the assembly can be viewed as “lubrication”, namely it does not drive assembly but rather facilitate proper packing of the hydrophobic surfaces in the final stages of the assembly. In some of the MD trajectories, a nanoscale dewetting (or drying) was also observed in which water expulsion preceded hydrophobic collapse, providing a strong driving force for the hydrophobic collapse and hydrophobic patch assembly. This can be attributed to the fact that when two strongly hydrophobic surfaces, greater than 1 nm in length, are brought together to a critical distance, a nanoscale drying might occur between the two hydrophobic surfaces, resulting in a strong hydrophobic collapse.42, 43 In the trajectories where no nanoscale drying was observed, water expulsion and hydrophobic collapse occurred roughly simultaneously (i.e., water acting as “lubricant”; Fig. 3).
Fig. 3.

(A) Aβ16–22 model protofilament (left panel). The initial structure used to start the MD trajectory with initial inter-β-sheet separation distance D0 of 1.28 nm. Front (a) and side (b) views are shown. Side chains are colored as follows: K) red, L) orange, V) yellow, F) green, A) blue, and E) violet. For clarity, only water molecules in the interpeptide region have been shown. (c) The same structure after 1,000 ps of unconstrained MD simulation at 300 K, started from the structure shown in (a) and (b). (d) A single Aβ16–22 peptide pair, one from each layer, is isolated from the protofilament shown in (c). (B) Number of interpeptide water molecules versus interpeptide distance (right panel). (a-d) Plots for each of the four trajectories at 300 K where D0= 1.28 nm. Trajectories (a) and (b) do not appear to show a dewetting transition, while trajectories (c) and (d) do. (e-h) The peptide-water van der Waals interaction is turned off, and D0 = 2.38 nm. (i-l) The peptide-water electrostatic interaction is turned off, and D0 = 1.28 nm. Reproduced with permission from ref. 41, copyright 2008 American Chemical Society.41
The authors also studied the interaction energy decomposition to explore the contributions from various forces.41 Interestingly, turning off the protein-water electrostatic interaction only slightly slowed down the assembly speed without significantly affecting the nanoscale drying (Fig. 3B). Conversely, if the protein-water van der Waals attraction was switched off, a strong dewetting transition and hydrophobic collapse takes place in every simulation (Fig. 3B). These predictions were later validated by experimental (and theoretical) studies of other proteins with large hydrophobic patches.44 Overall, these computer simulations demonstrate that in general, when attractive van der Waals forces exist between the solute and solvent, these forces, though individually small, can be sufficient to compensate for the loss of hydrogen bonds due to the confinement of water between the two plates. However, in extreme cases, such as those highly hydrophobic and rough surfaces in-between the amyloid protofilaments, some nanoscale dewetting might occur which can provide strong driving force for the hydrophobic collapse of amyloid peptides and their subsequent aggregation and fibril formation.45
3. Towards atomic structures of amyloid fibrils
As briefly introduced, amyloid fibrils are identified by a characteristic X-ray fibre diffraction pattern which is termed cross-β (Fig. 4). This pattern was first described for poached egg-white2 and later the data from the silk egg stalk from the green lacewing fly was interpreted to provide a detailed description of this repetitive structure.46 The cross-β diffraction pattern gives a strong, sharp diffraction signal at 4.76–4.78 Å on the meridional (vertical) axis, which was interpreted to arise from the distance between hydrogen bonded β-strands. On the equator, (horizontal axis) several signals may be observed but the dominant intensity is thought to arise from the spacing of several β-sheets.47, 48 For the silk from the egg stalk, the equatorial spacing was only 5 Å. However, the spacing arising from amyloid fibrils is more often larger to accommodate larger and more variable side chains, from around 8 Å for polyQ containing peptides49, 50 to 11–12 Å for those containing aromatic residues.51, 52
Fig. 4.

X-ray fibre diffraction provides the characteristic cross-β pattern for amyloid. Top panel shows a schematic showing the features of the cross-β pattern and structure. Lower panels show the cross-β diffraction patterns collected from amyloid fibrils formed by a diverse range of amyloidogenic proteins and peptides. Aβ11–25,72–74 AAAKKFFEAAAK,52 silk,75 hIAPP,76 NM Sup35,77 ccβ,78 Met30 TTR,79 Tau,80 Core PHF,81 β2M,82 Aβ42,73, 83 GNNQQNY,84 Fibrinogen,85 RVFNIM.86 Reproduced with permission from ref. 72, copyright 2000 The American Chemical Society.72 Reproduced with permission from ref. 73, copyright 2003 Elsevier.73 Reproduced with permission from ref. 74, copyright 2000 Elsevier.74 Reproduced with permission from ref. 52, copyright 2005 National Academy of Sciences.52 Reproduced with permission from ref. 75, copyright 2007 Wiley-VCH.75 Reproduced with permission from ref. 76, copyright 2004 Elsevier.76 Reproduced with permission from ref. 77, copyright 2000 American Association for the Advancement of Science.77 Reproduced with permission from ref. 78, copyright 2008 Elsevier.78 Reproduced with permission from ref. 79, copyright 1996 Ciba Foundation.79 Reproduced with permission from ref. 80, copyright 2003 National Academy of Sciences.80 Reproduced with permission from ref. 81, copyright 2017 Elsevier.81 Reproduced with permission from ref. 82, copyright 2008 American Society for Biochemistry and Molecular Biology.82 Reproduced with permission from ref. 87, copyright 2012 American Society for Biochemistry and Molecular Biology.87 Reproduced with permission from ref. 84, copyright 2010 Elsevier.84 Reproduced with permission from ref. 85, copyright 2007 Informa Healthcare.85 Reproduced with permission from ref. 86, copyright 2013 Portland Press.86
Transmission electron microscopy was a valuable asset and in 1959, Cohen and Calkins53 provided images of amyloid fibrils extracted from liver. Eanes and Glenner showed the cross-β diffraction pattern from amyloid extracted from liver and spleen1 and then created “amyloid” in vitro from Bence Jones proteins (excreted immunoglobulin light chains, LCs) or their fragments.54 This disease, arising from the formation of LC-derived amyloid fibrils, is now known as AL amyloidosis.55, 56 As early as 1946, Waugh reported precipitation of insulin under high temperature, acidic conditions.57 In 1972, synthetic amyloid fibrils were made from insulin by repeated heating and cooling under acidic conditions. This process generated long-straight, unbranching fibrils that resisted degradation58 and gave a cross-β diffraction pattern as well as the characteristic β-sheet signals by circular dichroism (CD) spectrophotometry and Fourier transform infrared (FTIR) spectroscopy. Following this advance, it became possible to create amyloid fibrils from many disease-related peptides such as Aβ,59, 60 hIAPP61 and peptides related to larger amyloidogenic precursors such as TTR.62 This paved the way for further structural characterisation to reinforce the description of the amyloid core cross-β structure. The cross-β pattern from ex-vivo Val30Met variant TTR provided a model structure, composed of repeating β-strands running perpendicular to the fibre axis and associated to from several sheets that twisted with a helical pitch of 115.5°.63 This model was found to be representative of a collection of extracted amyloid fibrils leading to the generic cross-β model for amyloid.64
Atomic force microscopy, transmission electron microscopy and cryo-transmission electron microscopy (AFM, TEM and cryoTEM) provided further macromolecular details and showed that amyloid fibrils were formed of individual protofilaments65–68 and that different precursor proteins may lead to different numbers of protofilaments. It was also becoming clear that synthetic fibrils grown under different conditions could lead to structural polymorphism. For example, insulin fibrils analyzed by cryoTEM showed multiple variations in the number of protofilaments from two to six,69 while Aβ40 was later shown to form even more different classes.70 AFM was instrumental in demonstrating the growth of the fibrils and generally showed that the diameters did not change, but that the growth was additive elongation at the growing ends.71
X-ray fibre diffraction from a short amyloidogenic region of Aβ11–25 gave exceptional detail and these fibrils were also analyzed by cryoTEM to directly visualize the cross-β structure74 (Fig. 4). High-resolution cryoTEM revealed striations that were 4.7 Å apart, reinforcing the previous interpretation from the X-ray data and providing new insights into the stability of the fibrils.74 4.7 Å appeared to be the largest repeating unit and no long-range repeat was apparent. Interestingly, later studies have shown quite considerable variation in the helical twist for fibrils and often very long-range repeats that can vary even within a single filament. This variation goes some way to explaining why longer-range repeats were not observed in diffraction patterns.
Amyloid fibrils are made from a large variety of precursor proteins, ranging from the β-sheet sandwich structures of TTR, immunoglobulin LC and β2 microglobulin, to the α+β structure of lysozyme and the α-rich structure of serum amyloid A.55 Natively unfolded proteins and peptides assemble in diseases such as T2D, AD and Parkinson’s disease (PD). Despite this diverse range of starting native structures, all amyloid fibrils share the cross-β structure and all precursor proteins, even those rich in β-sheet, undergo a significant conformational change upon forming amyloid. Early work assumed that antiparallel sheets were formed allowing different length polypeptide chains to access this repetitive structure.72 Intriguingly, electroparamagnetic resonance pointed to a parallel, in-register structure for amyloid fibrils formed from large proteins and this appeared to suggest that the proteins needed to unfold almost entirely to form a layer which then stacked to render the fibrils.88 It seemed improbable, but structural models were put forward showing a β-spine.89 It was not until the first solid-state NMR (ssNMR) models were provided of Aβ fibrils that it was shown that the β-sheets were formed by bending of two strands that stacked to assume a parallel, in register set of β-sheets.90, 91 This structure, held together by hydrogen bonding, provided a stack of identical amino acid side chains along the length of the fibrils. It was clear then, that the side chains played an important role in the structure. X-ray and electron diffraction were combined to provide a model for an amyloidogenic novel sequence AAAKKFFEAAAK, showing that the side chains associated across the sheets.52, 92
The first atomic-resolution X-ray crystal structures of amyloid-like fibrils, formed in vitro from short adhesive segments of amyloid-forming proteins, revealed the basis of amyloid stability and provided atomic level insights into the amyloid core.19, 94 The fibrils are formed from pairs of β-sheets, mated together by interdigitation of their amino acid sidechains. This zipper-like interdigitation of these structures suggested the term “steric zipper” for this motif, which has now been found in numerous X-ray, NMR, and cryoTEM structures19 (Fig. 5). In structures of full fibrils, steric zippers are frequently found at junctions of protofilaments (Fig. 5). Elsewhere in full fibrils, hetero zippers (formed by two different sequences) are found. Factors contributing to amyloid stability include: (1) the hydrophobic effect of releasing water molecules from the tight, dry interface between the sheets; (2) van der Waals stabilization of the interdigitating sidechains; (3) mutual polarization of stacked amide hydrogen-bonding groups parallel to the fibril axis;95 and (4) ladders of stacked sidechains such as the phenolic groups of tyrosine residues on the surface of the fibrils. The steric-zipper motif also explains the sequence specificity of amyloid formation: only compatible sequences can form steric zippers. Peptide inhibitors, designed on the basis of crystal structures of short segments, are effective in inhibiting aggregation and cell entry of full pathogenic amyloid fibrils.96
Fig. 5. Atomic-resolution crystal structures of two adhesive segments and two amyloid fibrils of the protein α-synuclein associated with PD.

Upper left: crystal structure of the PreNAC segment with sequence 47GVVHGVTTVA56 (the first T in this sequence is a hereditary early-onset disease mutation A52T). The upper view is down the axis of this steric zipper, showing atoms with van der Waals radii forming a tight, dry interface. Upper right: crystal structure of the NACore segment with sequence 68GAVVTGVTAVA78. The center shows two amyloid-like fibrils formed by α-synuclein (αS). In the top of the left center, one layer of the Rod polymorph is viewed down the fibril axis, showing that it contains two identical αS chains each bent into a double hairpin shape. The two chains meet at a steric zipper formed by the PreNAC segments of the two chains. Identical layers are stacked on each other, forming a two-protofilament, slowly twisting fibril. In the top right center, one layer of the Twister polymorph is viewed down the fibril axis. The fold of the two αS molecules is similar to that of the Rod polymorph but the two chains meet at a different point than those of the Rod polymorph. They meet at an interface similar to that of the NACore crystal structure. That is, the steric zipper interfaces that pair the β-sheets in the crystal structures are similar to the interfaces between paired protofilaments in the fibrils. Notice that the slowly twisting Twister fibril is formed by stacking identical layers on each other, with a slight twist. Reproduced with permission from ref. 93, copyright 2018 Springer Nature.93
The technical challenge of determination of X-ray crystal structures of amyloid fibrils is that the crystals are invariably no larger than several microns in size. The hypothesis for the small crystal size is that β-sheets normally exhibit a slow twist but are held in amyloid crystals in untwisted form, producing a strain that builds as the crystal grows, limiting size. In fact, the crystals of the 11-residue NACore segment of αS are only a few hundred nanometers in cross section, and hence invisible by light microscopy. Consequently their structure had to be determined by electron diffraction, for which small crystals are advantageous.93 Yet, as it will be discussed later, the untwisted form of amyloid crystals - compared to the twisted form of their homologue fibrils - opens for a different way to further decrease the overall free energy, placing them at the lowest minimum of the free energy landscape.20
Thanks to developments in ssNMR and cryoTEM,103, 104 numerous near-atomic-resolution structures of much longer segments of amyloid fibrils are now available (Figs. 5&6). ssNMR yielded structures for a 22-residue fragment of β2 microglobulin,105 Aβ40 by 2008,106 for the more toxic Aβ42 by 2016,97, 98 and for αS fibrils.107, 108 Further advances in cryoTEM, largely helped by the invention of direct detectors and the treatment of helical structures as single particles109, 110 have led to an explosion in atomic detail of amyloid structures. Near-atomic resolution structures were solved for amyloid fibrils of αS,111–113 Tau,101, 114 and TDP-43.115 Paired helical filaments and straight filaments from AD brain showed the parallel in register structure with further exciting details at the bends between the sheets. Tau filaments from chronic traumatic encephalopathy patients114 and from Picks disease patients116 show intricate differences that may give us clues regarding the differences between the diseases. Immunoglobulin LC amyloid,117 β2 microglobulin,118 acute phase protein amyloid A (AA)119, 120 form similar core structures. Many proteins in the amyloid state are able to assume a variety of structural folds termed polymorphs.
Fig. 6. Example ssNMR and cryoTEM structures for amyloid fibrils.

Upper panel: ssNMR structure of Aβ42.97, 98 Two S-shaped molecules of Aβ42 (black and gray) are related by a twofold axis (marked by a circle), which runs down the center of the fibril. The N-terminal 14 residues are disordered; one possible conformation is shown here by dotted lines. Many of the known hereditary mutations are carried by residues located on the outer surface (red). The surface hydrophobic patch formed by residues V40 and A42 (orange) may explain the greater rate of secondary nucleation by the 1–42 species compared with 1–40.99, 100 Bottom panel: CryoTEM structures of two amyloid fibrils of Tau.101 These two polymorphs of Tau amyloid fibrils were purified from the autopsied brains of AD patients. In both polymorphs, individual Tau proteins form C shapes, as shown by the cartoon ribbons with arrows that lie nearly in a plane perpendicular to the fibril axis. The protein layers are stacked up to form a protofilament. For each polymorph, there are two protofilaments, but they meet at different interfaces. Steric zippers are noted in the straight filament polymorph. The β-helical feature is enlarged in the right-hand panel where it is shown in yellow.102 Reproduced with permission from ref. 100, copyright 2016 National Academy of Sciences.100 Reproduced with permission from ref. 101, copyright 2017 Springer Nature.101 Reproduced with permission from ref. 102, copyright 2017 Annual Reviews.102
Despite the variety of molecular structures displayed by amyloid proteins, they show common features. The proteins mainly form extended β-strands, and these are bent into a series of hairpin-shaped β-arches, and are confined essentially in a 2-dimensional slab or “layer”. Backbone amide groups extend their hydrogen-bonding C=O and N-H groups up and down, parallel to the fibril axis, and the resulting hydrogen bonds stack the layers into slowly twisting protofilaments (Fig. 5). Most often, two or more protofilaments twist around each other, forming the fibril, but some fibrils are built from a single protofilament and some are formed from several protofilaments. The protein structures remain “cross-β” displaying the expected distance of 4.76–4.78 Å between the hydrogen bonded β-strands which generally run perpendicular to the fibre axis.
Whereas pathological amyloid fibrils tend to be so stable as to be irreversible, considerably more labile amyloid-like fibrils have been found to form from low-complexity domains of proteins that participate in hydrogels and liquid-liquid phase separation.121–123 These low-complexity domains are especially rich in Gly, Ser, and Tyr residues, and poor in most apolar residues. Short segments of these domains have been crystallized and the resulting structures (Fig. 7) are similar to steric zippers in that they show pairs of stacked β-strands. But these weakly adhesive elements differ from steric zippers in that the backbones are usually kinked, with more polar and apparently weaker interfaces that account for the reversibility of the fibrils. These mildly adhesive interfaces have been termed LARKS, an acronym for Low-complexity, Amyloid-like, Reversible, Kinked Segments. LARKS may contribute to the interactions between proteins with low-complexity domains that participate in transient subcellular bodies, such as stress granules.
Fig. 7.

Atomic-resolution crystal structures of five LARKS contrasted with the structure of a steric zipper from the segment with sequence NKGAII from Aβ. The right-hand column shows the paired β-sheets of the steric zipper at the top and of the five LARKS below. For each structure, five layers are shown of the thousands in the crystals, with the fibril axes vertical. The view in the middle column is down the fibril axis and shows all atoms of the interfaces. The view in the left column is also down the fibril axis and shows the tracings of the protein backbones. The tight interface of the steric zipper offers a strong interaction. The kinked interfaces of the LARKS are weaker. Each interface is characterized by its shape complementarity score (Sc = 1.0 for perfect complementarity) and buried solvent-accessible surface area (Aβ) in Å2 between the mated sheets. Nitrogen atoms are blue, and oxygen atoms are red.123 Reproduced with permission from ref. 123, copyright 2018 American Association for the Advancement of Science.123
Structural studies of amyloid and amyloid-like fibrils have opened understanding of these pathological and functional architectures at the atomic level. The hope is that continued studies will contribute to the development of diagnostics and therapies for the numerous diseases associated with these fibrils.
4. Mesoscopic structures of amyloids and their position in the energy landscape
The mesoscopic features of amyloids, obtained from rabbit and human kidney tissues affected by primary amyloidosis, were first described by Cohen and Calkins in 1959 with a negative-stain electron microscope.53 The authors marveled that “in the rabbit kidneys, the appearance of the amyloid was striking… (showing) delicate filaments”. Although limitation imposed by sectioning prevented the precise delineation of fibril dimensions, they appeared to range in length from 1,200 to 5,000 Å, and in width from 50 to 120 Å. The biopsy specimen of the patient with extensive primary amyloidosis also showed wavy bundles of delicate fibrils in the electron microscope. These correlated with the areas of amyloid as seen in the phase microscope and in stained sections. The dimensions were similar to the ones seen in the rabbit amyloid. The width varied from 70 to 140 Å and long strands up to 16,000 Å were measured. No cross-bundling was apparent. The amyloid in the kidney of the patient with parenchymatous involvement also demonstrated fine bundles of filaments similar to those noted above.”
The mesoscopic structure of amyloid fibrils has since been extensively characterized, primarily with TEM, cryoTEM and AFM, and similar fibrils have been documented for amyloids and amyloid-like entities including functional and pathological amyloids as well as engineered peptide and protein amyloids of various lengths as short as several amino acid residues, with di-phenylalanine being likely a minimum motif for fibrillization.7, 124–126
The repeating 3D structures of these amyloids are composed of many (usually hundreds to thousands) copies of a peptide/protein. As discussed earlier, at the atomic level amyloids are arranged in a one-dimensional ordered cross-β-sheet motif, which consists of two or more layers of intermolecular β-sheets that run along the fibril axis.127 The polypeptides often render unbranched fibrils, 6–12 nm in width and up to several micrometers in length,128 and are in general composed of several protofilaments.129 The protofilaments may twist around each other but not exclusively in a left-handed fashion. Straight fibrils composed of several filaments as well as right-handed fibrils have also been documented, though rarely for the latter.130 The left-handed twist is attributed to the underlying β-sheet secondary structure conformation composed of L-amino acid residues (correspondingly, amyloids composed of peptides of synthetic D-amino acids are usually right handed), although the transfer of chirality of amyloid fibrils across length scales is not conclusively solved, since protofilaments of a given handedness may merge to form mature amyloid fibrils of opposed handedness.131 Irrespective of the final handedness, a full rotation of a filament within a fibril may be in the order of tens to several hundred nanometers requiring a ~0 to a few degrees of rotation per β-strand. This imposes a limitation on the twist periodicity as the hydrogen bond network of the β-sheet important for the stabilization of the 3D structure is slightly perturbed by the twist.
In general, the twist of amyloid fibrils results from propagation of the chiral β-sheet secondary structure to higher hierarchies, and thus is intrinsically related to the topology of the fibrils; yet extrinsic parameters have also been found to contribute to the overall observed twist. For example, the charged side chains on the fibrillar surface induce a torsion per unit length which is directly proportional to the overall charge. Since the extent of this kind of charge repulsion can be tuned by salt concentration and composition of the buffer medium, the twist periodicity can be manipulated by the salt concentration of the system under study, as demonstrated for β-lactoglobulin first grown at a low ionic strength and then exposed to a high ionic strength post fibrillization.132, 133 According to this scenario, the “electrostatic” contribution to the twist can be relaxed by screening electrostatic charges via the presence of salts or buffers, until nearly complete untwist of the fibrils is achieved. However, a very different scenario may occur when the salt is already present at the fibrillization stage: a structural study on the functional amyloid hormone β-endorphin grown in the presence and absence of NaCl revealed that, while the fibrils were highly twisted when grown in salt, they appeared straight when grown in the absence of salt albeit displaying practically the same 3D atomic structure resolved by ssNMR spectroscopy.134 In this case, the presence of salt influenced not only the electrostatic interactions but also the fibrillization process itself.
The twist periodicity may vary between fibrils within the same sample and, to some extent, even within a single fibril. In the same sample there may be both straight fibrils as well as twisted fibrils, sheet-like structures, as well as helical ribbons (see below for definitions, as well as Fig. 8). These heterogeneous morphologies are at the core of the so-called “mesoscopic polymorphism”, which arises from distinct structures at the atomic level (also referred to as “molecular polymorphism”),113 distinct protofilament packings,113 local salt concentrations during the nucleation events, or distinct nucleation sites on heterogeneous surfaces. The origin of the polymorphisms is therefore attributed to the (local) environmental conditions, but may also indicate a kinetically trapped origin of the amyloid.135 Nonetheless, the large amount of polymorphism that can be observed at the mesoscopic level, as exemplified by Aβ40, is regarded both a typical property as well as a conundrum of amyloids.
Fig. 8.

Structure of amyloid fibrils as a function of the constitutive number of protofilaments in HEWL lysozyme as observed by AFM. The number of protofilaments is indicated in each panel. Up to 3 protofilaments, the fibrils remain straight and in a twisted ribbon configuration (H = 0). Starting from 4 protofilaments, amyloid fibrils change into a helical ribbon configuration, as revealed by the characteristic zig-zag contour shape associated with a non-zero mean curvature (H ≠ 0). Reproduced with permission from ref. 136, copyright 2011 American Chemical Society.136
In detail, the mesoscopic polymorphism of amyloid fibrils includes various topologies which can be classified directly by the Mean (H) and Gaussian (K) curvatures of the amyloid fibril surface, defined as:
where the two principal curvatures, c1 and c2, are the inverses of the main radii of curvatures R1 and R2 describing the surface at each point. Helical ribbons can approximately be wrapped around a cylinder of radius R and at every protofilament, the principal curvature are ; helical ribbons are therefore characterized by ; K ≈ 0. In twisted ribbons, the situation is quite different. Bending of protofilaments is very small, and external protofilaments must describe helical trajectories which introduce an increasingly large stretching when moving from the center to the external protofilaments, as recently described in the context of the morphogenesis of other topological objects, including plant leaves.137 Since the mesoscopic bending of twisted ribbon amyloids is minimal, these objects are well approximated by H ≈ 0, whereas K deviates from zero, due to a torsion which is a function of the width-to-thickness ratio of the ribbon.138 Thus, the twisted ribbon topology is well described by the geometry of a helicoid, i.e. the ruled minimal surface in between the helical trajectories of the two external protofilaments placed at a distance R from the central axis. For such a ruled minimal surface, H = 0 and , where P is the full pitch length (periodicity) of the twisted ribbon. Since in amyloid twisted ribbons generally P ≫ R,133, 139 this can be further approximated by H ≈ 0 and . The combined negative Gaussian curvature and zero mean curvature endow twisted ribbons with saddle-like topological features. In contrast, flat ribbons and amyloid crystals (achiral, no twist) both possess H = 0; K = 0 by definition. Nanotubes are topologically similar to helical ribbons, and the exact relation ; K = 0 holds for them.
The overall elastic energy per unit length of amyloid fibrils is a complex interplay of torsional and bending energies,20, 21 whose contributions change differently with the lateral dimensions of amyloid fibrils. As a consequence, different structures of amyloid fibrils are found as a function of the number of constitutive protofilaments: at a critical width to thickness ratio or for a specific number of protofilaments, a transition from twisted to helical ribbons occurs,136 in analogy to the behavior observed for chiral liquid crystalline films undergoing similar transitions at a critical film width138 or leaves undergoing identical twisted-helical ribbon transition for a critical differential strain.137 In other words, in a twisted ribbon morphology, the twist periodicity itself limits the number of protofilaments per fibril possible, as the outer protofilaments must go a longer way around the central one. A consequence of this fact is that the number of protofilaments per fibril in a twisted ribbon is approximately proportional to the twist periodicity and actually appears to be proportional to the twist periodicity for fibrils with several protofilaments.133, 139 Conversely, in a helical ribbon, no universal feature relating the periodicity to the number of protofilaments is observed. This is because helical ribbons can close into nanotubes, thereby reducing the line tension of external protofilaments by virtually maintaining their mean curvature H unchanged and rendering them a metastable precursor to nanotubes. Fibrils composed of two to several protofilaments have been documented. In the case of straight fibrils or helical ribbons, however, the number of protofilaments or ribbons may increase significantly towards a sheet-like entity composed of up to 10 or more filament entities,136, 140 with a record-large number of protofilaments in a single flat amyloid ribbon reported for the case of the R3 fragment of Tau protein.141
Recent studies of the elastic energies of twisted ribbons, helical ribbons, nanotubes, flat ribbons and crystals21 have allowed positioning each of these polymorphs in a relative scale of energy (Fig. 9, right panel). Specifically, the absolute minimum in the free energy of the protein folding landscape previously attributed to amyloid fibrils has been refined into a series of relative minima where each polymorph has a specific energy level.20, 21 Twisted ribbons occupy a relative minimum in the protein folding energy landscape and must overcome a precise energy barrier to fully untwist and enter the absolute minimum occupied by (achiral, untwisted) amyloid crystals; helical ribbons need to overcome a larger energy barrier to fully untwist and enter the same minimum as amyloid crystals: the extra energy barrier compared to the amyloid twisted ribbons is provided by the twist-bending coupling energetic term existing for helical ribbons but missing for twisted ribbons. Accordingly, no helical ribbon-amyloid crystals and their transitions have yet been observed, whereas twisted ribbon-amyloid crystals have been well documented.20 Because the energy level of a fully untwisted helical or twisted ribbon is equivalent, this places helical ribbons on a lower energy level than twisted ribbons. Thus, rather than overcoming this larger energy barrier, helical ribbons tend to further evolve by closing into nanotubes, which are further down the energy level reduced by the line tension associated with the external protofilaments found in helical ribbons. Only flat amyloid crystals, for which the translational symmetry associated with a lack of macroscopic chirality accepts reduction of surface tension by lateral aggregation, are allowed to (indefinitely) sink into an energy minimum funnel which is associated with the ground state of the protein folding landscape.
Fig. 9.

(Left) Schematic representation of the main mesoscopic polymorphs observed for amyloid fibrils and their approximate Mean (H) and Gaussian (K) curvatures. (Right) Sketch of the protein folding landscape in the region around the amyloid minimum: different polymorphs occupy different energy levels, with amyloid crystals populating the absolute minimum. The right panel is redrawn with permission from Adamcik et al.21 Reproduced with permission from ref. 21, copyright 2018 Wiley-VCH.21
A question rises spontaneously of why amyloid crystals which are postulated the ground state in the protein folding energy landscape, are so rarely observed in vivo. As already observed by Adamcik et al.,21 the protein folding process in vivo occurs in non-conservative energy ensembles, with energy injected into and/or dissipated by the system during biological processes and with chaperone proteins assisting protein folding. This is in stark contrast to in-vitro processes, where the lack of chaperone proteins and the closed (conservative) ensemble allow revealing the presence of amyloid crystals.
5. Primary and secondary nucleation
The formation of amyloid structures from a solution of peptide or protein molecules can be viewed as a phase transition where a more ordered phase is formed within a less ordered solution phase. Much attention has focused on the very early stages of the formation of the amyloid phase. In general, the formation of a new phase can be triggered either through spinodal decomposition or nucleation. Spinodal decomposition takes place under conditions where the solution phase is unstable and even small density fluctuations are amplified and the formation of a new phase takes place very rapidly. By contrast, nucleation takes place under conditions where the solution phase is metastable rather than unstable; this situation arises when the newly formed phase has a lower free energy than the soluble phase, but kinetic barriers slow down its initial formation. The early stages of amyloid formation have been found to follow the physics of nucleated processes.142, 143 The stability of the amyloid phase is determined by the thermodynamic solubility of the amyloid forming protein; this is the critical concentration Cc that remains in equilibrium with the amyloid phase, and is in turn directly related to the standard free energy -ΔG of transfer from the solution to the amyloid phase, Cc = exp(−ΔG/kBT), where T is the temperature and kB the Boltzmann constant. As such, when the concentration of soluble protein remains below the critical concentration, there is no thermodynamic driving force for forming the amyloid phase. When this threshold is exceeded, the amyloid phase is now more stable than the solution phase, and slow nucleation can take place. Once an initial fibril has been formed, further monomeric protein molecules can add on at a much faster rate, a feature which is common to nucleation-growth phenomena in nature, a special case of which is nucleated polymerization which results in elongated structures such as amyloid fibrils.144
There is a rich history of studies focusing on elucidating the principal features of nucleated polymerization. Much of the early work was carried out in the context of understanding the polymerization of cytoskeletal filaments, including actin and tubulin, which have a similar linear geometry to amyloid fibrils.146 Studies in the 1960s established the principal features of this type of process, including the fact that for early times t the increase in the aggregate mass M follows generically a polynomial behavior M ~ t n, where n = 2 for simple nucleated polymerization and can have a higher value when the nucleation process is multi-step in nature.146 An important feature of this type of classical nucleated polymerization is that there is only a weak lag phase due to the polynomial time dependence.
Commonly, however, for amyloid formation, the reaction starts with a very marked lag phase during which no or only very low concentrations of aggregates are detected. After the lag phase, the growth and formation of new amyloid fibrils takes place rapidly; this type of process has therefore the features of a highly cooperative transition, where the presence of aggregates facilitates the formation of further aggregates. A central challenge therefore in the mechanistic studies of amyloid formation is to relate the macroscopic observations of protein aggregation to the underlying microscopic mechanisms. A powerful tool in this context is chemical kinetics, a formalism that captures a series of molecular events into a rate law that describes the overall progress of the reaction (Fig. 10A). Application of chemical kinetics to protein aggregation has revealed that in many cases the apparent high level of cooperativity originates from a non-classical secondary nucleation process.16, 147, 148 In secondary nucleation, the existing amyloid fibrils act as catalytic surfaces for the formation of new amyloid nuclei which can then grow further themselves. This type of process was originally described for crystal nucleation where under many conditions growing crystal faces can favour the formation of new nuclei. Secondary nucleation was also found to be the key process controlling sickle haemoglobin polymerization,149 a non-amyloid related pathological protein assembly process. It has now been identified as a key mechanism for the formation of amyloid fibrils from systems as diverse as Aβ40,147 Aβ42,16 αS148 and hIAPP.150 Recent evidence suggests that the sites for secondary nucleation and growth are distinct and that secondary nucleation takes place preferentially at the sides of amyloid fibrils.151
Fig. 10.

Primary and secondary nucleation and their verification with microfluidics. (A) Illustration of the power of chemical kinetics to elucidate microscopic mechanisms. Experimental data for the aggregation of the Aβ42 peptide fitted to an integrated rate law where the dominant source of new aggregates is, from left to right, primary nucleation, fragmentation and secondary nucleation, respectively. (B) Schematic illustration of the microfluidic strategy to detect directly single primary nucleation events and monitor the aggregation reaction in both time and space. (C) Time-lapse microscopy of a single microdroplet trapped in the array shown in panel B. (D) Schematic illustration of the primary and secondary nucleation events and subsequent aggregate multiplication which can be measured directly in microfluidic experiments. Reproduced with permission from ref. 16, copyright 2013 National Academy of Sciences.16 Panels B-D adapted from ref. 145, copyright 2011 National Academy of Sciences.145
The existence of secondary nucleation challenges a number of intuitive assumptions about amyloid formation, and perhaps most strikingly that of the nature of the lag phase. Indeed, under conditions where secondary nucleation is a dominant factor, the lag phase is only very weakly dependent on the time to form the initial nuclei, but rather depends on the rate at which these nuclei can grow through elongation and multiply through secondary nucleation.152, 153 This observation implies that primary nucleation can be very challenging to study in bulk systems as it has only a very weak effect on the overall kinetics. This picture changes, however, when aggregation takes place in very small volumes, a regime that can be probed through droplet microfluidics (Fig. 10B–D). Microfluidic experiments have allowed the study of single nucleation events, as well as the rate at which amyloid conformations of proteins can propagate in space and time from the site of the original nucleation event.145
The role of secondary nucleation in the development of amyloid diseases remains an active area of investigation. There are indications that this process could be key in generating toxic oligomers that are responsible for neuronal death associated with the aggregation of the Aβ peptide in the central nervous system.154 Indeed, microscopy studies have revealed that the concentration of oligomers is highest in the vicinity of higher molecular weight aggregates such as plaques.155 If the formation of such oligomers was driven by primary nucleation, their concentration would be lowest in the vicinity of plaques as the latter can sequester monomer through their growth, thus leaving less monomer available for primary nucleation. Secondary nucleation, by contrast, is highest in locations which contain both monomer and aggregates,16 in agreement with experimental observations of oligomer localization in vivo. These considerations highlight secondary nucleation therefore as a potential new target for curtailing the accumulation of Aβ oligomers in vivo. Finally, it has become apparent that nature has evolved molecular chaperones that are able to inhibit secondary nucleation in a highly specific and effective manner.154 This inhibition has furthermore been shown to lead to a significant reduction in toxicity associated with protein aggregation, even when the overall concentration of aggregates is not affected, as it significantly reduces the concentration of oligomeric species.
6. The “oligomer hypothesis”
The lack of tools that allow visualizing the different stages of amyloid formation led initially to the thinking that amyloid formation was a two-state process that involved the conversion of soluble native proteins into highly ordered cross-β sheet fibrillar structures, similar to the polymerization of tubulin monomers into microtubules. Hence, the original versions of the amyloid hypothesis stipulated that amyloid diseases were caused by the formation and accumulation of amyloid fibrils in the brain or other affected organs.156 However, several consistent pathological observations suggested that amyloid fibrils may not be the culprits and led to reconsideration of this hypothesis.157–161 These observations include: (1) amyloid fibrils derived from different proteins were found in the post mortem tissues of individuals who died without exhibiting any symptoms of amyloid diseases;162 (2) the amyloid load did not always correlate with disease onset or severity; (3) several studies did not find a clear correlation between the extent of fibril formation and neurodegeneration in AD animal models;163–165 and (4) therapeutic interventions that successfully cleared amyloid plaques in humans did not result in reversal or improvement in clinical symptoms of AD.166, 167 The emergence of these findings coincided with reports from biophysical studies on Aβ, the key component of amyloid plaques, suggesting that amyloid fibril formation may be more complex than initially thought and involves the formation of protein assemblies other than the amyloid fibrils.
The ability to generate amyloid fibrils in cell-free systems has provided unique opportunities to investigate and dissect the mechanisms of amyloid formation. These studies, performed on Aβ peptides by the Teplow and Lansbury groups,168, 169 revealed for the first time that amyloid formation did not follow a two-state mechanism but rather occurred through a series of soluble oligomeric intermediates of variable size and morphologies. The observation that these oligomeric intermediates disappeared upon fibril formation suggested that they were on pathway to amyloid formation. As of today, oligomers have been observed during the fibrillization of nearly all amyloid-forming proteins, suggesting that they are obligate intermediates on pathway to amyloid formation.
Although the great majority of studies have focused on characterizing oligomers that form on the pathway to amyloid formation, increasing evidence suggests that oligomers could also form through fibril-mediated mechanisms or during processes aimed at promoting fibril clearance. Several studies have suggested that oligomers could form during the disassembly or fragmentation of fibrils or upon their interactions with membranes.170–172 The surfaces of fibrils have also been shown to nucleate the formation of oligomers via secondary nucleation mechanisms.173, 174 Furthermore, it has also been proposed that amyloid plaques and proteinaceous inclusions may also serve as reservoirs for toxic oligomers.175–179 However, whether oligomers are simply sequestered during the formation of amyloid-rich deposits/inclusions, represent the byproducts of cellular process aimed at dissociating and clearing fibrils, or are formed within these deposits/inclusions remains unknown.
Together, these observations sparked a huge interest in the field because they offered a possible explanation for the lack of correlation between amyloid load and disease onset or severity. This gave rise to an alternative amyloid hypothesis, the oligomer hypothesis, which stipulates that oligomeric prefibrillar intermediates, rather than the amyloid fibrils, are the primary cause of toxicity and cell death in AD, PD and systemic amyloid diseases.
Oligomeric intermediates on pathway to amyloid formation are by definition transient in nature, as already largely discussed above in the context of the protein folding landscape. They do not accumulate and are usually converted rapidly to higher order aggregates, and eventually to fibrils. Although it is possible to capture and detect oligomers during the process of amyloid formation using imaging techniques such as AFM or TEM, isolation of such oligomers during the fibrillization process has proven to be difficult for most proteins. To address this challenge, several protocols have been developed to enhance oligomer formation and/or slow their conversion to fibrils by manipulating solution condition or the use of mutant forms of the proteins that exhibit higher propensity to aggregation (e.g. Aβ42 vs Aβ40 and variants linked to early onset or severe forms of relevant amyloid disease). Other protocols relied on the use of chemical or radical-mediated cross-linking approaches to trap and/or stabilize transient oligomers to facilitate their characterization or isolation.180–186
At the structural level, on-pathway oligomers tend to exhibit a mixture of secondary structure contents,187–189 often dominated by β-sheet conformations.190 Compared to amyloid fibrils, oligomeric intermediates of most amyloid forming proteins exhibit weak binding to the amyloid specific dyes thioflavin T/S (ThT/S) and Congo red,191, 192 suggesting that they have not acquired the cross-β structure that is characteristic of amyloid fibrils, although studies on Aβ oligomers using X-ray fiber diffraction have suggested that some oligomers possess cross-β-like conformations.193 Unlike amyloid fibrils, which despite their polymorphism still share a common core structure, cross-β sheet, amyloid oligomers exhibit large differences in their dynamic properties and structural diversity, suggesting that it is unlikely that one specific molecule or antibody would recognize all types of oligomers formed by one protein. In 2003, Kayed et al. reported that it was possible to generate antibodies that not only recognized different types of oligomers and but also oligomers derived from different amyloid proteins (Aβ, hIAPP, αS and Tau) and suggested that amyloid oligomers derived from these proteins shared common structural features.194 This hypothesis was supported by subsequent findings showing that oligomeric preparations from these amyloidogenic proteins were toxic to cells and neurons. However, subsequent studies by Glabe and colleagues and other groups revealed that the different aggregate and oligomer specific antibodies stained different types of pathological aggregates in the brain and that there was no universal antibody capable of recognizing all type of Aβ oligomers.195–197
The heterogeneity and dynamic properties of the oligomers have thus precluded studies aimed at resolving their structural properties at the atomic level. Oligomers rapidly interconvert between different forms and exhibit high propensity to transition to higher order aggregates, thus making it difficult, if not virtually impossible, to isolate and investigate the structural, functional and toxic properties of a single oligomeric species. Several attempts have been made to achieve this goal, but without any success. The use of sequential chromatography separation methods or other protein separation techniques has enabled the generation of oligomer preparations that are enriched in specific morphologies,169, 187, 198–200, 201 but generation of homogeneous preparation consisting of one oligomeric species of a defined size and morphology has not been possible. This explains why, despite two decades of active research, it has not been possible to ascribe toxicity to a specific oligomeric entity or develop tools and strategies that target distinct types of oligomers. Furthermore, the diversity of the protocols used to produce oligomers, which leads to oligomer preparations of different size, structure and morphology distribution, combined with the lack of tools and methods that enable precise assessment of oligomer heterogeneity, has made it difficult to compare and reproduce results across different laboratories. Despite these challenges, such oligomer preparations have been used to gain insights into the dynamic properties of oligomers and to elucidate the sequence and structural determinants of oligomer formation and stability using solution and ssNMR, hydrogen deuterium exchange methods and other biophysical techniques.189, 202–208
6.1. The amyloid pore
Among all the different types of amyloid oligomers and prefibrillar aggregates that have been isolated, the only type of oligomers that suggest a specific mode of action and mechanism of toxicity are the annular pore-like oligomers, which have been observed for most amyloid forming proteins. Annular pore-like oligomers have been observed during the aggregation of both disease-associated (e.g. Aβ peptides18, 198, 209, αS, SOD1,209 exon1 of the Huntingtin protein, Tau,210 TTR211 and serum amyloid A212) and non-disease-associated amyloid forming proteins.213–218 They have been found in the absence of membranes and also upon addition to lipid bilayers or reconstitution of amyloid proteins and peptides with membranes. Furthermore, several AFM studies have provided direct evidence of amyloid-pore formation in synthetic vesicles or membrane mimics by several amyloid forming proteins.219–223 Their shape and dimensions, combined with extensive literature demonstrating that Aβ, hIAPP and other amyloid proteins exhibit channel-like activity on membranes,219, 222, 224–226 have led to the amyloid-pore/channel hypothesis, which suggests that channel/pore formation represents one of the key mechanisms by which oligomers cause toxicity and cell death in amyloid-related diseases. Evidence in support of this hypothesis comes primarily from in vitro studies. For example (1) mutations linked to early-onset AD and PD promote the formation of amyloid pores and increase the channel and membrane permeabilization activity of Aβ and αS; (2) mimicking cellular stress conditions associated with neurodegenerative diseases, such as oxidative stress and metal induced oxidation also promotes the formation of annular pore-like structure;209 and (3) several amyloid oligomers exhibited channel-like activity and size-selective membrane permeabilization.227, 228 Structurally, several studies have shown that amyloid pore oligomers or oligomers that exhibit channel-like activity exhibited β-sheet rich conformations that were distinct from that of mature fibrils.187, 229, 230 Although different types of oligomeric preparations of Aβ induced calcium uptake and disruption of ion homeostasis in cells, the exact mechanisms by which these preparations exerted their effects on cellular membranes remain unclear.
6.2. Toxic oligomers
The search for a toxic oligomer species has been the focus of active research in both academia and industry. The hope is that identifying a specific toxic species will pave the way for developing novel therapeutic drugs and antibodies that prevent their formation, induce their disassociation or block their activity. During the past two decades, many studies have shown that amyloid oligomers induce different types of toxic insults when added to different types of cells, organotypic slice cultures or injected into rodent brains. The extent and type of toxicity observed vary depending on the size distribution of oligomers and the assay and model systems used to assess their toxicity. However, for all amyloid-forming proteins, the nature of the oligomeric toxic species associated with each disease and their mechanism of action remain elusive. In addition to the complexity and heterogeneity of oligomer preparations, the lack of tools that allow monitoring amyloid oligomer formation and dynamics in cells makes it very difficult to attribute any toxic effects or phenotype directly to specific type of oligomers. The great majority of toxicity assays are based on addition, treatment or injection of in vitro oligomer preparations into culture media or directly into the brain, and toxicity is assessed hours to days or even months after treatment with oligomers. During this time, the extent to which the oligomer preparations retain their original properties or change their conformation and structural properties in response to changes in their environments remains unknown. Therefore, better understanding of the structure-function relationship of amyloid oligomers requires the deployment of assays that allow for rapid assessment of the cellular responses upon treatment with well-characterized preparations of different types of amyloid species.
6.3. Post-translational modifications
Post-translational modifications (PTMs) such as phosphorylation, proteolytic cleavage, nitration and ubiquitination play central roles in the aggregation and pathology formation in the majority of amyloid-related diseases, including AD, PD, Huntington’s disease (HD), and prion diseases. Amyloid fibrils, which are among the major constituents of these pathological inclusions are subjected to different types of PTMs, which very often co-occur on the same fibrils. Despite the fact that these modifications are used as pathological markers and antibodies, and assays targeting modified forms of pathological amyloid fibrils are commonly used to assess pathology formation and spread and to monitor disease progression, the role of PTMs in regulating the different steps along the amyloidosis pathway remains poorly understood. The roles of PTMs in amyloid oligomer formation, dynamics and the transition to fibrils have not been investigated. Indeed, all of the amyloid oligomer preparation protocols used to investigate amyloid oligomer structure and toxicity are devoid of PTMs because they are usually derived from recombinant and synthetic proteins. Although several studies have reported on the use of oligomers isolated from tissues, cells or model organisms, the biochemical properties of these oligomers and whether or not they are post-translationally modified have rarely been investigated. Given the increasing evidence demonstrating that PTMs could significantly influence oligomerization, amyloid formation and clearance, it is crucial to devote more attention and resources to map the PTM profiles of native oligomers from human tissues and biological fluids and to assess their effects on oligomer formation, structure and toxicity. It is reasonable to speculate that PTMs may act as molecular switches for regulating the equilibrium between different types of oligomers and/or transitions from oligomers to fibrils. Recent advances in protein synthesis of amyloid proteins have enabled site-specific introduction of single or multiple PTMs into amyloid proteins such as Aβ, αS, Tau, N-terminal fragments of the HTT protein and the prion protein, among others. Such homogeneously modified proteins can be prepared in milligram quantities, which should enable generation of modified amyloid oligomers with specific PTMs or patterns of PTMs, thus paving the way to elucidate the role of PTMs in regulating oligomer formation, stability, dynamics, and their transition to amyloid fibrils.
6.4. Evidence for oligomer formation in vivo
Unlike fibrils which can be easily visualized and characterized by several EM techniques in cells or in pathological inclusions,231–234 visualization of oligomers in pathological aggregates remains challenging, as protocols for specific and efficient immunolabelling of oligomers, including amyloid oligomer pores in vivo, in post-mortem brain tissues or on biological membranes, are lacking. Evidence for oligomers come primarily from: (1) studies demonstrating lack of correlation between amyloid fibril formation and toxicity, under conditions that favor fibrillization; (2) studies employing oligomer-specific antibodies; and (3) detection of HMW SDS-resistant oligomers by western blots.235 Even when solution-based methods such as size exclusion chromatography are used to isolate fractions rich in oligomers, estimations of the size of oligomers are then made on the basis of SDS-PAGE analysis of these fractions, due to the presence of other proteins. Furthermore, we have very limited insight into the biochemical and structural diversity of oligomers in vivo and it remains unclear to what extent the oligomers produced in vitro reproduce the landscape of conformational and quaternary structures of native amyloid oligomers. This is largely due to the fact that oligomers are (1) meta-stable; (2) present in low abundance; (3) heterogeneous; and (4) difficult to distinguish from other proteins in complex biological environments.
Several assays and methods have been developed to measure the level of oligomers in biological fluids, but the level of these oligomers is usually too low to allow interrogation of their size and conformational properties and thus these studies are usually limited to correlating oligomer concentrations to disease progression. One of the most commonly used oligomer-specific immunoassays is based on using the same antibody to capture and detect the amyloid protein of interest. However, these assays do not differentiate between oligomers and fibrils and may not provide an accurate quantitative assessment of oligomer levels due to the lack of the proper calibrants or calibrants that capture the diversity of oligomers in biological samples.
6.5. Targeting amyloid oligomers
The lack of correlation between amyloid fibril formation and disease onset and severity in several amyloid diseases combined with increasing evidence of amyloid oligomer toxicity has led to oligomers emerging as one of the primary targets for developing therapies to treat amyloid diseases such as AD and PD. The field of amyloid oligomers and the toxic oligomer hypothesis was initially driven by research on Aβ peptides mainly because (1) Aβ oligomers could be populated in large quantities; (2) validated protocols for producing Aβ oligomers were quickly developed and made accessible and; (3) the Aβ peptides were also readily accessible through commercial vendors. Despite this, Aβ preparations were still characterized by great variability in terms of their size, structure and morphology distribution. To reduce such variability or enrich oligomers of specific size or structure, heterogeneous preparations were further separated using different protein separation methods. Nonetheless, several academic labs and pharmaceutical companies used such oligomer preparations to develop large number of “Aβ oligomer-specific antibodies”, many of which advanced to clinical trials, but none of which has proven to be effective in significantly slowing or reversing the clinical course of AD.236–239 The same approaches have been extended to other amyloid proteins such as αS and Tau,240,241, 242, 243, 244 but with limited success thus far, although several antibodies are still being evaluated in different stages of clinical trials (more see section 7).239, 245, 246 One possible reason for the failure of oligomer-specific antibodies could be the fact that these antibodies may target only one oligomeric form or subset of the different types of oligomers that exist in vivo,247, 248 or that post-translational modifications on native oligomers may interfere with antibody recognition. To address these limitations, it is crucial to gain more insight into the biochemical and structural properties of native amyloid oligomers and develop strategies that enable isolation and characterization of as many native oligomeric species as possible from patient-derived tissues or biological fluids.
7. Amyloidosis mitigation in vitro and in vivo
Amyloidosis originates from protein misfolding, triggered by protein metastasis and abnormal physiological conditions and manifested by the production of protein aggregates possessing rich polymorphism and evolving physicochemical properties.249 Amyloid inhibition, in essence, works against the downward free-energy landscape of protein folding and aggregation250, 251 by stabilizing disordered monomers, driving toxic oligomers and protofibrils off pathway or sequestering them into elimination, and remodelling mature fibrils into biologically inert, amorphous aggregates.
The past decades have witnessed active development of mitigation strategies against amyloidosis, involving peptidomimetics (1990’s onwards),252 monoclonal antibodies (2000’s onwards),159 small molecules (2000’s onwards)253 and, more recently, organic and inorganic nanoparticles and nanocomposites (2010’s onwards) (Fig. 11).22, 254 Specifically, peptide inhibitors, such as the β-sheet breaker KLVFF,255 draw inspiration from the structural characteristics of Aβ256 to initiate hydrophobic interaction with the latter and act in a chaperone-like manner. However, enzymatic degradation and poor blood-brain-barrier translocation are the notable undoing of this strategy. Monoclonal antibodies, such as Bapineuzumab (Pfizer, J&J), Aducanumab (Biogen, Eisai), Solanzumab (Eli Lilly) and Ponezumab (Pfizer) targeting the N-terminus, amyloidogenic fragment and C-terminus of Aβ,257–259 failed to pass phase-II/III trials and evoked the question whether amyloidosis inhibition through peptide targeting and clearance is a viable strategy against neurological disorders. Small molecules identified by microarrays,260 such as 2,8-bis-(2,4-dihydroxy-phenyl)-7-hydroxy-phenoxazin-3-one (O4),253 or derived from natural compounds, such as epigallocatechin-3-gallate (EGCG),261, 262 ameliorated the toxicities of Aβ and hIAPP in vitro via inhibited (and, occasionally, accelerated) aggregation. A major shortcoming with small molecules, however, is their often limited water solubility and, hence, low bioavailability and poor delivery efficacy.
Fig. 11.

Major anti-amyloidosis strategies with peptidomimetics, antibodies, small molecules and nanoparticles/nanocomposites. The main purpose of such intervention is to stabilize the monomers, suppress the population of oligomers/protofibrils, or remodel amyloid fibrils. Such strategies have shown, to various degrees, potency and failures against amyloidosis and their associated toxicity in vitro and in vivo.
Nanomaterials and multifunctional nanocomposites are engineered/synthetic structures possessing versatile surface area, functionality and architecture, and can be tailor-designed to alter protein aggregation and match amyloid in size, morphology and physicochemical properties. As a result, amyloidosis inhibition with biocompatible/biomimetic nanoparticles and multifunctional nanocomposites has become an emerging frontier, driven by the rapid development of nanotechnology and accumulating knowledge on nano-bio interactions.254 Simple polymeric nanoparticles, such as dendrimers and star polymers, as well as condensed ceria nanocrystals, graphene quantum dots, graphene oxide, gold nanoparticles, carbon nanotubes, transition-metal dichalcogenide nanosheets (e.g., tungsten disulphide and molybdenum disulphide), multifunctional peptide-polymer nanosweepers, protein-KLVFF-polymers, hIAPP19–29S20G and mesoporous silica nanocomposites, have shown potency in mitigating the amyloidoses and pathogeneses of Aβ, αS and hIAPP in vitro and in AD, PD and T2D animal models.23, 263–272, 273 In recent years, it has been increasingly realized that, as with molecular inhibitors, the endpoint of amyloidosis mitigation with nanoparticles is not necessarily inhibition of protein fibrillization per se, but suppression of protein toxicity. Indeed, accelerated protein assembly may reduce the population of toxic oligomers and protofibrils,253, 274 analogous to Pmel17 aggregation in melanin synthesis in the skin.275 However, no systematic understanding is currently available to predict whether an exogenous substance, nanoparticles included, inhibits or accelerates protein aggregation, and if accelerated protein aggregation leads to a beneficial or a detrimental effect on cell viability.
Experimental studies of amyloidosis inhibition often involve a ThT or Congo red fluorescence assay to assess the cross-β content and kinetics in protein fibrillization;276 TEM or AFM to characterize the mesoscopic morphology of protein aggregates (see section 4); CD and FTIR spectroscopies to infer the peptide secondary structure and their time evolution; NMR and beam-diffraction (X-ray crystallography, EM) to reveal the atomic structures of amyloid fibrils (see section 3); gel electrophoresis, dot blotting and immunohistochemistry to confirm protein-inhibitor binding and amyloid/plaque formation; in vitro assays to quantify cell viability and mitochondrial activity in conjunction with alleviated production of reactive oxygen species (ROS); and in vivo assays to target and clear amyloid oligomers, fibrils and plaques via autophagy,277 improve cognitive function and mobility, and stimulate recovery of gene dysregulation.
Under in vivo conditions, inhibitors may be administered through systemic circulation, or via direct injection into the brain of transgenic mouse models in the cases of AD, PD and HD. The peripheral circulation of nanoparticle inhibitors, for example, may acquire a plasma protein “corona”278 through nonspecific adsorption, which ascribes a new biological identity to the nanoparticle inhibitors to evade opsonization. Here, the design usually aims at extending the circulation and improving the delivery of the inhibitors. Upon binding with amyloid proteins, nanoparticle inhibitors perturb the aggregation kinetics resulting from nanoparticle-protein interactions in competition with protein-protein self-assembly, mediated by hydrophobic interaction, hydrogen bonding and electrostatic interaction. A major indicator of strong amyloid protein-inhibitor interaction is changes induced in the protein secondary structure, which accordingly shift the high β-sheet contents of protofibrils and fibrils to render disordered structures, coils, or alpha helices. While this strategy may be easily demonstrated in test tubes or cell cultures, binding of inhibitors with amyloid proteins occurs far less frequently in vivo due to the much reduced concentrations of amyloid proteins and inhibitors, environmental pH, as well as binding of the interactants with other intra- or extracellular proteins, chaperones/chaperone-like proteins (such as serum albumin),279 enzymes, ligands, biometals, membranes and other cellular organelles. Each type of interaction may influence the conformation and/or toxicity of amyloid proteins and impact their associated pathogeneses downstream. Furthermore, it has been shown that hIAPP amyloid fibrils, owing to their strong capacity in initiating hydrogen bonding, can acquire a protein corona in culture medium, enriched mostly by linear proteins and multi-domain proteins of structural plasticity.280 The immune response to corona-shielded amyloids and their precursors, however, remains unclear. The nonspecific amyloid protein-environmental protein association, further complicated by the transient and heterogeneous nature of the toxic oligomeric species (see section 6) – with the exception of the structurally better defined β-barrels which are unfortunately of a small population281 – implies that morphology-based in vivo recognition of amyloid oligomers, fragments or plaques may be inherently problematic. In addition, the hallmarks of amyloid pathologies, such as inclusions and plaques, are highly heterogeneous enriched by tens to hundreds of types of proteins and metabolites,282–286 including extracellular matrix glycoprotein serum amyloid P (SAP), which is thought to stabilize amyloid from degradation,287–289 and membrane-bound heparan sulphate proteoglycans (HSPGs), which mediate Aβ aggregation and cell uptake.290, 291 The in vivo origin, dynamics and mechanisms of such hetero-aggregation and cross-seeding (see section 8) are largely unknown, posing a tremendous challenge to the design and implementation of amyloidosis inhibitors targeting multiple amyloid proteins.
In addition to the aforementioned strategies, chiral molecules292–294 or nanostructures,295 carbon nanotubes coated by sonicated fragments of whey protein β-lactoglobulin,23 gold nanoparticles coated with milk protein β-casein,296 and polyoxometalate-Dawson derivatives (POMds) functionalized with histidine-chelating metals (Cu, Fe, Ni, Co and Mn),297 have been utilized as inhibitors against amyloidosis in cell cultures and with zebrafish models (embryos, larvae and adults), exploiting the chirality of amyloid fibrils, protein-metal coordination, chaperone-like inhibition of protein aggregation, as well as functional-pathological double protein coronae mediated by hydrogen bonding and β-sheet stacking. Zebrafish, in particular, have been validated as a high-fecundity alternative to AD, PD and T2D (transgenic) mouse models,298–300 and are especially suited for testing a library of nanoparticle inhibitors to render significant statistics at low cost and high throughput. As multiple abnormalities occur in the homeostasis of essential endogenous biometals,301 cellular delivery or liberation of biometals (e.g., Zn2+ and Cu2+) with functional amyloids24 or metal-binding compounds302 may offer new breakthroughs against amyloid diseases. Although not directly targeting amyloidosis, personalized antisense oligonucleotide (ASO) therapy against a mutation in RNA-binding protein fused in sarcoma (FUS) is being implemented to ameliorate a severe form of ALS,303 a motor neuron amyloid disease. Furthermore, neurotrophic factor (NTF)-based therapies, such as the delivery of cerebral dopamine neurotrophic factor (CDNF), have shown promise in stopping and reversing neurodegeneration.304, 305
8. Cross-seeding of amyloid proteins
Cross-seeding refers to the stimulation of aggregation of one amyloid protein/peptide by another. Figure 12 contrasts self-seeding (homologous seeding) with cross-seeding (heterologous seeding). This subject has been discussed in previous reviews.306–309, 310 In the following, we highlight a number of studies which present important findings or address key challenges on this topic. Cross-seeding has been observed for several amyloid proteins including Aβ with αS, Tau and prion protein PrP. This may be relevant in vivo since co-deposition of Aβ with PrP occurs in amyloid plaques observed in brain sections from AD patients.311 Studies using transgenic mice that overexpress the amyloid precursor protein and which develop typical amyloid (Aβ-rich) plaques show that inoculation with prions leads to a significant enhancement of both the onset of prion disease symptoms and a concomitant increase in the level of misfolded prion protein along with a notable increase in amyloid plaque deposition.312 In the same study, in vitro fibrillization kinetics assays also showed substantial acceleration of Aβ42 fibrillization by PrPSc (aggregated prion protein).312 In an early study, double transgenic mice were developed that expressed Aβ and αS in neurons, displaying enhanced motor defects compared to single αS transgenic mice and more αS neuronal inclusions.313 Cell-free studies also showed that Aβ peptides promoted aggregation of αS, and intra-neuronal accumulation of αS in cell culture.313 In a similar fashion, exacerbated Aβ, Tau and αS pathologies have also been observed in studies using mice genetically engineered to exhibit both AD and DLB (dementia with Lewy bodies, which contain αS).314 This suggests that Aβ, Tau and αS interact in vivo to promote the aggregation and accumulation of each other and accelerate cognitive dysfunction.314 These studies indicate that Aβ peptides may contribute to the development of Lewy-body diseases by promoting αS aggregation and exacerbating αS-dependent neuronal pathologies.
Fig. 12.

Schematic contrasting homologous seeding with heterologous seeding where the seed oligomer (in red) stimulates the growth of oligomers and ultimately fibrils of a different protein or peptide.308 Reproduced with permission from ref. 308, copyright 2013 Morales et al.308
Different strains of αS (self-seeded or not) have a differential effect on Tau inclusion in neurons.315 Both Aβ and αS oligomers cause Tau aggregation and lead to the development of neurotoxic Tau oligomers that are rich in β-sheet structure.316 In vitro studies revealed that the A53T αS mutant enhanced the fibrillization of both Tau and αS itself.317 It was therefore proposed that such effects may be an important contributor to the heterogeneity in amyloid characteristics and symptoms among different individuals. Using the ThT amyloid fibrillization kinetic assay and EM, cross-seeding of Aβ40 or Aβ42 and αS has been examined.318 The greatest enhancement of fibrillization kinetics was observed for cross-seeding with fibrils of αS.318 Other amyloid forming proteins, such as the DNA-binding protein TDP-43, can cross-seed Aβ and suppress the aggregation of the latter into toxic amyloid oligomers.319 Such oligomers are found in the brains of frontotemporal lobar dementia, pointing to the important role of cross-seeding in pathogenesis in this neurodegenerative disease.319 Cross-seeding has been demonstrated in vivo with Aβ and hIAPP.320, 321 In vitro studies indicated that hIAPP fibrils are poor seeds for Aβ40 fibrillization (this paper also reported weak seeding efficiency of Aβ40 by a number of other peptides).320 On the other hand, preformed fibrils of Aβ42 were injected into hIAPP transgenic mice leading to hIAPP amyloid formation in the pancreas.321 Co-localization was also observed with hIAPP and pro-hIAPP (hIAPP precursor protein) being co-localized in cerebral and vascular Aβ deposits although in the converse situation, Aβ was not detected in islet amyloid from T2D patients.321 Cross-seeding effects have also been studied for the two different forms of Aβ, Aβ40 and Aβ4287, 322 and for point mutants of Aβ40 with Aβ40.320
Cross-seeding of prion proteins from one species to another has been suggested to be a mechanism behind the propagation of specific amyloid strains.323 This has also been observed for yeast prions such as Sup35 where the conformational switch that led to domains rich in glutamine and/or asparagine such as [PSI+] was promoted by heterologous proteins containing a similar domain (as in several other yeast prions) or by overexpression of proteins with prion-like Q-, N- or Q-/N- rich domains.324 Mutual cross-seeding has also been observed for Sup35 with the Rnq1 prion domain protein RndPD, with extended lag periods compared to the self-seeding processes.325 Hybrid morphologies of Sup35 fibrils sprouting from globular RnqPD structures and RnqpD spherical aggregates coated with Sup 35 seed fibrils were observed.325
Cross-seeding of subunits of insulin (A- or B-chain peptides) with the parent protein was found to be less efficient than self-seeding of the full-length insulin.326 The cross-seeded fibrils had features of the parent insulin protein, the morphology being distinct from that of the seeding peptides. Despite the observed cross-seeding, soluble forms of the A- and B-chain peptides were found to be able to inhibit insulin fibrillization.326 In another study, it was suggested that cross-seeding of hen lysozyme with other proteins was promoted by sequence similarity.327 The ThT fluorescence kinetics were enhanced for other forms of lysozyme compared to unrelated proteins (insulin and α-lactalbumin), even though, for example, hen lysozyme and α-lactalbumin share the same native state fold.327
9. Metabolite amyloidosis
While amyloid formation has been studied extensively with proteins and polypeptides and even ultrashort peptides, a major assumption in the field was that the minimal requirement for the formation of amyloid structures by a protein fragment is a dipeptide.328 This is due to the unique planar nature of the peptide bond, which stems from the electron resonance in the structure that results in a partial double bond between the alpha carbon and the amine nitrogen. To test this assumption and following the extensive work on diphenylalanine (FF)124 and other self-assembling ultrashort aromatic peptide such as triphenylalanine (FFF)329 and phenylalanine-tryptophan (FW),124 the ability of a single amino acid, phenylalanine, to form amyloid fibrils was evaluated.7 Surprisingly, it has been found that at concentrations above 1 mM phenylalanine readily forms toxic fibrillar assemblies in aqueous solution (as indicated by EM). The fibrils showed amyloid-characteristic binding to ThT and green-gold birefringence between crossed polarizers upon staining with Congo red.7 Moreover, it was shown that antibodies could be raised against the assemblies by immunization of rabbits and those antibodies could deplete the toxicity of the amyloid-like assemblies by pull-down of the toxic species.7
Following this unexpected discovery, there began an immediate quest to understand its significance, which was initially unclear considering the rather high concentration of phenylalanine required for the self-assembly of amyloid-like fibrils. While the normal concentration of phenylalanine in healthy individuals is only a few tens of μM, there are affected individuals with blood and tissue concertation of phenylalanine in the mM range. The medical condition is known as phenylketonuria (PKU), which is the result of a mutation in the gene coding for the phenylalanine hydroxylase enzyme.330 Individuals affected by the mutation are unable to convert the phenylalanine amino acid into tyrosine. This results in the accumulation of phenylalanine as well as an insufficient amount of tyrosine. Intriguingly, unless treated by a very strict diet, PKU patients show severe neurological symptoms similar to neurodegenerative patients.331 After years of follow-up on PKU patients and close monitoring of the correlation between phenylalanine levels and neurological abnormalities, it is clear that concentrations of phenylalanine above 0.5 mM are strongly associated with severe neurological damage. To validate the relevance of the observed phenylalanine fibrils to the disease, their occurrence in mice model of PKU has been tested. Mice have the same biosynthetic pathway for the production of tyrosine as humans and a mutation in the same enzyme results in the accumulation of the amino acid.332 It was indeed found that antibodies which recognized the fibrillar assemblies, just as in the case of the immunized rabbits, emerged spontaneously in the model mice.7 Moreover, antibodies that recognize the phenylalanine assemblies could allow the detection of phenylalanine deposits in the brains of PKU patients post mortem. These deposits are very similar to the amyloid deposits found in those suffering from neurodegenerative diseases such as AD, and could also be co-stained with the antibodies and Congo red.7 Taken together, these results suggested a typical amyloid etiology in PKU and an extension of the list of amyloid-associated disorders.
Follow-up studies have been conducted to understand the organization of phenylalanine into ordered assemblies and the possible occurrence of oligomeric structures of phenylalanine. Using mass spectrometry, Bowers and co-workers discovered that under physiological pH phenylalanine could form oligomeric structures, in which the phenyl groups were being exposed to the solution.333 This interesting organization could be associated with a tendency to interact with membranes as a result of the exposed hydrophobic patches in the oligomeric form, as has also been predicted theoretically by MD simulations,334 thus explaining the high toxicity of the assemblies. An additional immediate follow-up study by Salmona and co-workers indicates the organization of phenylalanine into order assemblies using small angle and wide angle X-ray scattering as well as AFM.335 Moreover, the researchers show that doxycycline, a known protein amyloid formation inhibitor, hinders effectively the formation of fibrillar assemblies by phenylalanine.335
While phenylalanine accumulation in the case of PKU is well-known, there are dozens of additional medical conditions known as inborn error of metabolism disorders, in which other metabolites are being accumulated.336 In order to test whether the formation of metabolite amyloids in PKU represents a unique case, a larger collection of other metabolites has been screened for the ability to form amyloid-like structures. It was revealed that additional metabolites, including tyrosine, orotic acid, adenine, uracil and cysteine could form fibrillar assemblies with amyloid-like morphology, which bound ThT and Congo red and presented notable cytotoxicity.337 Furthermore, this broader study also demonstrates that the mode of toxicity of these metabolite assemblies, as well as that of phenylalanine assemblies, is by late apoptosis, exactly as in the case of protein and polypeptide amyloids.337 Later studies indicated that additional metabolites, including tryptophan, glycine, and quinolinic acid, could form amyloid-typical fibrillar assemblies in aqueous solutions.338–340
Another interesting property of classical protein amyloids that has recently been discovered is their intrinsic fluorescence in the visible range of the electromagnetic spectrum, as was shown by Kaminsky and co-workers.341 The researchers explain the fluorescence by proton delocalization over terminal hydrogen bonds, which results in the formation of an essentially supramolecular emissive electronic state. Intriguingly, similar visible-range fluorescence has been observed for diphenylalanine nanostructures.342 It has recently been established that metabolite amyloids also possess such fluorescence properties, which also allow their detection in live cells.343 The observed fluorescence of the fluorescence in the context of hydrogen bond networks is also in line with the crystal structure of phenylalanine in its zwitterionic form at neutral pH, as was determined by synchrotron X-ray crystallography in 2014.344 The structure shows a network of hydrogen bonds and π-π interactions, layered with remarkably similar morphology and zig-zag arrangement as compared to protein β-sheet structures. Hydrogen bonds occur between polar layers and the charged moieties of the amino acids, while edge-to-face π-π interactions exist between the charged parts of the layers and parallel displaced π-π interactions between the aromatic side chains.344
Yet another property that is shared by protein and polypeptide amyloids and amyloid-like metabolite assemblies is the generic inhibition by polyphenols and other aromatic compounds.345, 346 This common feature is also consistent with the earlier observation concerning the inhibition of phenylalanine fibril formation by doxycycline.335 Additionally, acetylsalicylic acid, which does not affect the organization of protein amyloids, similarly has no inhibitory effect on metabolite fibril formation.346 The metabolite amyloids also exhibit the capacity to interact with membranes similarly to protein amyloids, as determined by a highly-characterized chromatic biomimetic membrane system containing phospholipids and polydiacetylene.347 Also in this sense, the similarity between metabolite and protein assemblies is remarkable.
The aggregation of metabolites appears to be directly linked with neurodegenerative disorders. An intriguing observation is related to the interplay between metabolite assemblies and the induction of protein amyloid formation in the context of seeding. It has been shown that phenylalanine fibrils could seed a large group of amyloidogenic proteins and polypeptides that are associated with amyloid diseases.348 It was suggested that the presence of metabolite seeds could have a triggering effect on the eruption of amyloid-associated neurodegenerative disorders.349 Indeed, it was shown that quinolinic acid, a well-established early marker of PD, could seed the aggregation of αS into amyloid fibrils.340 This may provide the missing link between the metabolite profile and the development of various degenerative processes and may clarify certain unexplained epidemiological associations.349
All of the experiments in metabolite amyloids until 2019 have been performed in vitro or in cell culture. It was recently demonstrated that a yeast model of metabolite aggregation could be constructed.350 By blockage of adenine salvage pathways, a yeast model in which adenine accumulates was obtained. Feeding these model yeast with adenine exerted a toxic effect in a non-linear sigmoidal-shaped dose-dependent manner.350 Moreover, the addition of polyphenol inhibitors of amyloid rescued the yeast cells without lowering the level of adenine, indicating that the formation of aggregates, rather than the presence of a high concentration of the metabolite, resulted in the cytotoxicity.350 As protein amyloid models in yeasts have been extensively explored by many groups flowing the pioneering work of the late Susan Lindquist,351 the current use of yeast models allows to compare the cellular mechanisms of protein homeostasis (proteostasis) and that of metabolite homeostasis (metabostasis). The yeast system will allow further analysis of the cellular machinery that is involved in the response to abnormal accumulation of metabolites with and without self-assembly into amyloid-like structures.
Finally, another interesting parallelism can be found between protein and metabolite amyloids in the sense that as with protein amyloids, functional assemblies that are composed of metabolites can be formed.352 One key example is the formation of tapetum lucidum, a retroreflector layer that facilitates night vision. These assemblies are formed by simple metabolites that are very similar to major human risk elements, including a nucleobase (guanine) in reptiles, an amino acid (cysteine coordinated with zinc) in dogs, and a vitamin (riboflavin) in cats and lemurs. Other studies indicated the materials-like properties of amino acid assemblies and the piezoelectric properties of amino acid crystals.353 The overall organization of the metabolites in those systems seems to be related to metabolite amyloids, highlighting once again the similarity between the protein and non-proteinaceous self-assembling systems.
Taken together, the spectrum of amyloid building blocks appears to include not only proteins, polypeptides and short peptides, but also a large number of metabolites (Fig. 13). The full molecular determinants that facilitate the ability to form amyloids are still unclear as very similar metabolites could either form or not form amyloid-like structures. For example, phenylalanine and tyrosine readily form amyloid-like structures, whereas the structurally similar 4-hydroxyphenylpyruvate does not. Additional work should be performed to fully understand the structural, functional, and pathological significance of these new type of non-proteinaceous amyloidal assemblies.
Fig. 13. Formation of ordered fibrils and oligomers by metabolites.

Malfunction of specific enzyme due to genetic mutation results in a significant increase in the amount of specific metabolite substrate in the blood and tissues and lack of the metabolic product. In the case of PKU, the increase in phenylalanine concertation is about 30–60 fold in PKU patients as compared to individuals with normal phenylalanine metabolism. The metabolite could form fibrils or oligomers under physiological pH and the process could be inhibited by doxycycline or generic polyphenol amyloid formation inhibitors. The assembled structures, most likely the oligomers, can interact with the membranes to lead to membrane destabilization and finally apoptosis and damage to cells and tissues.
10. Systemic amyloidosis
Systemic amyloidosis is defined as an amyloid disease, in which the synthesis of the fibril precursor protein and the deposition of the fibrils occur at different sites within the body.354 The fibril precursor protein is circulating in the blood, and the associated fibrils form deposits in multiple organs, such as heart, liver and kidneys. The deposits are often large-sized and exert physico-mechanical effects that are a major factor of pathogenicity.355 For example, cardiac amyloidosis can compromise the natural contractility and pumping function of the heart, leading to severe cardiomyopathy.356 There is, however, evidence that toxic fibrillization intermediates play a role in these diseases, similar to their involvement in neurodegeneration. Systemic amyloidosis can cause major impairments to the affected patients, if not death, and these diseases probably constituted the most abundant protein misfolding diseases until the mid of the 20th century. Nevertheless, they are nowadays much in the shadow of their neurodegenerative relatives.
Several types of systemic amyloidosis can be distinguished depending on the fibril precursor protein. This dependence is reflected by the disease nomenclature.56 Systemic AA amyloidosis arises from the misfolding of SAA protein. Systemic AL amyloidosis involves fibrils from immunoglobulin LCs, while systemic ATTR amyloidosis originates from TTR fibrillization. There are several commonalities among the different types of systemic amyloidosis. The precursor proteins are either constitutively or transiently present at high concentrations.354, 356 They are proteolytically truncated in the fibrils,354, 356, 357 and their deposits are associated with non-fibril components, such as glycosaminoglycans,358 lipids359 or non-fibril proteins,360 that are conserved across different diseases and possibly important for fibril formation358 or fibril stability in the tissue.355
Precisely 60 years after Cohen and Calkins revealed, with electron microscopy, the presence of fibrils in the tissue,53 cryoTEM structures became available for amyloid fibrils that were purified from the tissue of patients affected by systemic AA, AL or ATTR amyloidosis.120, 361–363 These structures revealed that the fibril protein folds are, in all cases, substantially different from their conformation in the respective native proteins. The implication of this observation is that the conformation of the native state has to be almost entirely unfolded in order to enable the reorganization of the polypeptide chain into the fibril protein fold. This conclusion holds even in cases where the fibril precursor is β-sheet rich as in TTR.362 The fibrils that can be extracted from a patient or animal are typically polymorphic with one dominating fibril morphology.120, 361–364 Consistent fibril morphologies are deposited at different sites/organs of the same patient or animal365 and even in different patients or animals, as long as they belong to the same subtype of systemic amyloidosis and possess the same sequence of the fibril precursor protein.119, 362, 365
Current ways of treatment of systemic amyloidosis are mostly focused on a reduction of the fibril precursor protein. Treatment in systemic AL amyloidosis typically involves a chemotherapy to remove the plasma cell that produces the amyloidogenic LC.356 Treatment in systemic AA amyloidosis involves anti-inflammatory treatments to reduce the serum levels of the acute phase protein SAA.354 Treatment of systemic ATTR amyloidosis can involve a liver transplantation to remove the main TTR-producing organ.366 In addition, there are recent advances to downregulate the expression of TTR protein by RNA interference therapy.366 These treatments, which target the precursor protein concentration, have to be combined, in some patients, with the removal of the compromised organ. An example hereof is the requirement of a heart transplantation in many patients with cardiac amyloidosis.356 Finally, certain forms of systemic ATTR amyloidosis can be pharmacologically treated with the drug tafamidis, which binds into the surface pocket of TTR, stabilizing the native state of this protein.367 In summary, systemic amyloidosis represents an interesting and unique model system for studying the principles of protein misfolding diseases and for developing new medical approaches.
11. Bacterial functional amyloid
The preceding ten sections have focused on pathological amyloid. However, there is growing awareness of the impressively useful roles that the amyloid motif plays in many different biological contexts. As discussed in a recent review,368 ‘functional’ amyloid examples are found in many organisms, ranging from mammals and insects369 to fungi and bacteria (though not yet plants – possibly due to the many metabolites in plants which are able to inhibit amyloid aggregation).370 Functional amyloids can participate in various cellular tasks, including serving as structural scaffolds, with examples like Pmel17 in melanosomes, curli and FapC in bacterial biofilms, and spidroins that enhance spider web tensile strength. Additionally, peptide hormones can be stored in an inert amyloid state,371 while amyloid formation by the P. anserina HET-s protein regulates heterokaryon incompatibility, transcription and translation can be regulated by prion proteins in yeast.372 Because space constraints prevent a more detailed exposition of every functional amyloid, here we will focus on the bacterial amyloid-forming proteins, CsgA and FapC. As functional amyloids, they are produced as the result of highly coordinated biosynthetic processes, and a great deal is now known about these biological systems as well as the principles promoting their optimized aggregation. Thus, functional amyloids serve as instructive examples of how nature can avoid the unwanted features of protein aggregation and instead exploit the amyloid fold for cellular good.
A critical component of bacterial biofilm is the extracellular matrix (ECM) that surrounds and protects cells. In many cases, the ECM is strengthened by bacterially produced amyloid.373, 374 The best-studied example is curli,375–377 produced by Enterobacteriaceae, such as Salmonella and Escherichia coli (E. coli), as well as hundreds of diverse Gram-negative bacteria.378 Unlike pathological amyloids, bacterial amyloids are functional (i.e. biologically useful) and therefore produced as the result of highly coordinated biosynthetic processes. Much can be learned about the nature of amyloid formation by studying this fascinating group of proteins.
Curli are required for proper biofilm development376, 379–381 and enhance biofilm strength, viscoelasticity, and resistance to strain.382 The main component of curli, CsgA, is a 120-residue IDP that is secreted into the extracellular milieu as an unfolded monomer and was the first ‘functional’ amyloid protein to be described.383, 384 The minor curli subunit CsgB, also an IDP, nucleates CsgA fibrillization as an amyloid template.385 Additional CsgA subunits are added to the growing fibril end in a seeding process.386, 387 CsgB also helps anchor the fibrils to the membrane surface via CsgF (in whose absence both proteins simply “escape” from the cell). Both CsgA and CsgB must be “chaperoned” to suppress their high aggregation propensity and promote their transport as monomers to and across the outer membrane prior to amyloid fibril formation. They are secreted to the cell surface through what is called the Type VIII secretion system (Fig. 14), in which CsgE targets them to the outer membrane secretion channel CsgG388–391 via the two proteins’ N-terminal 22 residues.392, 393 Once polymerized, fibrils can interact with the dyes Congo red and ThT, leading to a spectral change that is often used to monitor amyloidogenesis. The resulting CsgA fibrils are unusually durable. Not only do they resist heat, proteases, and denaturing agents such as sodium dodecyl sulfate (SDS),394 they only dissociate to monomers at very high concentrations (>50%) of the potent denaturant and solvent formic acid.395
Fig. 14.

Model of curli secretion. CsgE and CsgG facilitate secretion of unfolded CsgA across the outer membrane. Outside the cell, CsgA assembles into an amyloid via a nucleation precipitation reaction assisted by CsgB and CsgF. The CsgE and CsgC proteins have been shown to prevent CsgA aggregation in vitro. The CsgG nonamer is shown in purple, and one of the CsgG subunits is colored green.
CsgG is comprised of nine identical subunits, each lipidated at the N-terminal cysteine residue to promote localization and assembly in the outer membrane (Fig. 14).389, 391 Each CsgG subunit within the membrane-embedded CsgG homononamer contributes four β-strands to a 36-strand transmembrane β-barrel, which sits atop a ring of globular periplasmic domains. The structure contains two vestibules separated by a series of constriction loops that restrict the pore diameter to 9 Å.390, 396 The first vestibule, formed by the periplasmic domains, encloses ~24,000 Å3, roughly the predicted volume occupied by a single unfolded CsgA monomer. The second vestibule formed by the β-strands spanning the outer membrane remains permanently open to the extracellular milieu, enclosing ~41,000 Å3. This is consistent with the need for subunits to be largely unfolded during secretion into the extracellular space.397 Besides binding and inhibiting the folding of CsgA, CsgE also forms a functional nonameric cap that binds to the CsgG pore and prevents unregulated transport across the outer membrane.388, 390, 398 CsgE contains a mix of three stacked β-strands, 2 α-helices, and a prominent IDR.398 Once CsgA is confined to the space formed by periplasmic vestibule of CsgG and the CsgE cap, it is thought that the resulting entropic free energy gradient favors CsgA expulsion into the extracellular space. CsgF and CsgB are also apparently secreted through CsgG. A partial NMR structure of CsgF suggests it to be conformationally dynamic with exposed hydrophobic regions. CsgF contains an N-terminal unstructured region, a 21-residue α-helix, and a C-terminal antiparallel 4 β-strands.399
According to “fishing expeditions” with amyloid-specific antibodies, many bacteria produce amyloid,400 and numerous operons coding for functional amyloid have been discovered since curli (see an overview in reference 401). Despite differences in detail and independent evolutionary origins, there are common features. These include one main amyloid component and a secretion system to transport them to the outer membrane in a monomeric state that is ready for extracellular assembly. For example, the fap operon in Pseudomonas species directs the formation of fibrils mainly consisting of FapC,402 assisted by the (likely) nucleator FapB and transported through the outer membrane by FapF.403 The ensuing fibrils increase biofilm rigidity and resistance to drying out through increased hydrophobicity.404 However, given their independent origins, there will always be distinguishing features in each system. For example, the membrane-embedded part of FapF is a trimeric porin gated by a helical plug (much simpler than CsgG), while its periplasmic coiled-coil domain could potentially guide transport of FapC to the FapF channel as a dedicated “amyloid shunt”.405
The core of both CsgA and CsgB protein sequences is composed of 5 imperfect repeated sequences (R1-R5), each of around 20 residues.375 Peptides corresponding to R1, R5 and R3 assemble into amyloid fibrils that are morphologically indistinguishable from those produced by WT CsgA,406, 407 whereas R2 and R4, which are largely dispensable for fibril formation in vivo,406 do not fibrillate on their own. This indicates that CsgA consists of multiple amyloid-forming units, and indeed a combination of NMR data and secondary structure predictions suggest that each repeat sequence in CsgA forms strand-loop-strand motifs.393, 408, 409 Subsequently co-evolutionary analysis combined with computational force field analysis led to an atomic-level structure prediction in which the 5 hairpins stack on top of each other to form an α-helix,410 although the handedness of this helix still is unresolved in the absence of experimental restraints. The repeats in CsgA are distinguished by the sequence Ser-X5-Gln-X4-Asn-X5-Gln;393, 406 the Gln and Asn residues in R1 and R5 are critical for efficient amyloid formation393, 406, 411, 412 while “gatekeeper” residues in R2, R3 and R4 slow CsgA amyloid formation both in vivo and in vitro.411 The structural convergence of CsgA and CsgB is rooted in amino acid sequence similarities, including regularly spaced Ser, Gln and Asn residues.413
A similar situation is found with FapC, although it differs from CsgA in that it contains 3 longer imperfect repeats (~35 residues), separated by linkers of variable length. A computational-bioinformatic structure prediction, similar to that performed for CsgA, suggests that the FapC repeats form hairpin loops, while the linker regions constitute disordered regions that may lead to a “hydrophilic halo” around the fibril.414 The advantage of this repeat-based design, both for FapC and CsgA, is that each protein monomer constitutes an amyloid-forming unit, which will facilitate rapid initiation of fibrillization. No stable oligomers of either CsgA or FapC have been isolated (except when assisted by fibrillization inhibitors such as polyphenols415, 416). This indicates that the repeat structure efficiently drives fibrillization from a small and stable nucleus and that there are no competing non-fibrillar structures, unlike e.g. the stable αS oligomer.417, 418 Consistent with this, fibrillization of both CsgA and FapC follows the simplest possible model, namely primary nucleation followed by elongation.419 This mechanism is ideal for the nucleation-initiated elongation of bacterial amyloid fibrils in vivo, where long (i.e. not fragmented) fibrils are likely required to strengthen the ECM. Remarkably, step-wise deletion of the repeats of FapC gradually increases the tendency of the fibrils to fragment, and this means that elongation of the new growing ends generated by fragmentation (rather than elongation of the nuclei formed by primary nucleation) becomes the dominant contribution to fibril growth.420 The resulting fibrils are also more prone to dissociation in formic acid, a direct indication of their reduced stability, and complete removal of all repeats, while not preventing formation of β-sheet rich fibrillary aggregates, catastrophically destabilizes the fibrils in terms of sensitivity to denaturants such as formic acid and surfactants.421 A further twist on in vivo fibrillization is that biomolecules in the milieu close to the outer membrane of Pseudomonas, such as the biosurfactant rhamnolipid and the outer membrane component lipopolysaccharide, strongly accelerate FapC aggregation and suppress accumulation of a transient aggregation intermediate.416 Thus, conditions have truly been optimized to facilitate rapid and direct fibrillization of these proteins.
Amyloidogenic proteins present significant challenges to the cell because of their strong tendency to aggregate. Uncontrolled amyloid formation can upset proteostasis, and is associated with significant cytotoxicity.422 Therefore, a fundamentally important question is: “How can aggregation-prone proteins such as CsgA be guided from the inside of the cell, through the periplasmic space, and across the outer membrane so that amyloid fibrils only form outside the cell?” The answer is: accessory proteins such as CsgC, a small periplasmic protein made by curli-producing bacteria that helps discourage intracellular amyloid formation by CsgA and CsgB. Purified CsgC can inhibit amyloid formation at extremely substoichiometric 1:500 (CsgC:CsgA) molar ratio, both for WT CsgA, R1, R3 or R5 CsgA deletion mutants, along with synthetic peptides corresponding to R1 and R5. CsgC also inhibits amyloid assembly of E. coli CsgA and CsgB homologs, but not the AD-associated protein Aβ42, suggesting some specificity.423 However, CsgC does inhibit amyloid assembly by the PD-associated protein αS,423 possibly due to a 8–9 amino acid motif (D-Q-Φ-X0,1-G-K-N-ζ) shared by both CsgA and αS.423 Structural homologs of CsgC are found in almost all bacteria.424 The CsgC structure can even be seen in the human amyloid-forming protein TTR. Like CsgC, TTR can discourage CsgA and αS amyloid formation.425 Therefore, it is likely that nature has many anti-amyloid chaperone proteins at its disposal that can help modulate amyloid formation.
Functional amyloids have much to offer going forward. Amyloid-associated diseases affect an estimated 50 million people annually. We need better approaches for combating diseases such as AD and PD, which are twice as prevalent today as they were 20 years ago, a number that could quadruple by 2050. The rules governing amyloid fibril formation have been mostly defined through in vitro observations, because most disease-associated amyloids aggregate inefficiently or sporadically in vivo. It is not unreasonable to expect that next generation amyloid therapeutics will be inspired by the mechanisms that functional amyloid systems employ to temporally and spatially control amyloid formation. At the very least, the N22-based display system can be circumvented to target other amyloidogenic proteins to the CsgG secretion channel and provide a bacterial selection system for e.g. aggregation inhibitors.426 Furthermore, the robust fibrils produced by these bacteria may serve both as selectable display vehicles for e.g. peptides with different binding properties427 and even as components in underwater glue when fused with mineral-binding proteins such as mussel foot proteins.428
12. Artificial amyloids and engineering opportunities
In stark contrast to their original negative implications in neurodegenerative diseases, but in line with the positive role played by functional amyloids, artificial amyloids, that is amyloid fibrils artificially produced in vitro from non-toxic affordable proteins, have become a well-accepted building block of next generation functional materials and nanotechnologies.26, 429 Particularly noteworthy for advanced materials and technologies are the following physical-chemical features of amyloid fibrils: the physical-chemical features of amyloid fibrils: (1) their extreme aspect ratio; (2) the amino acid functionalities of their surface chemistry; and (3) their Young’s modulus comparable to high performance commodity plastics. These features have enabled unique and original applications of these systems into a multitude of applications, some of which have now entered the commercial realm. While the use of amyloid fibrils as templates for innovative materials, devices and nanotechnologies has already been discussed in detail in separate comprehensive reviews,26, 429, 430 here we highlight briefly how the features (1) to (3) of artificial amyloids allow their use in innovative material design.
The extreme aspect ratio of amyloid fibrils provides unique opportunities for assisting the self-assembly of materials templated by liquid crystalline interactions, for example. This has allowed the design of 2D materials for optoelectronic and biosensing applications, among others. The general strategy followed is that the 1D excluded volume interactions of amyloid fibrils can be exploited to drive the self-assembly of other higher dimensionality anisotropic objects, such as 2D organic and inorganic materials, starting at concentrations of the 2D objects well below their intrinsic isotropic-nematic transition composition. In other words, 2D objects can be ordered in their dilute, isotropic state, when amyloid fibrils above their isotropic-nematic transitions are present. In one example, amyloid fibrils above the isotropic-nematic transition were used to drive the alignment and stacking of graphene nanoflakes into layered structures and finally 2D nanocomposities with unconventional sensing, shape-memory and biodegradable properties.431 This offered the first opportunity to design amyloid-based devices for enzymatic biosensing.431 Similarly, replacing graphene nanoflakes with gold 2D single crystals allowed generating hybrids materials with innovative optoelectronic properties, conductivity and plasmonic properties.432 In a further example, inorganic gold single crystals were replaced by ad-hoc synthesized 2D hydroxyapatite platelets, which once aligned by excluded volume interactions into layered composites were able to mimic bone structure, with amyloids replacing collagen fibrils.433 The further growth of osteoblasts onto these 2D hybrid materials brought the templates one step closer to mimic real human bones.433 In the dense phase, liquid crystalline interactions of amyloid fibrils were further used to design solid films with anisotropic absorption and fluorescence properties;434 furthermore, although not directly demonstrated, the role of highly anisotropic interactions and structure in amyloid-based films may lead to improved quantum efficiency of semiconductive polymers when embedded in amyloid solid matrices,435 as extensively demonstrated by the Inganas group,436–438 among others.
In the liquid state, the liquid crystalline interactions of the amyloids combined with their chiral nature have been recently shown to be responsible for the formation of chiral nematic phases, or cholesteric liquid crystals, in both a continuum (bulk cholesteric phase) and dispersed state (i.e. cholesteric droplets dispersed in a continuum isotropic phase).439 In both the solid and liquid states, the concepts discussed above can open the path to optical devices where the anisotropic nature of the amyloid-based materials can be used to alter the orientation of an electromagnetic field, as in filters for polarized fluorescence and low-power liquid crystal displays, or the collective diffusion of excitons or photons, in photovoltaic and organic light-emitting diode/polymer light-emitting diode (OLED/PLED) applications, respectively.
An alternative way to exploit the extreme aspect ratio of amyloid fibrils is to generate gels, rather than liquid crystals, where the high aspect ratio is now used to reach a gel phase at extremely small percolation thresholds. This can be done by simply altering the interactions among amyloids fibrils in the colloidal state, by for example favoring attractive interactions via changes in ionic strength (charge screening) or pH (alteration of linear charge density).440–442 Amyloid-based gels of this type have been shown to work as efficient scaffolds for cell culture and growth, due to a noteworthy low cytotoxicity in the greatest majority of cases investigated.443–446 Remarkably, this spans over a wide range of amyloid building blocks, ranging from the 10-residue 105–115 fragment from the protein TTR,444, 445 modified and engineered to carry the RGD integrin binding sequence of fibronectin, to the much simpler amyloid gels based on food proteins, such as lysozyme.447, 448 These gels have been demonstrated to be efficient means for cell proliferation, to mimic the ECM environment, to perform tissue engineering, and, when stem cell precursors are used, to promote cell differentiation, as in the case of neural progenitor cells differentiating into mature neurons during cell growth.449 Of particular significance in this case is the fact that small variations in pH, ionic strength and concentration can result in gels with significant changes in mechanical and rheological properties.450 This is understood to be the playground in which stem cell differentiation in vitro and in vivo occurs, and has been demonstrated to date in several types of amyloid-based gels of different amyloid sources, including peptides based on Aβ451 and αS.452
The amino acid chemistry decorating the surface of amyloid fibrils further enables unique applications, some of which are highly specific to these systems. Indeed, the presence of peptide bonds opens for both versatile covalent and supramolecular chemical approaches. Examples of covalent grafting of organic moieties to amyloid fibrils include PNIPAM brushes grown from β-lactoglobulin amyloid fibrils to produce injectable temperature-responsive hydrogels;453 photo-induced cross-linking of the lipase enzyme onto amyloid fibrils to produce biocatalysts with enhanced enzyme stability;454 and DNA-amyloid conjugates in which DNA provides the possibility of origami-driven colloidal assembly of amyloid fibrils.455 Supramolecular interactions between metal ions and amyloid fibrils have been shown to be particularly promising for applications. Metal-ligand supramolecular binding is at the origin of the strong adsorption of metal ions observed on the surface of amyloid fibrils, mediated by strong binding to a multitude of amino acids (see Fig. 15 for a schematic cartoon).25, 456, 457 By exploiting this effect, Bolisetty et al. were able to produce amyloid-based membranes for adsorption of heavy metals and radionuclides for efficient water purification.25 Due to the simultaneous presence of diverse amino acids and thus, a multitude of metal-ligand pair binding constants, these membranes act as “universal sponges”, challenging the highly specific exchange resins traditionally used in water purification, for example. This has led to a commercial application of membranes based on artificial amyloids capable to remove a wide range of heavy metal ions such as gold, platinum, silver, copper, lead, mercury, uranium, aluminium, and even metalloids such arsenic.25, 456, 458
Fig. 15.

Schematic illustration of the binding process of gold ions with the β-sheets of amyloid fibrils. The β-sheet structure is based on the molecular dynamic simulations of the self-assembly process of the LACQCL sequence, a fragment found on the β-lactoglobulin amyloid fibrils used for water purification properties.25 Image rights: R. Mezzenga.
The concept has further been extended to remove halogen contaminants from water, such as in the case of fluoride, by first capturing transition metals on the fibrils, then growing them in-situ into fine transition metal nanoparticles, which finally, by adhering to the amyloid surface, are capable of binding targeted fluoride contaminants.459
The growth of metal ions on the surface of amyloids into metal nanoparticles conjugated to the amyloid fibrils can be further exploited for applications as diverse as heterogeneous catalysis460 and human nutrition. For example, by growing iron oxide nanoparticles from Fe2+ bound on the surface of β-lactoglobulin amyloid fibrils, Shen et al.24 showed that the resulting hybrids maintained iron at the right oxidation state (i.e., the II form, which is more bioavailable than the III form), which could then be used to target iron deficiency in vivo. The authors further demonstrated that the β-lactoglobulin amyloid fibril-iron oxide nanoparticle hybrids could be used as efficient iron fortificants for nutrition purposes, with outstanding bioavailability features. Alternatively, by growing noble metal ions such as gold and palladium into nanoparticles, and using β-lactoglobulin amyloid fibrils as a sole reducing agent, the ensued metal nanoparticle-amyloid hybrids could be used to design membranes for continuous heterogeneous flow catalysis.460
The scope of artificial amyloids can be expanded significantly by combining some of the unique features discussed above: amyloid gels can be combined with the reducing properties of the amyloid fibrils to template gold aerogels with ultra-light density;461 the reducing properties of amyloids can be used to generate other types of oxide nanoparticles, such as TiO2 for dye cell units for energy conversion;462 engineered amyloid fibrils with specific binding sites can be used to target analytes and specific molecules; for example, the Eisenberg group463, 464 has shown how amyloid templates can be used for efficient CO2 adsorption, thereby addressing another pressing environmental issue. It is the multitude of features highly specific to amyloid fibrils, which, when exploited synergistically, can give rise to an extremely vast range of beneficial applications ranging from the biomedical field, to environment preservation, energy management and alike, serving modern society and shifting paradigms away from the adverse tinge that amyloids have been mostly known for.
13. Summary and future work
Much progress has been made in the field of amyloid science. The atomic structures of extensively studied amyloid peptides/proteins Aβ42,465 Aβ40,466 Tau101 and αS93, 107, 112 have been resolved with improved in vitro fibril preparation and breakthroughs in cryo-TEM and ssNMR technologies. Such progress, however, is contrasted by the lack of success in anti-amyloidosis clinical trials and highlights this persistent question: just how much amyloidosis contributes to cell degeneration in amyloid diseases, alongside ageing, deficiencies in microglial activation and peripheral clearance,467, 468 as well as other environmental and familial factors? The structural characteristics of amyloid fibrils are essential for clinical classification and, hence, the treatment of amyloid diseases. Indeed, while distinct prion strains are associated with different clinical and pathological phenotypes,469–471 variations in AD phenotype correlate with variations in Aβ fibril structure.472–475 For example, among AD clinical subtypes, the rapidly progressive form possesses rich polymorphism of Aβ40 fibrils, while the posterior cortical atrophy variant and typical prolonged-duration form shows dominance of a specific Aβ40 fibril structure from seeded growth of AD brain cortex extracts. On the other hand, all three subtypes displayed structural heterogeneity for Aβ42 fibrils.476 Clearly, amyloid structure offers a basis for deciphering the often nuanced clinical manifestations of amyloid diseases.
Amyloidosis occurs across different organs and biological barriers in vivo, as exemplified by systemic ATTR, AL and AA amyloidoses. On the other hand, amyloid proteins such as Aβ, αS and hIAPP have been detected in systemic circulation, fuelling the hypotheses of cross-seeding, inflammation, the gut microbiota and metabolite amyloid as causative to amyloid diseases.5, 477–479 Furthermore, Aβ oligomers have shown a negative effect on constricting human capillaries in AD via signalling to pericytes.480 Accordingly, the structure and toxicity profiles of amyloid proteins may be examined across different compartments, physiological conditions and pathologies.
A major challenge in the clinical treatment of amyloidosis is to ensure early diagnosis of the disease. In T2D, for example, beta-cell mass accounts for only 1–2% of the pancreas but is a key indicator of the onset and severity of the disease. Accordingly, the development of molecular imaging probes and biomarkers based on antibodies/nanobodies, RNA sequencing and multimodal detection is an identified area of clinical importance.481–483 Aside from technological development, and in addition to solubility and efficacy in delivery, the dose and timing of therapeutics administration are crucial parameters for the treatment of amyloid diseases. This is because amyloid proteins in vivo undergo conformational changes on timescales vastly different from in vitro test conditions, depending upon the age, mutation, anatomy and body mass of the animal models. Fundamentally, this challenge arises from the transient and highly heterogeneous nature of toxic oligomers and protofibrils, compounded by cell membranes, pH, ligands and chaperones to influence the protein aggregation energy landscape and toxicity. The evolution of amyloid fibrils in physical and toxicological properties484 adds another element of complexity for amyloidosis inhibition.
It should be noted that significant progress has been made in recent years with regard to the clinical diagnosis and effective management of systemic AL and ATTR amyloidoses. This is achieved by mass spectroscopy identification, chemotherapy, liver transplantation, gene sequencing, small-molecule (e.g., tafamidis and diflunisal) stabilization, antisense oligonucleotide therapy as well as RNAi to mitigate the production or misfolding pathways of amyloid proteins.485–487
Much remains to be understood concerning the structure-function-pathogenesis relationship of amyloid proteins and their structural derivatives, as well as the co-aggregation and cross-seeding of amyloid proteins with environmental proteins. A large body of experimental evidence, particularly from genetics, indicates that the proteins associated with amyloidosis are the central player in pathogeneses. The toxicities of the oligomers and protofibrils are indisputable facts to implicate amyloid proteins, whose amyloid plaques first observed by Alzheimer in 1907 and their cross-β structure first revealed by Eanes and Glenner a half century ago,1 as a major culprit for a wide range of debilitating human diseases which remain to be deciphered and, hopefully, eradicated in the coming decades.
Amyloids in current science and technology have evolved significantly beyond their original strictly pathological roles, taking up a novel original profile and illuminating new opportunities which were unthinkable only two decades ago. Two new classes of amyloids have emerged, the functional amyloids and the artificial amyloids, performing challenging and remarkably important roles in vivo as well as in modern nanotechnologies. The unique structural and physicochemical properties shared by pathological, functional and artificial amyloids, have evolved from the debilitating role of the former class in neurodegenerative diseases, to the beneficial tasks played by the two latter classes of amyloids, with a demonstrated and emerging role in modern technologies, and entailing a wealth of applications in environmental remediation, health, composite nanomaterials, energy devices, biosensors, soft matter and 3D printing.26, 428–430, 488
Acknowledgements
We thank our funding sources for support (HHMI, NIH, and NSF for DSE, BBSRC, MRC, Alzheimer’s Society and Alzheimer’s Research UK for LCS, ARC for TPD and PCK, AFTAM Research Collaboration Award to TPD and EG, EPSRC for a Platform grant EP/L020599/1 to IWH, Independent Research Council Denmark-Natural Sciences grant 8021-00208B and Independent Research Council Denmark-Technical Sciences grant 6111-00241 to DEO, R01GM118651 from the NIH to MRC) and our co-workers for discussions. We thank the unanimous reviewers for their insights, opinions and comments which made our review more comprehensive. We thank Aparna Nandakumar, Aleksandr Kakinen and Kairi Koppel for their assistance with the manuscript preparation.
Footnotes
Conflicts of interest
DSE is SAB chair and equity holder in ADDRx, Inc.
This article is dedicated to the memory of our dear colleague Sir Christopher Dobson. Chris was an exceptional scientist, scholar, and a true pioneer of the field of amyloid studies as well as a wonderful mentor to his colleagues and students. He has inspired a whole generation of scientists and enriched the lives of so many people. He was widely admired and will always be remembered for his formidable contributions to the field of Chemistry, his sharp wit, and also his kindness and generosity.
References
- 1.Eanes ED and Glenner GG, J. Histochem. Cytochem, 1968, 16, 673–677. [DOI] [PubMed] [Google Scholar]
- 2.Astbury WT, Woods HJ and Bragg WL, Philos. Trans. R. Soc. A, 1934, 232, 333–394. [Google Scholar]
- 3.Westermark P, Sletten K, Johansson B and Cornwell GG 3rd, Proc. Natl. Acad. Sci. U.S.A, 1990, 87, 2843–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colon W and Kelly JW, Biochemistry, 1992, 31, 8654–8660. [DOI] [PubMed] [Google Scholar]
- 5.Horvath I and Wittung-Stafshede P, Proc. Natl. Acad. Sci. U.S.A, 2016, 113, 12473–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R and Wüthrich K, Nature, 1996, 382, 180–182. [DOI] [PubMed] [Google Scholar]
- 7.Adler-Abramovich L, Vaks L, Carny O, Trudler D, Magno A, Caflisch A, Frenkel D and Gazit E, Nat. Chem. Biol, 2012, 8, 701–706. [DOI] [PubMed] [Google Scholar]
- 8.Kaelberer MM, Buchanan KL, Klein ME, Barth BB, Montoya MM, Shen X and Bohórquez DV, Science, 2018, 361, 6408, eaat5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet M-F, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R and Mazmanian SK, Cell, 2016, 167, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, Wu H, Carreras A, Jeong H, Olofsson LE, Bergh P-O, Gerdes V, Hartstra A, de Brauw M, Perkins R, Nieuwdorp M, Bergström G and Bäckhed F, Cell, 2018, 175, 947–961. [DOI] [PubMed] [Google Scholar]
- 11.Meijnikman AS, Gerdes VE, Nieuwdorp M and Herrema H, Endocr. Rev, 2017, 39, 133–153. [DOI] [PubMed] [Google Scholar]
- 12.Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA and Costa-Mattioli M, Neuron, 2019, 101, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valles-Colomer M, Falony G, Darzi Y, Tigchelaar EF, Wang J, Tito RY, Schiweck C, Kurilshikov A, Joossens M, Wijmenga C, Claes S, Van Oudenhove L, Zhernakova A, Vieira-Silva S and Raes J, Nat. Microbiol, 2019, 4, 623–632. [DOI] [PubMed] [Google Scholar]
- 14.Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carrá A, Brennan CA, Chun E, Ngo L, Samson LD, Engelward BP, Garrett WS, Balbo S and Balskus EP, Science, 2019, 363, 6428, eaar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iadanza MG, Jackson MP, Hewitt EW, Ranson NA and Radford SE, Nat. Rev. Mol. Cell Biol, 2018, 19, 755–773. [DOI] [PubMed] [Google Scholar]
- 16.Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM and Knowles TPJ, Proc. Natl. Acad. Sci. U.S.A, 2013, 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira ST and Klein WL, Neurobiol. Learn. Mem, 2011, 96, 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lashuel HA, Hartley D, Petre BM, Walz T and Lansbury PT Jr., Nature, 2002, 418, 291. [DOI] [PubMed] [Google Scholar]
- 19.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AØ, Riekel C and Eisenberg D, Nature, 2007, 447, 453–457. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds NP, Adamcik J, Berryman JT, Handschin S, Zanjani AAH, Li W, Liu K, Zhang A and Mezzenga R, Nat. Commun, 2017, 8, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamcik J and Mezzenga R, Angew. Chem. Int. Ed. Engl, 2018, 57, 8370–8382. [DOI] [PubMed] [Google Scholar]
- 22.Ke PC, Sani M-A, Ding F, Kakinen A, Javed I, Separovic F, Davis TP and Mezzenga R, Chem. Soc. Rev, 2017, 46, 6492–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Javed I, Yu T, Peng G, Sánchez-Ferrer A, Faridi A, Kakinen A, Zhao M, Mezzenga R, Davis TP, Lin S and Ke PC, Nano Lett, 2018, 18, 5797–5804. [DOI] [PubMed] [Google Scholar]
- 24.Shen Y, Posavec L, Bolisetty S, Hilty FM, Nystrom G, Kohlbrecher J, Hilbe M, Rossi A, Baumgartner J, Zimmermann MB and Mezzenga R, Nat. Nanotechnol, 2017, 12, 642–653. [DOI] [PubMed] [Google Scholar]
- 25.Bolisetty S and Mezzenga R, Nat. Nanotechnol, 2016, 11, 365–376. [DOI] [PubMed] [Google Scholar]
- 26.Wei G, Su Z, Reynolds NP, Arosio P, Hamley IW, Gazit E and Mezzenga R, Chem. Soc. Rev, 2017, 46, 4661–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anfinsen CB, Science, 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- 28.Dobson CM, Nat. Rev. Drug. Discov, 2003, 2, 154–160. [DOI] [PubMed] [Google Scholar]
- 29.Teplow DB, Amyloid, 1998, 5, 121–142. [DOI] [PubMed] [Google Scholar]
- 30.Dobson CM, Trends Biochem. Sci, 1999, 24, 329–332. [DOI] [PubMed] [Google Scholar]
- 31.Crick SL, Jayaraman M, Frieden C, Wetzel R and Pappu RV, Proc. Natl. Acad. Sci. U.S.A, 2006, 103, 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoop CL, Lin H-K, Kar K, Magyarfalvi G, Lamley JM, Boatz JC, Mandal A, Lewandowski JR, Wetzel R and van der Wel PCA, Proc. Natl. Acad. Sci. U.S.A, 2016, 113, 1546–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levinthal C, Mossbauer Spectroscopy in Biological Systems: Proceedings of a meeting held at Allerton House, Monticello, Illinois, 1969, 22–24. [Google Scholar]
- 34.Hartl FU and Hayer-Hartl M, Nat. Struct. Mol. Biol, 2009, 16, 574–581. [DOI] [PubMed] [Google Scholar]
- 35.Onuchic JN and Wolynes PG, Curr. Opin. Struct. Biol, 2004, 14, 70–75. [DOI] [PubMed] [Google Scholar]
- 36.Bryngelson JD, Onuchic JN, Socci ND and Wolynes PG, Proteins, 1995, 21, 167–195. [DOI] [PubMed] [Google Scholar]
- 37.Dill KA and Chan HS, Nat. Struct. Biol, 1997, 4, 10–19. [DOI] [PubMed] [Google Scholar]
- 38.Chan HS and Dill KA, Proteins, 1998, 30, 2–33. [DOI] [PubMed] [Google Scholar]
- 39.Clark PL, Trends Biochem. Sci, 2004, 29, 527–534. [DOI] [PubMed] [Google Scholar]
- 40.Zhou R, Proc. Natl. Acad. Sci. U.S.A, 2003, 100, 13280–13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krone MG, Hua L, Soto P, Zhou R, Berne BJ and Shea JE, J. Am. Chem. Soc, 2008, 130, 11066–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu P, Huang X, Zhou R and Berne BJ, Nature, 2005, 437, 159–162. [DOI] [PubMed] [Google Scholar]
- 43.Zhou R, Huang X, Margulis CJ and Berne BJ, Science, 2004, 305, 1605–1609. [DOI] [PubMed] [Google Scholar]
- 44.Das P, Kapoor D, Halloran KT, Zhou R and Matthews CR, J. Am. Chem. Soc, 2013, 135, 1882–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berne BJ, Weeks JD and Zhou R, Annu. Rev. Phys. Chem, 2009, 60, 85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geddes AJ, Parker KD, Atkins EDT and Beighton E, J. Mol. Biol, 1968, 32, 343–358. [DOI] [PubMed] [Google Scholar]
- 47.Makin OS and Serpell LC, FEBS J, 2005, 272, 5950–5961. [DOI] [PubMed] [Google Scholar]
- 48.Morris KL and Serpell LC, Methods Mol. Biol, 2012, 849, 121–135. [DOI] [PubMed] [Google Scholar]
- 49.Perutz MF, Finch JT, Berriman J and Lesk A, Proc. Natl. Acad. Sci. U.S.A, 2002, 99, 5591–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sikorski P and Atkins E, Biomacromolecules, 2005, 6, 435–432. [DOI] [PubMed] [Google Scholar]
- 51.Fandrich M and Dobson CM, Embo J, 2002, 21, 5682–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Makin OS, Atkins E, Sikorski P, Johansson J and Serpell LC, Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen AS and Calkins E, Nature, 1959, 183, 1202–1203. [DOI] [PubMed] [Google Scholar]
- 54.Glenner GG, Ein D, Eanes ED, Bladen HA, Terry W and Page DL, Science, 1971, 174, 712–714. [DOI] [PubMed] [Google Scholar]
- 55.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ and Westermark P, Amyloid, 2014, 21, 221–224. [DOI] [PubMed] [Google Scholar]
- 56.Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, Sipe JD and Westermark P, Amyloid, 2018, 25, 215–219. [DOI] [PubMed] [Google Scholar]
- 57.Waugh DF, J. Am. Chem. Soc, 1946, 68, 247–250. [Google Scholar]
- 58.Burke M and Rougvie M, Biochemistry, 1972, 11, 2435–2439. [DOI] [PubMed] [Google Scholar]
- 59.Kirschner DA, Inouye H, Duffy LK, Sinclair A, LInd M and Selkoe DJ, Proc. Natl. Acad. Sci. U.S.A, 1987, 84, 6953–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fraser PE, Nguyen JT, Inouye H, Surewicz WK, Selkoe DJ, Podlisny MB and Kirschner DA, Biochemistry, 1992, 31, 10716–10723. [DOI] [PubMed] [Google Scholar]
- 61.Clark A, Cooper G, Lewis C, Morris J, Willis A, Reid K and Turner R, Lancet, 1987, 2, 231–234. [DOI] [PubMed] [Google Scholar]
- 62.Jarvis J, Craik D and Wilce M, BBRC, 1993, 192, 991–998. [DOI] [PubMed] [Google Scholar]
- 63.Blake C and Serpell L, Structure, 1996, 4, 989–998. [DOI] [PubMed] [Google Scholar]
- 64.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB and Blake CC, J. Mol. Biol, 1997, 273, 729–739. [DOI] [PubMed] [Google Scholar]
- 65.Serpell LC, Sunde M, Fraser PE, Luther PK, Morris EP, Sangren O, Lundgren E and Blake CC, J. Mol. Biol, 1995, 254, 113–118. [DOI] [PubMed] [Google Scholar]
- 66.Serpell L, Sunde M, Benson M, Tennent G, Pepys M and Fraser P, J. Mol. Biol, 2000, 300, 1033–1039. [DOI] [PubMed] [Google Scholar]
- 67.Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P, Muller SA, Kistler J, Cooper GJ and Aebi U, J. Struct. Biol, 2000, 130, 352–362. [DOI] [PubMed] [Google Scholar]
- 68.Jimenez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M and Saibil HR, Embo J, 1999, 18, 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM and Saibil HR, Proc. Natl. Acad. Sci. U.S.A, 2002, 99, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meinhardt J, Sachse C, Hortschansky P, Grigorieff N and Fandrich M, J. Mol. Biol, 2009, 386, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldsbury C, Aebi U and Frey P, Trends Mol. Med, 2001, 7, 582. [DOI] [PubMed] [Google Scholar]
- 72.Serpell LC, Blake CCF and Fraser PE, Biochemistry, 2000, 39, 13269–13275. [DOI] [PubMed] [Google Scholar]
- 73.Sikorski P, Atkins ED and Serpell LC, Structure 2003, 11, 915–926. [DOI] [PubMed] [Google Scholar]
- 74.Serpell LC and Smith JM, J. Mol. Biol, 2000, 299, 255–231. [DOI] [PubMed] [Google Scholar]
- 75.Slotta U, Hess S, Spiess K, Stromer T, Serpell L and Scheibel T, Macromol. Biosci, 2007, 7, 183–188. [DOI] [PubMed] [Google Scholar]
- 76.Makin OS and Serpell LC, J. Mol. Biol, 2004, 335, 1279–1288. [DOI] [PubMed] [Google Scholar]
- 77.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF and Lindquist SL, Science, 2000, 289, 1317–1321. [DOI] [PubMed] [Google Scholar]
- 78.Steinmetz MO, Gattin Z, Verel R, Ciani B, Stromer T, Green JM, Tittmann P, Schulze-Briese C, Gross H, van Gunsteren WF, Meier BH, Serpell LC, Muller SA and Kammerer RA, J. Mol. Biol, 2008, 376, 898–912. [DOI] [PubMed] [Google Scholar]
- 79.Blake CCF, Serpell LC, Sunde M, Sandgren O and Lundgren E, Ciba foundation symposia, 1996, 199, 6–21. [DOI] [PubMed] [Google Scholar]
- 80.Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M and Crowther RA, Proc. Natl. Acad. Sci. U.S.A, 2003, 100, 9034–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Al-Hilaly YK, Pollack SJ, Vadukul DM, Citossi F, Rickard JE, Simpson M, Storey JMD, Harrington CR, Wischik CM and Serpell LC, J. Mol. Biol, 2017, 429, 3650–3665. [DOI] [PubMed] [Google Scholar]
- 82.Jahn TR, Tennent GA and Radford SE, J. Biol. Chem, 2008, 283, 17279–17286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pauwels K, Williams TL, Morris KL, Jonckheere W, Vandersteen A, Kelly G, Schymkowitz J, Rousseau F, Pastore A, Serpell LC and Broersen K, J. Biol. Chem, 2012, 287, 5650–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marshall KE, Hicks MR, Williams TL, Hoffmann SV, Rodger A, Dafforn TR and Serpell LC, Biophys. J, 2010, 98, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Serpell LC, Benson M, Liepnieks JJ and Fraser PE, Amyloid, 2007, 14, 199–203. [DOI] [PubMed] [Google Scholar]
- 86.Morris KL, Rodger A, Hicks MR, Debulpaep M, Schymkowitz J, Rousseau F and Serpell LC, Biochem. J, 2013, 450, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pauwels K, Williams TL, Morris KL, Jonckheere W, Vandersteen A, Kelly G, Schymkowitz J, Rousseau F, Pastore A, Serpell LC and Broersen K, J. Biol. Chem, 2012, 287, 5650–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG and Langen R, J. Biol. Chem, 2002, 277, 40810–40815. [DOI] [PubMed] [Google Scholar]
- 89.Kajava AV, Aebi U and Steven AC, J. Mol. Biol, 2005, 348, 247–252. [DOI] [PubMed] [Google Scholar]
- 90.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM and Griffin RG, Proc. Natl. Acad. Sci. U.S.A, 2004, 101, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F and Tycko R, Proc. Natl. Acad. Sci. U.S.A, 2002, 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Diaz-Avalos R, Long C, Fontano E, Balbirnie M, Grothe R, Eisenberg D and Caspar DLD, J. Mol. Biol, 2003, 330, 1165–1175. [DOI] [PubMed] [Google Scholar]
- 93.Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S, Eisenberg DS, Zhou ZH and Jiang L, Nat. Commun, 2018, 9, 3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R and Eisenberg D, Nature, 2005, 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsemekhman K, Goldschmidt L, Eisenberg D and Baker D, Protein Sci, 2007, 16, 761–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seidler PM, Boyer DR, Murray KA, Yang TP, Bentzel M, Sawaya MR, Rosenberg G, Cascio D, Williams CK, Newell KL, Ghetti B, DeTure MA, Dickson DW, Vinters HV and Eisenberg DS, J. Biol. Chem, 2019, 294, 16451–16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Walti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Bockmann A, Guntert P, Meier BH and Riek R, Proc. Natl. Acad. Sci. U.S.A, 2016, 113, E4976–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S and Griffin RG, J. Am. Chem. Soc, 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moshe A, Landau M and Eisenberg D, Methods Mol. Biol, 2016, 1345, 201–210. [DOI] [PubMed] [Google Scholar]
- 100.Eisenberg DS and Sawaya MR, Proc. Natl. Acad. Sci. U.S.A, 2016, 113, 9398–9400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M and Scheres SHW, Nature, 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eisenberg DS and Sawaya MR, Annu. Rev. Biochem, 2017, 86, 69–95. [DOI] [PubMed] [Google Scholar]
- 103.Nogales E and Scheres SH, Molecular Cell, 2015, 58, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tycko R, Annu. Rev. Phys. Chem, 2011, 62, 279–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iwata K, Fujiwara T, Matsuki Y, Akutsu H, Takahashi S, Naiki H and Goto Y, Proc. Natl. Acad. Sci. U.S.A, 2006, 103, 18119–18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paravastu AK, Leapman RD, Yau WM and Tycko R, Proc. Natl. Acad. Sci. U.S.A, 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM and Rienstra CM, Nat. Struct. Mol. Biol, 2016, 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH and Melki R, Nat. Commun, 2013, 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Egelman EH, Ultramicroscopy, 2000, 85, 225–234. [DOI] [PubMed] [Google Scholar]
- 110.Scheres SH, J. Struct. Biol, 2012, 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, Zhang X, Li D, Liu C and Li X, Cell Res, 2018, 28, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Jiang L, Arbing MA, Nannenga BL, Hattne J, Whitelegge J, Brewster AS, Messerschmidt M, Boutet S, Sauter NK, Gonen T and Eisenberg DS, Nature, 2015, 525, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M and Stahlberg H, Elife, 2018, 7, e36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M and Scheres SHW, Nature, 2019, 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cao Q, Boyer DR, Sawaya MR, Ge P and Eisenberg DS, Nat. Struct. Mol. Biol, 2019, 26, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres S and Goedert M, Nature, 2018, 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Radamaker L, Lin YH, Annamalai K, Huhn S, Hegenbart U, Schonland SO, Fritz G, Schmidt M and Fandrich M, Nat. Commun, 2019, 10, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iadanza MG, Silvers R, Boardman J, Smith HI, Karamanos TK, Debelouchina GT, Su Y, Griffin RG, Ranson NA and Radford SE, Nat. Commun, 2018, 9, 4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liberta F, Rennegarbe M, Rosler R, Bijzet J, Wiese S, Hazenberg BPC and Fandrich M, Amyloid, 2019, 26, 164–170. [DOI] [PubMed] [Google Scholar]
- 120.Liberta F, Loerch S, Rennegarbe M, Schierhorn A, Westermark P, Westermark GT, Hazenberg BPC, Grigorieff N, Fandrich M and Schmidt M, Nat. Commun, 2019, 10, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R and McKnight SL, Cell, 2012, 149, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL and Tycko R, Cell, 2017, 171, 615–627 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hughes MP, Sawaya MR, Boyer DR, Goldschmidt L, Rodriguez JA, Cascio D, Chong L, Gonen T and Eisenberg DS, Science, 2018, 359, 698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reches M and Gazit E, Science, 2003, 300, 625–627. [DOI] [PubMed] [Google Scholar]
- 125.Greenwald J and Riek R, J. Mol. Biol, 2012, 421, 417–426. [DOI] [PubMed] [Google Scholar]
- 126.Gazit E, Annu. Rev. Biochem, 2018, 87, 533–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Astbury WT, Dickinson S and Bailey K, Biochem. J, 1935, 29, 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sunde M and Blake C, Adv. Prot. Chem, 1997, 50, 123–159. [DOI] [PubMed] [Google Scholar]
- 129.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R and Meier BH, Science, 2008, 319, 1523–1526. [DOI] [PubMed] [Google Scholar]
- 130.Lara C, Reynolds NP, Berryman JT, Xu A, Zhang A and Mezzenga R, J. Am. Chem. Soc, 2014, 136, 4732–4739. [DOI] [PubMed] [Google Scholar]
- 131.Usov I, Adamcik J and Mezzenga R, ACS Nano, 2013, 7, 10465–10474. [DOI] [PubMed] [Google Scholar]
- 132.Adamcik J and Mezzenga R, Soft Matter, 2011, 7, 5437–5443. [Google Scholar]
- 133.Assenza S, Adamcik J, Mezzenga R and De Los Rios P, Phys. Rev. Lett, 2014, 113, 268103. [DOI] [PubMed] [Google Scholar]
- 134.Seuring C, Verasdonck J, Ringler P, Cadalbert R, Stahlberg H, Böckmann A, Meier BH and Riek R, J. Phys. Chem. B, 2017, 121, 1783–1792. [DOI] [PubMed] [Google Scholar]
- 135.Verel R, Tomka IT, Bertozzi C, Cadalbert R, Kammerer RA, Steinmetz MO and Meier BH, Angew. Chem. Int. Ed. Engl, 2008, 47, 5842–5845. [DOI] [PubMed] [Google Scholar]
- 136.Lara C, Adamcik J, Jordens S and Mezzenga R, Biomacromolecules, 2011, 12, 1868–1875. [DOI] [PubMed] [Google Scholar]
- 137.Huang C, Wang Z, Quinn D, Suresh S and Hsia KJ, Proc. Natl. Acad. Sci. U.S.A, 2018, 115, 12359–12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sawa Y, Ye F, Urayama K, Takigawa T, Gimenez-Pinto V, Selinger RLB and Selinger JV, Proc. Natl. Acad. Sci. U.S.A, 2011, 108, 6364–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Adamcik J, Jung J-M, Flakowski J, De Los Rios P, Dietler G and Mezzenga R, Nat. Nanotechnol, 2010, 5, 423–428. [DOI] [PubMed] [Google Scholar]
- 140.Zhang S, Andreasen M, Nielsen JT, Liu L, Nielsen EH, Song J, Ji G, Sun F, Skrydstrup T, Besenbacher F, Nielsen NC, Otzen DE and Dong M, Proc. Natl. Acad. Sci. U.S.A, 2013, 110, 2798–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Adamcik J, Sánchez‐Ferrer A, Ait‐Bouziad N, Reynolds NP, Lashuel HA and Mezzenga R, Angew. Chem. Int. Ed. Engl, 2016, 55, 618–622. [DOI] [PubMed] [Google Scholar]
- 142.Lomakin A, Chung DS, Benedek GB, Kirschner DA and Teplow DB, Proc. Natl. Acad. Sci. U.S.A, 1996, 93, 1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jarrett JT and Lansbury PT Jr., Cell, 1993, 73, 1055–1058. [DOI] [PubMed] [Google Scholar]
- 144.Cohen SI, Vendruscolo M, Welland ME, Dobson CM, Terentjev EM and Knowles TP, J. Chem. Phys, 2011, 135, 065105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Knowles TP, White DA, Abate AR, Agresti JJ, Cohen SI, Sperling RA, De Genst EJ, Dobson CM and Weitz DA, Proc. Natl. Acad. Sci. U.S.A, 2011, 108, 14746–14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oosawa F and Kasai M, J. Mol. Biol, 1962, 4, 10–21. [DOI] [PubMed] [Google Scholar]
- 147.Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SI, Dobson CM, Linse S and Knowles TP, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TPJ, Sparr E and Linse S, Q. Rev. Biophys, 2017, 50, e6. [DOI] [PubMed] [Google Scholar]
- 149.Ferrone FA, Hofrichter J and Eaton WA, J. Mol. Biol, 1985, 183, 611–631. [DOI] [PubMed] [Google Scholar]
- 150.Ruschak AM and Miranker AD, Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 12341–12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Scheidt T, Łapińska U, Kumita JR, Whiten DR, Klenerman D, Wilson MR, Cohen SIA, Linse S, Vendruscolo M, Dobson CM, Knowles TPJ and Arosio P, Sci. Adv, 2019, 5, eaau3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Arosio P, Knowles TP and Linse S, Phys. Chem. Chem. Phys, 2015, 17, 7606–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Arosio P, Cukalevski R, Frohm B, Knowles TP and Linse S, J. Am. Chem. Soc, 2014, 136, 219–225. [DOI] [PubMed] [Google Scholar]
- 154.Cohen SIA, Arosio P, Presto J, Kurudenkandy FR, Biverstal H, Dolfe L, Dunning C, Yang X, Frohm B, Vendruscolo M, Johansson J, Dobson CM, Fisahn A, Knowles TPJ and Linse S, Nat. Struct. Mol. Biol, 2015, 22, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT and Spires-Jones TL, Proc. Natl. Acad. Sci. U.S.A, 2009, 106, 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hardy JA and Higgins GA, Science, 1992, 256, 184–185. [DOI] [PubMed] [Google Scholar]
- 157.Selkoe DJ and Hardy J, EMBO Mol. Med, 2016, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hardy J, J. Alzheimers Dis, 2006, 9, 151–153. [DOI] [PubMed] [Google Scholar]
- 159.Lansbury PT and Lashuel HA, Nature, 2006, 443, 774–779. [DOI] [PubMed] [Google Scholar]
- 160.Cline EN, Bicca MA, Viola KL and Klein WL, J. Alzheimers Dis, 2018, 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kametani F and Hasegawa M, Front. Neurosci, 2018, 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH and Aronson MK, Neurobiol. Aging, 1992, 13, 179–189. [DOI] [PubMed] [Google Scholar]
- 163.Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR and Hayden MR, Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 11402–11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Arrasate M, Mitra S, Schweitzer ES, Segal MR and Finkbeiner S, Nature, 2004, 431, 805–810. [DOI] [PubMed] [Google Scholar]
- 165.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M and Arendash GW, Nature, 2000, 408, 982–985. [DOI] [PubMed] [Google Scholar]
- 166.Harrison JR and Owen MJ, Br. J. Psychiatry, 2016, 208, 1–3. [DOI] [PubMed] [Google Scholar]
- 167.Makin S, Nature, 2018, 559, S4–S7. [DOI] [PubMed] [Google Scholar]
- 168.Harper JD, Wong SS, Lieber CM and Lansbury PT, Chem. Biol, 1997, 4, 119–125. [DOI] [PubMed] [Google Scholar]
- 169.Walsh DM, Lomakin A, Benedek GB, Condron MM and Teplow DB, J. Biol. Chem, 1997, 272, 22364–22372. [DOI] [PubMed] [Google Scholar]
- 170.Shahnawaz M and Soto C, J. Biol. Chem, 2012, 287, 11665–11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stefani M, FEBS J, 2010, 277, 4602–4613. [DOI] [PubMed] [Google Scholar]
- 172.Tipping KW, Karamanos TK, Jakhria T, Iadanza MG, Goodchild SC, Tuma R, Ranson NA, Hewitt EW and Radford SE, Proc. Natl. Acad. Sci. U.S.A, 2015, 112, 5691–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cremades N, Cohen SIA, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TPJ, Dobson CM and Klenerman D, Cell, 2012, 149, 1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mahul-Mellier AL, Vercruysse F, Maco B, Ait-Bouziad N, De Roo M, Muller D and Lashuel HA, Cell Death Differ, 2015, 22, 2107–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dorostkar MM, Burgold S, Filser S, Barghorn S, Schmidt B, Anumala UR, Hillen H, Klein C and Herms J, Brain, 2014, 137, 3319–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT and Spires-Jones TL, Proc. Natl. Acad. Sci. U.S.A, 2009, 106, 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Roher A, Chaney M, Kuo Y-M, Webster S, Stine W, Haverkamp L, Woods A, Cotter R, Tuohy J, Krafft G, Bonnell B and Emmerling M, J. Biol. Chem, 1996, 271, 20631–20635. [DOI] [PubMed] [Google Scholar]
- 178.Upadhaya AR, Lungrin I, Yamaguchi H, Fändrich M and Thal DR, J. Cell. Mol. Med, 2012, 16, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yang T, Li S, Xu H, Walsh DM and Selkoe DJ, J. Neurosci, 2017, 37, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Borsarelli CD, Falomir-Lockhart LJ, Ostatna V, Fauerbach JA, Hsiao HH, Urlaub H, Palecek E, Jares-Erijman EA and Jovin TM, Free Radic. Biol. Med, 2012, 53, 1004–1015. [DOI] [PubMed] [Google Scholar]
- 181.Al-Hilaly YK, Williams TL, Stewart-Parker M, Ford L, Skaria E, Cole M, Bucher WG, Morris KL, Sada AA, Thorpe JR and Serpell LC, Acta Neuropathol. Commun, 2013, 1, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chang YJ and Chen YR, FEBS J, 2014, 281, 2674–2687. [DOI] [PubMed] [Google Scholar]
- 183.Cline EN, Das A, Bicca MA, Mohammad SN, Schachner LF, Kamel JM, DiNunno N, Weng A, Paschall JD, Bu RL, Khan FM, Rollins MG, Ives AN, Shekhawat G, Nunes-Tavares N, de Mello FG, Compton PD, Kelleher NL and Klein WL, J. Neurochem, 2019, 148, 822–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Piening N, Weber P, Hogen T, Beekes M, Kretzschmar H and Giese A, Amyloid, 2006, 13, 67–77. [DOI] [PubMed] [Google Scholar]
- 185.Teruya K, Nishizawa K, Oguma A, Sakasegawa Y, Kitamoto T and Doh-Ura K, Biochim. Biophys. Acta Gen. Subj, 2019, 1863, 384–394. [DOI] [PubMed] [Google Scholar]
- 186.Vollers SS, Teplow DB and Bitan G, Methods Mol. Biol, 2005, 299, 11–18. [DOI] [PubMed] [Google Scholar]
- 187.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T and Lansbury PT Jr., J. Mol. Biol, 2002, 322, 1089–1102. [DOI] [PubMed] [Google Scholar]
- 188.Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB and Wetzel R, J. Mol. Biol, 2012, 415, 881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kotler SA, Brender JR, Vivekanandan S, Suzuki Y, Yamamoto K, Monette M, Krishnamoorthy J, Walsh P, Cauble M, Holl MM, Marsh EN and Ramamoorthy A, Sci. Rep, 2015, 5, 11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sun Y, Kakinen A, Xing Y, Pilkington EH, Davis TP, Ke PC and Ding F, Biochim. Biophys. Acta Mol. Basis Dis, 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Younan ND and Viles JH, Biochemistry, 2015, 54, 4297–4306. [DOI] [PubMed] [Google Scholar]
- 192.Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, Muhs A and Lashuel HA, J. Biol. Chem, 2011, 286, 8585–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Stroud JC, Liu C, Teng PK and Eisenberg D, Proc. Natl. Acad. Sci. U.S.A, 2012, 109, 7717–7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW and Glabe CG, Science, 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- 195.Glabe CG, J. Biol. Chem, 2008, 283, 29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Breydo L, Kurouski D, Rasool S, Milton S, Wu JW, Uversky VN, Lednev IK and Glabe CG, Biochem. Biophys. Res. Commun, 2016, 477, 700–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kayed R, Canto I, Breydo L, Rasool S, Lukacsovich T, Wu J, Albay R 3rd, Pensalfini A, Yeung S, Head E, Marsh JL and Glabe C, Mol. Neurodegener, 2010, 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T and Lansbury PT Jr., J. Mol. Biol, 2003, 332, 795–808. [DOI] [PubMed] [Google Scholar]
- 199.Jan A, Hartley DM and Lashuel HA, Nat. Protoc, 2010, 5, 1186–1209. [DOI] [PubMed] [Google Scholar]
- 200.Serra-Batiste M, Ninot-Pedrosa M, Puig E, Ciudad S, Gairi M and Carulla N, Methods Mol. Biol, 2018, 1779, 13–22. [DOI] [PubMed] [Google Scholar]
- 201.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea J-E, Ruotolo BT, Robinson CV and Bowers MT, Nat. Chem, 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Singh J, Sabareesan AT, Mathew MK and Udgaonkar JB, J. Mol. Biol, 2012, 423, 217–231. [DOI] [PubMed] [Google Scholar]
- 203.Serra-Vidal B, Pujadas L, Rossi D, Soriano E, Madurga S and Carulla N, ACS Chem. Biol, 2014, 9, 2678–2685. [DOI] [PubMed] [Google Scholar]
- 204.Paslawski W, Mysling S, Thomsen K, Jorgensen TJ and Otzen DE, Angew. Chem. Int. Ed. Engl, 2014, 53, 7560–7563. [DOI] [PubMed] [Google Scholar]
- 205.Kheterpal I, Lashuel HA, Hartley DM, Walz T, Lansbury PT Jr. and Wetzel R, Biochemistry, 2003, 42, 14092–14098. [DOI] [PubMed] [Google Scholar]
- 206.Scheidt HA, Morgado I and Huster D, J. Biol. Chem, 2012, 287, 22822–22826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Potapov A, Yau WM, Ghirlando R, Thurber KR and Tycko R, J. Am. Chem. Soc, 2015, 137, 8294–8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.van der Wel PCA, Solid State Nucl. Magn. Reson, 2017, 88, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chung J, Yang H, de Beus MD, Ryu CY, Cho K and Colon W, Biochem. Biophys. Res. Commun, 2003, 312, 873–876. [DOI] [PubMed] [Google Scholar]
- 210.Lasagna-Reeves CA, Sengupta U, Castillo-Carranza D, Gerson JE, Guerrero-Munoz M, Troncoso JC, Jackson GR and Kayed R, Acta Neuropathol. Commun, 2014, 2, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dasari AKR, Hughes RM, Wi S, Hung I, Gan Z, Kelly JW and Lim KH, Sci. Rep, 2019, 9, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang L, Lashuel HA, Walz T and Colon W, Proc. Natl. Acad. Sci. U.S.A, 2002, 99, 15947–15952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Oropesa-Nunez R, Seghezza S, Dante S, Diaspro A, Cascella R, Cecchi C, Stefani M, Chiti F and Canale C, Oncotarget, 2016, 7, 44991–45004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Diociaiuti M, Polzi LZ, Valvo L, Malchiodi-Albedi F, Bombelli C and Gaudiano MC, Biophys. J, 2006, 91, 2275–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bhattacharya M, Jain N, Dogra P, Samai S and Mukhopadhyay S, J. Phys. Chem. Lett, 2013, 4, 480–485. [DOI] [PubMed] [Google Scholar]
- 216.Bhattacharya M and Dogra P, Langmuir, 2015, 31, 8911–8922. [DOI] [PubMed] [Google Scholar]
- 217.Arya S, Kumari A, Dalal V, Bhattacharya M and Mukhopadhyay S, Phys. Chem. Chem. Phys, 2015, 17, 22862–22871. [DOI] [PubMed] [Google Scholar]
- 218.Ghosh DK, Roy A and Ranjan A, J. Mol. Biol, 2018, 430, 963–986. [DOI] [PubMed] [Google Scholar]
- 219.Lee J, Kim YH, T. AF, Gillman AL, Jang H, Kagan BL, Nussinov R, Yang J and Lal R, ACS Chem. Neurosci, 2017, 8, 1348–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lin H, Bhatia R and Lal R, FASEB J, 2001, 15, 2433–2444. [DOI] [PubMed] [Google Scholar]
- 221.Lin H, Zhu YJ and Lal R, Biochemistry, 1999, 38, 11189–11196. [DOI] [PubMed] [Google Scholar]
- 222.Patel N, Ramachandran S, Azimov R, Kagan BL and Lal R, Biochemistry, 2015, 54, 7320–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J and Lal R, Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hirakura Y, Azimov R, Azimova R and Kagan BL, J. Neurosci. Res, 2000, 60, 490–494. [DOI] [PubMed] [Google Scholar]
- 225.Hirakura Y, Carreras I, Sipe JD and Kagan BL, Amyloid, 2002, 9, 13–23. [DOI] [PubMed] [Google Scholar]
- 226.Mirzabekov TA, Lin MC and Kagan BL, J. Biol. Chem, 1996, 271, 1988–1992. [DOI] [PubMed] [Google Scholar]
- 227.Anguiano M, Nowak RJ and Lansbury PT Jr., Biochemistry, 2002, 41, 11338–11343. [DOI] [PubMed] [Google Scholar]
- 228.Volles MJ and Lansbury PT Jr., Biochemistry, 2002, 41, 4595–4602. [DOI] [PubMed] [Google Scholar]
- 229.Kim HY, Cho MK, Kumar A, Maier E, Siebenhaar C, Becker S, Fernandez CO, Lashuel HA, Benz R, Lange A and Zweckstetter M, J. Am. Chem. Soc, 2009, 131, 17482–17489. [DOI] [PubMed] [Google Scholar]
- 230.Kayed R, Pensalfini A, Margol L, Sokolov Y, Sarsoza F, Head E, Hall J and Glabe C, J. Biol. Chem, 2009, 284, 4230–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bäuerlein FJB, Saha I, Mishra A, Kalemanov M, Martínez-Sánchez A, Klein R, Dudanova I, Hipp MS, Hartl FU, Baumeister W and Fernández-Busnadiego R, Cell, 2017, 171, 179–187. [DOI] [PubMed] [Google Scholar]
- 232.Goedert M, Jakes R and Spillantini MG, J. Parkinsons Dis, 2017, 7, S51–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Goedert M and Spillantini MG, in Basic Neurochemistry (Eighth Edition), eds. Brady ST, Siegel GJ, Albers RW and Price DL, Academic Press, New York, 2012, pp. 829–843. [Google Scholar]
- 234.Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW and Lashuel HA, Proc. Natl. Acad. Sci. U.S.A, 2020, 117, 4971–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL and Selkoe DJ, Nat. Med, 2008, 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Panza F, Lozupone M, Dibello V, Greco A, Daniele A, Seripa D, Logroscino G and Imbimbo BP, Immunotherapy, 2019, 11, 3–6. [DOI] [PubMed] [Google Scholar]
- 237.de la Torre JC, Engl N. J. Med, 2014, 370, 1459–1460. [DOI] [PubMed] [Google Scholar]
- 238.Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, Basun H and Lannfelt L, Alzheimers Res. Ther, 2016, 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cummings J, Lee G, Ritter A, Sabbagh M and Zhong K, Alzheimers Dement., 2019, 5, 272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett AD, Dineley KT, Jackson GR and Kayed R, J. Neurosci, 2014, 34, 4260–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gerson JE, Farmer KM, Henson N, Castillo-Carranza DL, Carretero Murillo M, Sengupta U, Barrett A and Kayed R, Mol. Neurodegener, 2018, 13, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gustafsson G, Lindstrom V, Rostami J, Nordstrom E, Lannfelt L, Bergstrom J, Ingelsson M and Erlandsson A, J. Neuroinflammation, 2017, 14, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.El-Agnaf O, Overk C, Rockenstein E, Mante M, Florio J, Adame A, Vaikath N, Majbour N, Lee SJ, Kim C, Masliah E and Rissman RA, Neurobiol. Dis, 2017, 104, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lindstrom V, Fagerqvist T, Nordstrom E, Eriksson F, Lord A, Tucker S, Andersson J, Johannesson M, Schell H, Kahle PJ, Moller C, Gellerfors P, Bergstrom J, Lannfelt L and Ingelsson M, Neurobiol. Dis, 2014, 69, 134–143. [DOI] [PubMed] [Google Scholar]
- 245.Salloway S, Honigberg LA, Cho W, Ward M, Friesenhahn M, Brunstein F, Quartino A, Clayton D, Mortensen D, Bittner T, Ho C, Rabe C, Schauer SP, Wildsmith KR, Fuji RN, Suliman S, Reiman EM, Chen K and Paul R, Alzheimers Res. Ther, 2018, 10, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yang T, Dang Y, Ostaszewski B, Mengel D, Steffen V, Rabe C, Bittner T, Walsh DM and Selkoe DJ, Ann. Neurol, 2019, 86, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rosenblum WI, Neurobiol. Aging, 2014, 35, 969–974. [DOI] [PubMed] [Google Scholar]
- 248.Mably AJ, Liu W, Mc Donald JM, Dodart JC, Bard F, Lemere CA, O’Nuallain B and Walsh DM, Neurobiol. Dis, 2015, 82, 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Knowles TPJ, Vendruscolo M and Dobson CM, Nat. Rev. Mol. Cell Biol, 2014, 15, 384–396. [DOI] [PubMed] [Google Scholar]
- 250.Zheng W, Tsai M-Y, Chen M and Wolynes PG. Proc. Natl. Acad. Sci. U.S.A, 2016, 113, 11835–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Balchin D, Hayer-Hartl M and Hartl FU, Science, 2016, 353, aac4354. [DOI] [PubMed] [Google Scholar]
- 252.McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MHL, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HTJ, Przybylski M and St George-Hyslop P, Nat. Med, 2002, 8, 1263–1269. [DOI] [PubMed] [Google Scholar]
- 253.Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Günther S, Walsh DM and Wanker EE, Nat. Chem. Biol, 2011, 8, 93–101. [DOI] [PubMed] [Google Scholar]
- 254.Ke PC, Pilkington EH, Sun Y, Javed I, Kakinen A, Peng G, Ding F and Davis TP, Adv. Mater, 2019, DOI: 10.1002/ADMA.201901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Soto C, Sigurdsson EM, Morelli L, Asok Kumar R, Castaño EM and Frangione B, Nat. Med, 1998, 4, 822–826. [DOI] [PubMed] [Google Scholar]
- 256.Krysmann MJ, Castelletto V, Kelarakis A, Hamley IW, Hule RA and Pochan DJ, Biochemistry, 2008, 47, 4597–4605. [DOI] [PubMed] [Google Scholar]
- 257.Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H and Mohs R, N. Eng. J. Med, 2014, 370, 311–321. [DOI] [PubMed] [Google Scholar]
- 258.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R and Brashear HR, N. Eng. J. Med, 2014, 370, 322–333. [Google Scholar]
- 259.Feuerstein A, STATNEWS.COM, https://www.statnews.com/2019/03/21/biogen-eisai-alzheimer-trial-stopped/, March 21, 2019.
- 260.Chen J, Armstrong AH, Koehler AN and Hecht MH, J. Am. Chem. Soc, 2010, 132, 17015–17022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K and Wanker EE, Proc. Natl. Acad. Sci. U.S.A, 2010, 107, 7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Palhano FL, Lee J, Grimster NP and Kelly JW, J. Am. Chem. Soc, 2013, 135, 7503–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Chen Q, Du Y, Zhang K, Liang Z, Li J, Yu H, Ren R, Feng J, Jin Z, Li F, Sun J, Zhou M, He Q, Sun X, Zhang H, Tian M and Ling D, ACS Nano, 2018, 12, 1321–1338. [DOI] [PubMed] [Google Scholar]
- 264.Gladytz A, Abel B and Risselada HJ, Angew. Chem. Int. Ed. Engl, 2016, 55, 11242–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Gurzov EN, Wang B, Pilkington EH, Chen P, Kakinen A, Stanley WJ, Litwak SA, Hanssen EG, Davis TP, Ding F and Ke PC, Small, 2016, 12, 1615–1626. [DOI] [PubMed] [Google Scholar]
- 266.Kim D, Yoo JM, Hwang H, Lee J, Lee SH, Yun SP, Park MJ, Lee M, Choi S, Kwon SH, Lee S, Kwon S-H, Kim S, Park YJ, Kinoshita M, Lee Y-H, Shin S, Paik SR, Lee SJ, Lee S, Hong BH and Ko HS, Nat. Nanotechnol, 2018, 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li M, Zhao A, Dong K, Li W, Ren J and Qu X, Nano Research, 2015, 8, 3216–3227. [Google Scholar]
- 268.Luo Q, Lin Y-X, Yang P-P, Wang Y, Qi G-B, Qiao Z-Y, Li B-N, Zhang K, Zhang J-P, Wang L and Wang H, Nat. Commun, 2018, 9, 1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Mahmoudi M, Akhavan O, Ghavami M, Rezaee F and Ghiasi SMA, Nanoscale, 2012, 4, 7322–7325. [DOI] [PubMed] [Google Scholar]
- 270.Wang J, Liu L, Ge D, Zhang H, Feng Y, Zhang Y, Chen M and Dong M, Chem. Eur. J, 2018, 24, 3397–3402. [DOI] [PubMed] [Google Scholar]
- 271.Wang M, Sun Y, Cao X, Peng G, Javed I, Kakinen A, Davis TP, Lin S, Liu J, Ding F and Ke PC, Nanoscale, 2018, 10, 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Zhao Y, Cai J, Liu Z, Li Y, Zheng C, Zheng Y, Chen Q, Chen H, Ma F, An Y, Xiao L, Jiang C, Shi L, Kang C and Liu Y, Nano Lett, 2019, 19, 674–683. [DOI] [PubMed] [Google Scholar]
- 273.Kakinen A, Xing Y, Hegoda Arachchi N, Javed I, Feng L, Faridi A, Douek AM, Sun Y, Kaslin J, Davis TP, Higgins MJ, Ding F and Ke PC, Nano Lett, 2019, 19, 6535–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Pilkington EH, Lai M, Ge X, Stanley WJ, Wang B, Wang M, Kakinen A, Sani M-A, Whittaker MR, Gurzov EN, Ding F, Quinn JF, Davis TP and Ke PC, Biomacromolecules, 2017, 18, 4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE and Kelly JW, PLoS Biol, 2006, 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hamley IW, Angew. Chem. Int. Ed. Engl, 2007, 46, 8128–8147. [DOI] [PubMed] [Google Scholar]
- 277.Nixon RA, J. Cell Sci, 2007, 120, 4081–4091. [DOI] [PubMed] [Google Scholar]
- 278.Cedervall T, Lynch I, Lindman S, Berggård T, Thulin E, Nilsson H, Dawson KA and Linse S, Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Finn TE, Nunez AC, Sunde M and Easterbrook-Smith SB, J. Biol. Chem, 2012, 287, 21530–21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Pilkington EH, Gustafsson OJR, Xing Y, Hernandez-Fernaud J, Zampronio C, Kakinen A, Faridi A, Ding F, Wilson P, Ke PC and Davis TP, ACS Nano, 2018, 12, 6066–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Sun Y, Kakinen A, Xing Y, Pilkington EH, Davis TP, Ke PC and Ding F, BBA Mol. Basis of Disease, 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Layfield R, Fergusson J, Aitken A, Lowe J, Landon M and Mayer RJ, Neurosci. Lett, 1996, 209, 57–60. [DOI] [PubMed] [Google Scholar]
- 283.Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI and Peng J, J. Biol. Chem, 2004, 279, 37061–37068. [DOI] [PubMed] [Google Scholar]
- 284.Perry G, Friedman R, Shaw G and Chau V, Proc. Natl. Acad. Sci. U.S.A, 1987, 84, 3033–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Perry G, Siedlak S, Richey P, Kawai M, Cras P, Kalaria R, Galloway P, Scardina J, Cordell B and Greenberg B, J. Neurosci, 1991, 11, 3679–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wakabayashi K, Tanji K, Mori F and Takahashi H, Neuropathology, 2007, 27, 494–506. [DOI] [PubMed] [Google Scholar]
- 287.Pepys MB, Philos. Trans. R. Soc. Lond. B. Biol. Sci, 2001, 356, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Tennent GA, Lovat LB and Pepys MB, Proc. Natl. Acad. Sci. U. S. A, 1995, 92, 4299–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Alexandrescu AT, Protein Sci, 2005, 14, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Fu Y, Zhao J, Atagi Y, Nielsen HM, Liu C-C, Zheng H, Shinohara M, Kanekiyo T and Bu G, Mol. Neurodegener, 2016, 11, 37–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Christianson HC and Belting M, Matrix Biology, 2014, 35, 51–55. [DOI] [PubMed] [Google Scholar]
- 292.Li M, Howson SE, Dong K, Gao N, Ren J, Scott P and Qu X, J. Am. Chem. Soc, 2014, 136, 11655–11663. [DOI] [PubMed] [Google Scholar]
- 293.Malishev R, Arad E, Bhunia SK, Shaham-Niv S, Kolusheva S, Gazit E and Jelinek R, Chem. Commun, 2018, 54, 7762–7765. [DOI] [PubMed] [Google Scholar]
- 294.Wei W, Xu C, Gao N, Ren J and Qu X, Chem. Sci, 2014, 5, 4367–4374. [Google Scholar]
- 295.Faridi A, Sun Y, Okazaki Y, Peng G, Gao J, Kakinen A, Faridi P, Zhao M, Javed I, Purcell AW, Davis TP, Lin S, Oda R, Ding F and Ke PC, Small, 2018, 14, e1802825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Javed I, Peng G, Xing Y, Yu T, Zhao M, Kakinen A, Faridi A, Parish CL, Ding F, Davis TP, Ke PC and Lin S, Nat. Commun, 2019, 10, 3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Gao N, Sun H, Dong K, Ren J, Duan T, Xu C and Qu X, Nat. Commun, 2014, 5, 3422. [DOI] [PubMed] [Google Scholar]
- 298.Best JD and Alderton WK, Neuropsychiatr. Dis. Treat, 2008, 4, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kalueff AV, Stewart AM and Gerlai R, Trends Pharmacol. Sci, 2014, 35, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.McGrath P and Li C-Q, Drug Discov. Today, 2008, 13, 394–401. [DOI] [PubMed] [Google Scholar]
- 301.Barnham KJ and Bush AI, Chem. Soc. Rev, 2014, 43, 6727–6749. [DOI] [PubMed] [Google Scholar]
- 302.Geng J, Li M, Wu L, Ren J and Qu X, J. Med. Chem, 2012, 55, 9146–9155. [DOI] [PubMed] [Google Scholar]
- 303.Arnold C, Nat. Med, 2019, DOI: 10.1038/d41591-019-00013-w. [DOI] [Google Scholar]
- 304.Lindholm P, Voutilainen MH, Laurén J, Peränen J, Leppänen V-M, Andressoo J-O, Lindahl M, Janhunen S, Kalkkinen N, Timmusk T, Tuominen RK and Saarma M, Nature, 2007, 448, 73. [DOI] [PubMed] [Google Scholar]
- 305.Lindahl M, Saarma M and Lindholm P, Neurobiol. Dis, 2017, 97, 90–102. [DOI] [PubMed] [Google Scholar]
- 306.Derkatch IL and Liebman SW, Prion, 2007, 1, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ma BY and Nussinov R, J. Mol. Biol, 2012, 421, 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Morales R, Moreno-Gonzalez I and Soto C, PLoS Pathog, 2013, 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Spires-Jones TL, Attems J and Thal DR, Acta Neuropathol, 2017, 134, 187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Hamley IW, Chemical Reviews, 2012, 112, 5147–5192. [DOI] [PubMed] [Google Scholar]
- 311.Ferrer I, Blanco R, Carmona M, Puig B, Ribera R, Rey MJ and Ribalta T, Acta Neuropathol, 2001, 101, 49–56. [DOI] [PubMed] [Google Scholar]
- 312.Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J and Soto C, J. Neurosci, 2010, 30, 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M and Mucke L, Proc. Natl. Acad. Sci. U.S.A, 2001, 98, 12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ and LaFerla FM, J. Neurosci, 2010, 30, 7281–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ and Lee VMY, Cell, 2013, 154, 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Munoz MJ, Jackson GR and Kayed R, Biochemistry, 2010, 49, 10039–10041. [DOI] [PubMed] [Google Scholar]
- 317.Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ and Lee VMY, Exp. Neurol, 2004, 187, 279–288. [DOI] [PubMed] [Google Scholar]
- 318.Ono K, Takahashi R, Ikeda T and Yamada M, J. Neurochem, 2012, 122, 883–890. [DOI] [PubMed] [Google Scholar]
- 319.Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, Wu CC, Liao JY, Chou SC, Lin V, Jin LW, Yuan HS, Cheng IH, Tu PH and Chen YR, Nat. Commun, 2014, 5, 13. [DOI] [PubMed] [Google Scholar]
- 320.O’Nuallain B, Williams AD, Westermark P and Wetzel R, J. Biol. Chem, 2004, 279, 17490–17499. [DOI] [PubMed] [Google Scholar]
- 321.Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P and Westermark GT, Am. J. Pathol, 2015, 185, 834–846. [DOI] [PubMed] [Google Scholar]
- 322.Tran J, Chang D, Hsu F, Wang HS and Guo ZF, FEBS Lett, 2017, 591, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Jones EM and Surewicz WK, Cell, 2005, 121, 63–72. [DOI] [PubMed] [Google Scholar]
- 324.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL and Liebman SW, Proc. Natl. Acad. Sci. U.S.A, 2004, 101, 12934–12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Vitrenko YA, Gracheva EO, Richmond JE and Liebman SW, J. Biol. Chem, 2007, 282, 1779–1787. [DOI] [PubMed] [Google Scholar]
- 326.Devlin GL, Knowles TPJ, Squires A, McCammon MG, Gras SL, Nilsson MR, Robinson CV, Dobson CM and MacPhee CE, J. Mol. Biol, 2006, 360, 497–509. [DOI] [PubMed] [Google Scholar]
- 327.Krebs MRH, Morozova-Roche LA, Daniel K, Robinson CV and Dobson CM, Protein Sci, 2004, 13, 1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Reches M and Gazit E, Isr. J. Chem, 2005, 45, 363–371. [Google Scholar]
- 329.Tamamis P, Adler-Abramovich L, Reches M, Marshall K, Sikorski P, Serpell L, Gazit E and Archontis G, Biophys. J, 2009, 96, 5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Al Hafid N and Christodoulou J, Transl. Pediatr, 2015, 4, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.MacLeod EL and Ney DM, Annales Nestlé (English ed.), 2010, 68, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Embury JE, Charron CE, Martynyuk A, Zori AG, Liu B, Ali SF, Rowland NE and Laipis PJ, Brain Res, 2007, 1127, 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Do TD and Bowers MT, Anal. Chem, 2015, 87, 4245–4252. [DOI] [PubMed] [Google Scholar]
- 334.German HW, Uyaver S and Hansmann UH, J. Phys. Chem. A, 2014, 119, 1609–1615. [DOI] [PubMed] [Google Scholar]
- 335.De Luigi A, Mariani A, De Paola M, Depaolini AR, Colombo L, Russo L, Rondelli V, Brocca P, Adler-Abramovich L and Gazit E, Sci. Rep, 2015, 5, 15902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Mak CM, Lee H-CH, Chan AY-W and Lam C-W, Crit. Rev. Clin. Lab. Sci, 2013, 50, 142–162. [DOI] [PubMed] [Google Scholar]
- 337.Shaham-Niv S, Adler-Abramovich L, Schnaider L and Gazit E, Sci. Adv, 2015, 1, e1500137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Banik D, Roy A, Kundu N and Sarkar N, J. Phys. Chem. B, 2016, 120, 11247–11255. [DOI] [PubMed] [Google Scholar]
- 339.Shaham‐Niv S, Rehak P, Vuković L, Adler‐Abramovich L, Král P and Gazit E, Isr. J. Chem, 2017, 57, 729–737. [Google Scholar]
- 340.Tavassoly O, Sade D, Bera S, Shaham-Niv S, Vocadlo DJ and Gazit E, J. Mol. Biol, 2018, 430, 3847–3862. [DOI] [PubMed] [Google Scholar]
- 341.Pinotsi D, Buell AK, Dobson CM, Kaminski Schierle GS and Kaminski CF, ChemBioChem, 2013, 14, 846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Amdursky N, Molotskii M, Aronov D, Adler-Abramovich L, Gazit E and Rosenman G, Nano Lett, 2009, 9, 3111–3115. [DOI] [PubMed] [Google Scholar]
- 343.Shaham‐Niv S, Arnon ZA, Sade D, Lichtenstein A, Shirshin EA, Kolusheva S and Gazit E, Angew. Chem. Int. Ed. Engl, 2018, 57, 12444–12447. [DOI] [PubMed] [Google Scholar]
- 344.Mossou E, Teixeira SC, Mitchell EP, Mason SA, Adler-Abramovich L, Gazit E and Forsyth VT, Acta Cryst. C, 2014, 70, 326–331. [DOI] [PubMed] [Google Scholar]
- 345.Porat Y, Abramowitz A and Gazit E, Chem. Biol. Drug Des, 2006, 67, 27–37. [DOI] [PubMed] [Google Scholar]
- 346.Shaham-Niv S, Rehak P, Zaguri D, Levin A, Adler-Abramovich L, Vuković L, Král P and Gazit E, Commun. Chem, 2018, 1, 25. [Google Scholar]
- 347.Shaham-Niv S, Rehak P, Zaguri D, Kolusheva S, Král P and Gazit E, Chem. Commun, 2018, 54, 4561–4564. [DOI] [PubMed] [Google Scholar]
- 348.Anand BG, Dubey K, Shekhawat DS and Kar K, Sci. Rep, 2017, 7, 11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Sade D, Shaham-Niv S, Arnon ZA, Tavassoly O and Gazit E, Open biology, 2018, 8, 170229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Laor D, Sade D, Shaham-Niv S, Zaguri D, Gartner M, Basavalingappa V, Raveh A, Pichinuk E, Engel H and Iwasaki K, Nat. Commun, 2019, 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Outeiro TF and Lindquist S, Science, 2003, 302, 1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Aizen R, Tao K, Rencus-Lazar S and Gazit E, J. Nanopart. Res, 2018, 20, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Guerin S, Tofail SAM and Thompson D, NPG Asia Materials, 2019, 11, 10. [Google Scholar]
- 354.Westermark GT, Fändrich M and Westermark P, Annu. Rev. Pathol. Mech, 2015, 10, 321–344. [DOI] [PubMed] [Google Scholar]
- 355.Pepys MB, Ann. Rev. Med, 2006, 57, 223–241. [DOI] [PubMed] [Google Scholar]
- 356.Merlini G, Dispenzieri A, Sanchorawala V, Schönland SO, Palladini G, Hawkins PN and Gertz MA, Nat. Rev. Dis. Primers, 2018, 4, 38. [DOI] [PubMed] [Google Scholar]
- 357.Suhr OB, Lundgren E and Westermark P, J. Intern. Med, 2017, 281, 337–347. [DOI] [PubMed] [Google Scholar]
- 358.Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG and Szarek WA, Nat. Med, 1995, 1, 143–148. [DOI] [PubMed] [Google Scholar]
- 359.Gellermann GP, Appel TR, Tannert A, Radestock A, Hortschansky P, Schroeckh V, Leisner C, Lütkepohl T, Shtrasburg S, Röcken C, Pras M, Linke RP, Diekmann S and Fändrich M, Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 6297–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Winter M, Tholey A, Kristen A and Röcken C, Proteomics, 2017, 17, 1700236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Swuec P, Lavatelli F, Tasaki M, Paissoni C, Rognoni P, Maritan M, Brambilla F, Milani P, Mauri P, Camilloni C, Palladini G, Merlini G, Ricagno S and Bolognesi M, Nat. Commun, 2019, 10, 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Schmidt M, Wiese S, Adak V, Engler J, Agarwal S, Fritz G, Westermark P, Zacharias M and Fändrich M, Nat. Commun, 2019, 10, 5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Radamaker L, Lin Y-H, Annamalai K, Huhn S, Hegenbart U, Schönland SO, Fritz G, Schmidt M and Fändrich M, Nat. Commun, 2019, 10, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Annamalai K, Gührs K-H, Koehler R, Schmidt M, Michel H, Loos C, Gaffney PM, Sigurdson CJ, Hegenbart U, Schönland S and Fändrich M, Angew. Chem. Int. Ed. Engl, 2016, 55, 4822–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Annamalai K, Liberta F, Vielberg M-T, Close W, Lilie H, Gührs K-H, Schierhorn A, Koehler R, Schmidt A, Haupt C, Hegenbart U, Schönland S, Schmidt M, Groll M and Fändrich M, Angew. Chem. Int. Ed. Engl, 2017, 56, 7510–7514. [DOI] [PubMed] [Google Scholar]
- 366.Gertz MA, Mauermann ML, Grogan M and Coelho T, Brain Behav, 2019, 9, e01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Bulawa CE, Connelly S, DeVit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW and Labaudinière R, Proc. Natl. Acad. Sci. U.S.A, 2012, 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Otzen D and Riek R, Cold Spring Harb. Perspect. Biol, 2019, 11, a033860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Hervas R, Rau MJ, Park Y, Zhang W, Murzin AG, Fitzpatrick JAJ, Scheres SHW and Si K, Science, 2020, 367, 1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Mohammad-Beigi H, Kjaer L, Eskandari H, Aliakbari F, Christiansen G, Ruvo G, Ward JL and Otzen DE, Front. Plant Sci, 2019, 10, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Riek R and Eisenberg DS, Nature, 2016, 539, 227–235. [DOI] [PubMed] [Google Scholar]
- 372.Wickner RB, Edskes HK, Shewmaker FP, Kryndushkin D, Nemecek J, McGlinchey R and Bateman D, Wiley Interdiscip. Rev. RNA, 2010, 1, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Shewmaker F, P McGlinchey R, Thurber K, McPhie P, Dyda F, Tycko R and B Wickner R, J. Biol. Chem, 2009, 284, 25065–25076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S and Hultgren SJ, Science, 2002, 295, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Blanco LP, Evans ML, Smith DR, Badtke MP and Chapman MR, Trends Microbiol, 2012, 20, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Evans ML and Chapman MR, Biochimica et biophysica acta, 2014, 1843, 1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Van Gerven N, Klein RD, Hultgren SJ and Remaut H, Trends Microbiol, 2015, 23, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Dueholm MS, Albertsen M, Otzen D and Nielsen PH, Plos One, 2012, 7, e51274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Barnhart MM and Chapman MR, Annu. Rev. Microbiol, 2006, 60, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Tursi SA and Tükel Ç, Microbiol. Mo.l Biol. Rev, 2018, 82, e00028–00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Kikuchi T, Mizunoe Y, Takade A, Naito S and Yoshida S. i., Microbiol. Immunol, 2005, 49, 875–884. [DOI] [PubMed] [Google Scholar]
- 382.DePas WH, Hufnagel DA, Lee JS, Blanco LP, Bernstein HC, Fisher ST, James GA, Stewart PS and Chapman MR, Proc. Natl. Acad. Sci. U.S.A, 2013, 110, 2629–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S and Hultgren SJ, Science, 2002, 295, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Buxbaum JN and Linke RP, J. Mol. Biol, 2012, 421, 142–159. [DOI] [PubMed] [Google Scholar]
- 385.Shu Q, Crick SL, Pinkner JS, Ford B, Hultgren SJ and Frieden C, Proc. Natl. Acad. Sci. U.S.A, 2012, 109, 6502–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Brandl MT, Carter MQ, Parker CT, Chapman MR, Huynh S and Zhou Y, Plos One, 2011, 6, e25553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Zhou Y, Smith D, Leong BJ, Brannstrom K, Almqvist F and Chapman MR, J. Biol. Chem, 2012, 287, 35092–35103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Nenninger AA, Robinson LS, Hammer ND, Epstein EA, Badtke MP, Hultgren SJ and Chapman MR, Mol. Microbiol, 2011, 81, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Epstein EA, Reizian MA and Chapman MR, J. Bacteriol, 2009, 191, 608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Goyal P, Krasteva PV, Van Gerven N, Gubellini F, Van den Broeck I, Troupiotis-Tsailaki A, Jonckheere W, Pehau-Arnaudet G, Pinkner JS, Chapman MR, Hultgren SJ, Howorka S, Fronzes R and Remaut H, Nature, 2014, 516, 250–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Robinson LS, Ashman EM, Hultgren SJ and Chapman MR, Mol. Microbiol, 2006, 59, 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Hammer ND, Wang X, McGuffie BA and Chapman MR, J. Alzheimers Dis, 2008, 13, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Collinson SK, Parker JMR, Hodges RS and Kay WW, J. Mol. Biol, 1999, 290, 741–756. [DOI] [PubMed] [Google Scholar]
- 394.DePas WH and Chapman MR, Res. Microbiol, 2012, 163, 592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Collinson SK, Emody L, Muller KH, Trust TJ and Kay WW, J. Bact, 1991, 173, 4773–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Cao B, Zhao Y, Kou Y, Ni D, Zhang XC and Huang Y, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, E5439–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Gibson DL, White AP, Rajotte CM and Kay WW, Microbiology, 2007, 153, 1131–1140. [DOI] [PubMed] [Google Scholar]
- 398.Shu Q, Krezel AM, Cusumano ZT, Pinkner JS, Klein R, Hultgren SJ and Frieden C, Proc. Natl. Acad. Sci. U.S.A, 2016, 113, 7130–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Schubeis T, Spehr J, Viereck J, Köpping L, Nagaraj M, Ahmed M and Ritter C, FEBS Lett, 2018, 592, 1020–1029. [DOI] [PubMed] [Google Scholar]
- 400.Larsen P, Dueholm M, Christiansen G, Nielsen JL, Otzen DE and Nielsen PH, Env. Microbiol, 2007, 9, 3077–3090. [DOI] [PubMed] [Google Scholar]
- 401.Christensen LFB, Schafer N, Wolf-Perez AM, Madsen DJ and Otzen DEO, in Biological and Bio-inspired Nanomaterials: Assembly Mechanisms and Properties, eds. Perrett S, Buell AK and Knowles TJ, Springer Press, 2019, vol. Chapter 4. [Google Scholar]
- 402.Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, Hein KL, Enghild JJ, Nielsen JL, Nielsen KL, Halkjær PH and Otzen DE, Mol. Microbiol, 2010, 77, 1009–1020. [DOI] [PubMed] [Google Scholar]
- 403.Rouse SL, Hawthorne WJ, Berry JL, Chorev DS, Ionescu SA, Lambert S, Stylianou F, Ewert W, Mackie U, Morgan RML, Otzen DE, Herbst FA, Nielsen PH, Dueholm M, Bayley H, Robinson CV, Hare S and Matthews S, Nat. Commun, 2017, 8, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Zeng G, Vad BS, Dueholm MS, Christiansen G, Nilsson M, Tolker-Nielsen T, Nielsen PH, Meyer RL and Otzen DE, Front. Microbiol, 2015, 6, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Rouse SL, Stylianou F, Wu HYG, Berry JL, Sewell L, Morgan RML, Sauerwein AC and Matthews S, J. Mol. Biol, 2018, 430, 3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Wang X, Hammer ND and Chapman MR, J. Biol. Chem, 2008, 283, 21530–21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Wang X, Smith DR, Jones JW and Chapman MR, J. Biol. Chem, 2007, 282, 3713–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Barnhart MM, Lynem J and Chapman MR, J. Bacteriol, 2006, 188, 5212–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Shewmaker F, McGlinchey RP, Thurber KR, McPhie P, Dyda F, Tycko R and Wickner RB, J. Biol. Chem, 2009, 284, 25065–25076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Tian P, Boomsma W, Wang Y, Otzen DE, Jensen MH and Lindorff-Larsen K, J. Am. Chem. Soc, 2015, 137, 22–25. [DOI] [PubMed] [Google Scholar]
- 411.Wang X, Zhou Y, Ren J-J, Hammer ND and Chapman MR, Proc. Natl. Acad. Sci. U.S.A, 2010, 107, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Wang X and Chapman MR, Prion, 2008, 2, 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Hammer ND, Schmidt JC and Chapman MR, Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 12494–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Rouse SL, Matthews SJ and Dueholm MS, J. Mol. Biol, 2018, 430, 3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Stenvang M, Dueholm MS, Vad BS, Seviour T, Zeng G, Geifman-Shochat S, Søndergaard MT, Christiansen G, Meyer RL, Kjelleberg S, Nielsen PH and Otzen DE, J. Biol. Chem, 2016, 291, 26540–26553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Najarzadeh Z, Mohammad-Beigi H, Pedersen JN, Christiansen G, Sønderby TV, Shojaosadati S, Morshedi D, Strømgaard K, Meisl G, Sutherland D, Pedersen JS and Otzen DE, Biomolecules., 2019, 9, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Lorenzen N, Nielsen SB, Buell AK, Kaspersen JD, Arosio P, Vad BS, Paslawski W, Christiansen G, Valnickova-Hansen Z, Andreasen M, Enghild JJ, Pedersen JS, Dobson CM, Knowles TJ and Otzen DE, J. Am. Chem. Soc, 2014, 136, 3859–3868. [DOI] [PubMed] [Google Scholar]
- 418.Paslawski W, Mysling S, Thomsen K, Jørgensen TJD and Otzen DE, Angew. Chem. Int. Ed. Engl, 2014, 53, 7560–7563. [DOI] [PubMed] [Google Scholar]
- 419.Andreasen M, Meisl G, Taylor JD, Michaels TCT, Otzen DE, Chapman M, Dobson CM, Matthews S and Knowles TPJ, mBio, 2018, 10, e02279–02218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Rasmussen C, Christiansen G, Vad BS, Enghild JJ, Lynggaard C, Andreasen M and Otzen DE, Prot. Sci, 2019, 28, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Christensen LFB, Nowak JS, Sønderby TV, Frank SA and Otzen DE, bioRxiv, 2020, DOI: 10.1101/2020.03.09.983882. [DOI] [Google Scholar]
- 422.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM and Stefani M, Nature, 2002, 416, 507–511. [DOI] [PubMed] [Google Scholar]
- 423.Evans ML, Chorell E, Taylor JD, Aden J, Gotheson A, Li F, Koch M, Sefer L, Matthews SJ, Wittung-Stafshede P, Almqvist F and Chapman MR, Mol. Cell, 2015, 57, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Taylor JD, Hawthorne WJ, Lo J, Dear A, Jain N, Meisl G, Andreasen M, Fletcher C, Koch M, Darvill N, Scull N, Escalera-Maurer A, Sefer L, Wenman R, Lambert S, Jean J, Xu Y, Turner B, Kazarian SG, Chapman MR, Bubeck D, de Simone A, Knowles TPJ and Matthews SJ, Sci. Rep, 2016, 6, 24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Jain N, Aden J, Nagamatsu K, Evans ML, Li X, McMichael B, Ivanova MI, Almqvist F, Buxbaum JN and Chapman MR, Proc. Natl. Acad. Sci. U.S.A, 2017, 114, 12184–12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Sivanathan V and Hochschild A, Nat. Protoc, 2013, 8, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Nguyen PQ, Botyanszki Z, Tay PK and Joshi NS, Nat. Commun, 2014, 5, 4945. [DOI] [PubMed] [Google Scholar]
- 428.Zhong C, Gurry T, Cheng AA, Downey J, Deng Z, Stultz CM and Lu TK, Nat. Nanotechnol, 2014, 9, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Knowles TPJ and Mezzenga R, Adv. Mater, 2016, 28, 6546–6561. [DOI] [PubMed] [Google Scholar]
- 430.Hauser CAE, Maurer-Stroh S and Martins IC, Chem. Soc. Rev, 2014, 43, 5326–5345. [DOI] [PubMed] [Google Scholar]
- 431.Li CX, Adamcik J and Mezzenga R, Nat. Nanotechnol, 2012, 7, 421–427. [DOI] [PubMed] [Google Scholar]
- 432.Li CX, Bolisetty S and Mezzenga R, Adv. Mater, 2013, 25, 3694–3700. [DOI] [PubMed] [Google Scholar]
- 433.Li CX, Born AK, Schweizer T, Zenobi-Wong M, Cerruti M and Mezzenga R, Adv. Mater, 2014, 26, 3207–3215. [DOI] [PubMed] [Google Scholar]
- 434.Knowles TPJ, Oppenheim TW, Buell AK, Chirgadze DY and Welland ME, Nat. Nanotechnol, 2010, 5, 204–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Aslund A, Herland A, Hammarstrom P, Nilsson KPR, Jonsson BH, Inganas O and Konradsson P, Bioconjug. Chem, 2007, 18, 1860–1868. [DOI] [PubMed] [Google Scholar]
- 436.Rizzo A, Solin N, Lindgren LJ, Andersson MR and Inganas O, Nano Lett, 2010, 10, 2225–2230. [DOI] [PubMed] [Google Scholar]
- 437.Herland A, Bjork P, Hania PR, Scheblykin IG and Inganas O, Small, 2007, 3, 318–325. [DOI] [PubMed] [Google Scholar]
- 438.Herland A, Bjork P, Nilsson KPR, Olsson JDM, Asberg P, Konradsson P, Hammarstrom P and Inganas O, Adv. Funct. Mater, 2005, 17, 1466–1471. [Google Scholar]
- 439.Nystrom G, Arcari M and Mezzenga R, Nat. Nanotechnol, 2018, 13, 330–336. [DOI] [PubMed] [Google Scholar]
- 440.Boothroyd S, Miller AF and Saiani A, Faraday Discuss, 2013, 166, 195–207. [DOI] [PubMed] [Google Scholar]
- 441.Gao J, Tang C, Elsawy MA, Smith AM, Miller AF and Saiani A, Biomacromolecules, 2017, 18, 826–834. [DOI] [PubMed] [Google Scholar]
- 442.Bolisetty S, Harnau L, Jung JM and Mezzenga R, Biomacromolecules, 2012, 13, 3241–3252. [DOI] [PubMed] [Google Scholar]
- 443.Jayawarna V, Ali M, Jowitt TA, Miller AE, Saiani A, Gough JE and Ulijn RV, Adv. Mater, 2006, 18, 611–620. [Google Scholar]
- 444.Bongiovanni MN and Gras SL, Biomaterials, 2015, 46, 105–116. [DOI] [PubMed] [Google Scholar]
- 445.Bongiovanni MN, Scanlon DB and Gras SL, Biomaterials, 2011, 32, 6099–6110. [DOI] [PubMed] [Google Scholar]
- 446.Gras SL, Tickler AK, Squires AM, Devlin GL, Horton MA, Dobson CM and MacPhee CE, Biomaterials, 2008, 29, 1553–1562. [DOI] [PubMed] [Google Scholar]
- 447.Reynolds NP, Charnley M, Mezzenga R and Hartley PG, Biomacromolecules, 2014, 15, 599–608. [DOI] [PubMed] [Google Scholar]
- 448.Reynolds NP, Styan KE, Easton CD, Li YL, Waddington L, Lara C, Forsythe JS, Mezzenga R, Hartley PG and Muir BW, Biomacromolecules, 2013, 14, 2305–2316. [DOI] [PubMed] [Google Scholar]
- 449.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA and Stupp SI, Science, 2004, 303, 1352–1355. [DOI] [PubMed] [Google Scholar]
- 450.Cao YP, Bolisetty S, Adamcik J and Mezzenga R, Phys. Rev. Lett, 2018, 120. [DOI] [PubMed] [Google Scholar]
- 451.Jacob RS, Ghosh D, Singh PK, Basu SK, Jha NN, Das S, Sukul PK, Patil S, Sathaye S, Kumar A, Chowdhury A, Malik S, Sen S and Maji SK, Biomaterials, 2015, 54, 97–105. [DOI] [PubMed] [Google Scholar]
- 452.Das S, Zhou K, Ghosh D, Jha NN, Singh PK, Jacob RS, Bernard CC, Finkelstein DI, Forsythe JS and Maji SK, NPG Asia Materials, 2016, 8, e304. [Google Scholar]
- 453.Li CX, Alam MM, Bolisetty S, Adamcik J and Mezzenga R, Chem. Commun, 2011, 47, 2913–2915. [DOI] [PubMed] [Google Scholar]
- 454.Chaves S, Pera LM, Avila CL, Romero CM, Baigori M, Vieyra FEM, Borsarelli CD and Chehin RN, RSC Adv, 2016, 6, 8528–8538. [Google Scholar]
- 455.Udomprasert A, Bongiovanni MN, Sha RJ, Sherman WB, Wang T, Arora PS, Canary JW, Gras SL and Seeman NC, Nat. Nanotechnol, 2014, 9, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Bolisetty S, Reinhold N, Zeder C, Orozco MN and Mezzenga R, Chem. Commun, 2017, 53, 5714–5717. [DOI] [PubMed] [Google Scholar]
- 457.Peydayesh M, Bolisetty S, Mohammadi T and Mezzenga R, Langmuir, 2019, 35, 4161–4170. [DOI] [PubMed] [Google Scholar]
- 458.Peydayesh M, Pauchard M, Bolisetty S, Stellacci F and Mezzenga R, Chem. Commun, 2019, 55, 11143–11146. [DOI] [PubMed] [Google Scholar]
- 459.Zhang QR, Bolisetty S, Cao YP, Handschin S, Adamcik J, Peng QM and Mezzenga R, Angew. Chem. Int. Ed. Engl, 2019, 58, 6012–6016. [DOI] [PubMed] [Google Scholar]
- 460.Bolisetty S, Arcari M, Adamcik J and Mezzenga R, Langmuir, 2015, 31, 13867–13873. [DOI] [PubMed] [Google Scholar]
- 461.Nystrom G, Fernandez-Ronco MP, Bolisetty S, Mazzotti M and Mezzenga R, Adv. Mater, 2016, 28, 472–484. [DOI] [PubMed] [Google Scholar]
- 462.Bolisetty S, Adamcik J, Heier J and Mezzenga R, Adv. Funct. Mater, 2012, 22, 3424–3428. [Google Scholar]
- 463.Li D, Furukawa H, Deng HX, Liu C, Yaghi OM and Eisenberg DS, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Li D, Jones EM, Sawaya MR, Furukawa H, Luo F, Ivanova M, Sievers SA, Wang WY, Yaghi OM, Liu C and Eisenberg DS, J. Am. Chem. Soc, 2014, 136, 18044–18051. [DOI] [PubMed] [Google Scholar]
- 465.Antzutkin ON, Leapman RD, Balbach JJ and Tycko R, Biochemistry, 2002, 41, 15436–15450. [DOI] [PubMed] [Google Scholar]
- 466.Sachse C, Xu C, Wieligmann K, Diekmann S, Grigorieff N and Fändrich M, J. Mol. Biol, 2006, 362, 347–354. [DOI] [PubMed] [Google Scholar]
- 467.Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, Fote GM, Lau L, Weinger JG, Lane TE, Inlay MA, Poon WW and Blurton-Jones M, Proc. Natl. Acad. Sci. U.S.A, 2016, 113, E1316–E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP and Sabbagh M, Neurobiol. Aging, 2006, 27, 1733–1739. [DOI] [PubMed] [Google Scholar]
- 469.Bessen RA and Marsh RF, J. Virol, 1994, 68, 7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Collinge J, Sidle KCL, Meads J, Ironside J and Hill AF, Nature, 1996, 383, 685–690. [DOI] [PubMed] [Google Scholar]
- 471.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE and Prusiner SB, Nat. Med, 1998, 4, 1157–1165. [DOI] [PubMed] [Google Scholar]
- 472.Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, Lissemore F, Shea M, Cohen Y, Chen W, Blevins J, Appleby BS, Surewicz K, Surewicz WK, Sajatovic M, Tatsuoka C, Zhang S, Mayo P, Butkiewicz M, Haines JL, Lerner AJ and Safar JG, Brain, 2015, 138, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC and Jucker M, J. Neurosci, 2011, 31, 14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Stöhr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, DeArmond SJ, Giles K, DeGrado WF and Prusiner SB, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 10329–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret J-M, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC and Jucker M, Science, 2006, 313, 1781–1784. [DOI] [PubMed] [Google Scholar]
- 476.Qiang W, Yau W-M, Lu J-X, Collinge J and Tycko R, Nature, 2017, 541, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Friedland RP and Chapman MR, PLoS Pathog, 2017, 13, e1006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Lundmark K, Westermark GT, Olsén A and Westermark P, Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 6098–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Wang M-M, Miao D, Cao X-P, Tan L and Tan L, Ann Transl Med, 2018, 6, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H and Attwell D, Science, 2019, 365, eaav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Eriksson O, Korsgren O, Selvaraju RK, Mollaret M, de Boysson Y, Chimienti F and Altai M, Acta Diabetol, 2018, 55, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Hampe CS, Wallen AR, Schlosser M, Ziegler M and Sweet IR, Exp. Clin. Endocrinol. Diabetes, 2005, 113, 381–387. [DOI] [PubMed] [Google Scholar]
- 483.Chau Y-FC, Wang C-K, Shen L, Lim CM, Chiang H-P, Chao C-TC, Huang HJ, Lin C-T, Kumara NTRN and Voo NY, Sci. Rep, 2017, 7, 16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Kakinen A, Sun Y, Javed I, Faridi A, Pilkington EH, Faridi P, Purcell AW, Zhou R, Ding F, Lin S, Ke PC and Davis TP, Sci. Bull, 2019, 64, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Ankarcrona M, Winblad B, Monteiro C, Fearns C, Powers ET, Johansson J, Westermark GT, Presto J, Ericzon BG and Kelly JW, J. Intern. Med, 2016, 280, 177–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Falk RH, Alexander KM, Liao R and Dorbala S, J. Am. Coll. Cardiol, 2016, 68, 1323–1341. [DOI] [PubMed] [Google Scholar]
- 487.Adams D, Koike H, Slama M and Coelho T, Nat. Rev. Neurol, 2019, 15, 387–404. [DOI] [PubMed] [Google Scholar]
- 488.Latza V, Guerette PA, Ding D, Amini S, Kumar A, Schmidt I, Keating S, Oxman N, Weaver JC, Fratzl P, Miserez A and Masic A, Nat. Commun, 2015, 6, 8313–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]