

Abstract
Colicins are antimicrobial proteins produced by Escherichia coli, which, upon secretion from the host, kill non-host E. coli strains by forming pores in the inner membrane and degrading internal cellular components such as DNA and RNA. Due to their unique cell-killing activities, colicins are considered viable alternatives to conventional antibiotics. Recombinant production of colicins requires co-production of immunity proteins to protect host cells; otherwise, the colicins are lethal to the host. In this study, we used cell-free protein synthesis (CFPS) to produce active colicins without the need for protein purification and co-production of immunity proteins. Cell-free synthesized colicins were active in killing model E. coli cells with different modes of cytotoxicity. Pore-forming colicins E1 and nuclease colicin E2 killed actively growing cells in a nutrient-rich medium, but the cytotoxicity of colicin Ia was low compared to E1 and E2. Moreover, colicin E1 effectively killed cells in a nutrient-free solution, while the activity of E2 was decreased compared to nutrient-rich conditions. Both colicins E1 and E2 decreased the level of persister cells (metabolically dormant cell populations that are insensitive to antibiotics) by up to six orders of magnitude compared to that of the rifampin pretreated persister cells. This study finds that colicins can eradicate non-growing cells including persisters, and that CFPS is a promising platform for rapid production and characterization of toxic proteins.
Keywords: colicins, cell-free protein synthesis, antimicrobials, persister cells
1. Introduction
Colicins are a type of bacteriocin produced by Escherichia coli which kill non-host E. coli strains by forming pores in the inner membrane, inhibiting cell-wall synthesis and degrading nucleic acids (1). All colicins must bind to and cross the outer membrane of target cells, and some colicins must cross the inner membrane, depending on their mode of cytotoxicity (2). For transport, colicins use nutrient transporters located at the outer membrane and a group of inner membrane and periplasmic proteins. Colicins consist of three functional domains, including a central receptor binding domain, N-terminal translocation domain and C-terminal cytotoxicity domain. The central receptor binding domain helps colicins to recognize target cells by binding to a cell surface receptor protein with high affinity, the N-terminal translocation domain enables the colicins to cross the outer membrane into or through the periplasm and the C-terminal cytotoxicity domain disrupts the target components via pore formation or enzymatic degradation (3). Colicin transport into cells requires energy during release from the outer membrane receptor and transfer of the colicin polypeptide across the outer membrane through the translocator. Additionally, nuclease colicins require energy during extracellular release of their bound immunity protein (2). Such energy is supplied via TolAQR (for Group A colicin) or TonB/ExbB/ExbD (for Group B colicin) proteins which are responsible for maintaining membrane integrity and for transducing energy from the inner membrane to the outer membrane for substrate uptake into the E. coli cell (2).
Upon colicin release by cell lysis induced by the cellular stress response, the colicin invades the target cell by binding to the receptor protein of the cell, resulting in target cell killing. During growth, the colicin-producing host cells are protected by an immunity protein that blocks the activity of its cognate colicin (4). The immunity proteins of nuclease colicins form a complex with colicins by binding to their cytotoxicity domains, neutralizing their catalytic activity. The immunity proteins of pore-forming colicins localize in the inner membrane, disabling pore formation by the colicin. Over 20 colicins have been studied and the functional diversity of colicins offers unique cell-killing mechanisms (1). For example, colicins Ia (5) and E1 (6) form pores on the inner membrane, colicin M inhibits cell-wall biosynthesis by degrading an undecaprenyl phosphate-linked peptidoglycan precursor (7), colicin E2 degrades DNA templates (8) and colicin E3 degrades 16S ribosomal RNA resulting in slow translation (9). Therefore, colicins can target specific microbes by penetrating cells and disrupting cellular components.
Colicins are considered viable alternatives to antibiotics (10) for several compelling reasons. First, the cell-killing activities of colicins that physically disrupt cellular components are distinct from most conventional antibiotics that disable bacterial growth by inhibiting protein synthesis (e.g. kanamycin), cell-wall biosynthesis (e.g. penicillin) and DNA replication/repair (e.g. ciprofloxacin) (11). Colicins may have the ability to kill non-growing cells known as persisters that can survive under antibiotic treatments by entering a metabolically dormant state (12). Second, colicins may not be toxic to humans because their cytotoxicity is only effective on bacteria that produce receptor proteins such as BtuB, Cir, FhuA and OmpF (1), which are not present in human cells. This hypothesis is supported by reports that some colicins including E1, E3 and E7 demonstrate little toxicity to mammalian cells (13). Third, the cell-killing kinetics of colicins are fast, which may eradicate harmful bacteria during early growth stage (6, 14), preventing the formation of persistent or resistant bacteria. Lastly, multiple domains of colicins provide flexibility to engineer new types of colicins by combining them with other bacteriocins. For example, chimeric forms of bacteriocins have been synthesized by exchanging domains between colicins (E2 and E3) of E. coli and pyocins (S1 and S2) of Pseudomonas aeruginosa (15). Hence, further engineering of colicins may provide new proteins with novel functional and structural properties that can be applied to control bacterial infections.
Cell-free protein synthesis (CFPS) is a promising platform for flexible expression and screening of enzymes (16). CFPS has been used to construct antimicrobial peptide libraries (17), characterize enterocins L50A and L50B (18), and identify colicin E1 production from Col E1 plasmid DNA (19, 20). CFPS was also applied to produce toxic proteins such as the cancer therapeutic onconase (21). Compared to the live-cell systems, CFPS is a more flexible and controllable experimentation platform, providing several important advantages for researchers (22). First, the open reaction environment brings an unprecedented level of control and freedom of design for a newly synthesized protein (23). Second, cell-free system can obviate the need for time-consuming cloning steps by using linear DNA templates generated by polymerase chain reaction (PCR) (24–26). Third, cell-free systems can produce toxic proteins without cell viability restraints (27). As a rapid protein prototyping method, CFPS can use small-scale batch reactions; however, CFPS has also been shown to scale linearly over six orders of magnitude to provide a cost-effective, industrial protein production platform (28). Typically, bacterial extract gives higher protein yield than eukaryotic cell extract, but has limitations in the production of active and soluble mammalian therapeutics such as antibodies (29). To address these limitations while utilizing the advantages of bacterial cell-free systems, additional bacterial CFPS technologies have been developed that enhance protein folding (28) and introduce post-translational modifications (30), thereby enabling the synthesis of active therapeutic proteins.
In this study, we used E. coli-based CFPS systems for rapid synthesis and characterization of colicins. We examined the cytotoxicity of the cell-free synthesized colicins M, Ia, E1 and E2 with different mechanisms on a model E. coli K361 strain and investigated the activity of colicins in killing non-growing bacterial populations including persister cells. We report, for the first time, that colicins E1 and E2 effectively eradicate persister cells. This study further demonstrates the feasibility of CFPS systems for toxic protein synthesis and characterization and provides a new platform for developing colicins as next-generation antimicrobials.
2. Materials and methods
2.1 Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table 1. We used E. coli K361 strain as an indicator for colicin cell-killing activity (31) and BL21(DE3) Star strain for making cell extracts for CFPS. Kanamycin (50 µg/ml), ampicillin (100 µg/ml) and streptomycin (100 µg/ml) were used to maintain plasmids as necessary.
Table 1.
Strains and plasmids used in this study. StrR, KmR, AmpR are streptomycin, kanamycin, ampicillin resistance, respectively
| Strains and plasmids | Genotype/relevant characteristics | Source |
|---|---|---|
| Strains | ||
| E. coli K361 | Wild type W3110 strain with StrR | (31) |
| E. coli BL21(DE3) Star | F-ompT, hsdSB(), gal, dcm, rne131 (DE3) | Invitrogen |
| E. coli K964 | K91 containing pColE2 | (52) |
| E. coli TG1 | Strain containing colicin plasmid | (6) |
| Plasmids | ||
| pJL1-sfGFP | KmR, PT7::sfGFP, C-terminal Strep-tag, pY71-sfGFP | (53) |
| pJL1-cma | KmR, PT7::cma encoding colicin M | This study |
| pCC1-cia | AmpR, PT7::cia encoding colicin Ia | This study |
| pJL1-imm | KmR, PT7::cma encoding E2 immunity | This study |
| pSL101 | StrR, containing luxABCDE operon and axe-txe cassette | (54) |
| pKSJ331 | AmpR, ColE1 operon | (6) |
| pKSJ340 | AmpR, ColIa operon | (5) |
2.2 Crude extract preparation
Escherichia coli cell extract was prepared from BL21(DE3) Star strain using a high-throughput sonication method (32). Overnight culture was diluted 1000 times with 1.0 L of 2xYTPG media (16 g/l tryptone, 10 g/l yeast extract, 5 g/l NaCl, 7 g/l K2HPO4, 3 g/l KH2PO4 and 18 g/l glucose; adjusted pH to 7.2 with KOH) in a 2.5 L of Tunair flask. Cells were grown to a turbidity at 600 nm (OD600nm) of 0.5 in 1.0 L of 2xYTPG media at 37°C 220 rpm, and T7 RNA polymerase production was induced by adding 1 mM of isopropyl β-d-1-thiogalactopyranoside. The cells were further grown until OD600nm of 3.0. Cells were pelleted by centrifuging for 15 min at 5000×g at 4°C, washed twice with cold S30 buffer [10 mM tris-acetate pH 8.2, 14 mM magnesium acetate, 60 mM potassium acetate and 1 mM dithiothreitol (DTT)] and stored at −80°C. Thawed cells were suspended in 1 ml of S30 buffer per gram of cells and lysed on ice using a Q125 sonicator (Qsonica, Newtown, CT, USA) with 45 s pulses and 60 s intervals three times at 50% amplitude setting. Cell debris and insoluble components were removed by two rounds of centrifugation for 10 min at 14 000×g at 4°C. In order to completely remove the unlysed remaining cells, the crude extract was filtered by a 0.2 µm sterile syringe filter (Corning, NY, USA). The total protein concentration of the extracts was approximately 50 mg/ml, as measured by Quick-Start Bradford protein assay kit (Bio-Rad, Hercules, CA, USA). The ‘cell-free’ crude extract was stored at −80°C until use.
2.3 Gibson assembly
Gibson assembly (33) was used to construct a plasmid containing a gene cma encoding colicin M. The DNA sequence of cma was synthesized by Integrated DNA Technologies (Coralville, IA, USA) and amplified using GAcol-F and GAcol-R primers (Supplementary Table S1), and the pJL1 vector was amplified from pJL1-sfGFP using pJL1-F and pJL1-R primers (Supplementary Table S1). The plasmid pJL1-cma was confirmed by DNA sequencing using T7-Pro and T7-Ter primers (Supplementary Table S1).
2.4 Linear DNA template amplification of colicin genes
Colicin genes were amplified by PCR. The cma gene encoding colicin M was amplified from pJL1-cma plasmid using GAcol-F and GAcol-R, cia encoding colicin Ia was amplified from pKSJ340 plasmid using ColIa-F and ColIa-R, cea encoding colicin E1 was amplified from pKSJ331 plasmid using ColE1-F and ColE1-R and col encoding colicin E2 was amplified from K964 using ColE2-F and ColE2-R. PCR was performed using Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA) at 98°C for 30 s, with 30 cycles of 98°C for 10 s, 55 or 60°C for 30 s and 72°C for 2 min 30 s, and a final extension at 72°C for 10 min. Three rounds of PCR were performed to insert T7 promotor and T7 terminator sequences for genes of colicin M, Ia, E1 and E2. The first PCR was used to amplify the colicin genes with the addition of a Strep-tag (Trp-Ser-His-Pro-Gln-Phe-Glu-Lys) at the C-terminus. The second PCR was used to add the T7 promotor sequence and some overlapping portion before the T7 terminator. The third PCR added the T7 terminator sequence. All the primers used in this study are listed in Supplementary Table S1. DNA sequences of all colicin genes are listed in Supplementary Table S2. E.Z.N.A. Cycle Pure kit (Omega Bio-Tek, Norcross, GA, USA) was used to purify the PCR products before adding to CFPS.
2.5 E2 immunity protein preparation
E2 immunity gene imm was amplified from K964 using E2Imm-F and E2Imm-R containing a Strep-tag at the C-terminus and NdeI and SalI restriction enzyme sites. The PCR fragment was double digested by NdeI and SalI and ligated into pJL1-backbone at the same restriction sites. The ligation mix was electroporated into BL21(DE3) Star competent cell. To produce E2 immunity protein, overnight culture of E. coli BL21(DE3) Star/pJL1-imm was diluted 100 times in 250 ml of Luria-Bertani (LB) media with 50 µg/ml of kanamycin in 1 L flask and incubated at 37°C at 220 rpm until OD600nm ∼ 0.5. Then, 1 mM of isopropyl β-d-1-thiogalactopyranoside was added to induce E2 immunity protein production and incubated at 37°C at 220 rpm for another 5 h. E2 immunity protein was purified by a Strep-Tactin column by following the manufacturer’s suggested protocol (IBA, Göttingen, Germany). The concentration of the purified E2 immunity protein was 520 µg/ml, measured by Quick-Start Bradford protein assay kit (Bio-Rad, Hercules, CA, USA).
2.6 Colicin E2 nuclease activity assay with E2 immunity protein
Cell-free reaction product containing colicin E2 or no colicin (5 µl) was pre-incubated at 37°C for 30 min with and without E2 immunity protein (70 µg/ml). Upon addition of pSL101 plasmid (13 277 bp, 1000 ng) (Table 1) into each reaction tube, the mixture (15 µl reaction volume) was incubated at 37°C for 3 h in a 15 µl reaction. Then, DNA in the reaction mixture was purified with E.Z.N.A. Cycle Pure kit (Omega Bio-Tek, Norcross, GA, USA) and loaded on 1% agarose gel to check the amount of DNA degradation.
2.7 CFPS reaction
CFPS reactions were performed to produce colicins. Fifteen microlitre of CFPS reaction in a 1.5 ml microcentrifuge tube was prepared by mixing the following components: 1.2 mM ATP; 0.85 mM each of GTP, UTP and CTP; 34.0 µg/ml folinic acid; 200 ng plasmid or linear DNA template; 100 µg/ml T7 RNA polymerase; 2 mM each of 20 standard amino acids; 0.33 mM nicotinamide adenine dinucleotide (NAD); 0.27 mM coenzyme-A (CoA); 1.5 mM spermidine; 1 mM putrescine; 4 mM sodium oxalate; 130 mM potassium glutamate; 10 mM ammonium glutamate; 12 mM magnesium glutamate; 33 mM phosphoenolpyruvate (PEP); 4 µl of ‘cell-free’ extract. The sample was incubated for 20 h at 30°C.
2.8 Radioactive 14C-Leu assay
To quantify colicins, total and soluble protein yields were measured by determining radioactive 14C-Leu incorporation (34). Briefly, triplicate CFPS reactions were supplemented with 10 µM 14C-leucine (Perkin Elmer, Waltham, MA, USA) and incubated at 30°C for specified periods of time. When the reaction was complete, proteins were precipitated and washed three times with 5% trichloroacetic acid (TCA), washed in 100% ethanol, and quantified using a Microbeta2 liquid scintillation counter (Perkin Elmer, Waltham, MA, USA). Soluble fractions were taken after centrifugation at 12 000×g for 15 min at 4°C. Background radioactivity and protein synthesis from a no plasmid control has been subtracted.
2.9 Autoradiogram
CFPS reactions were supplemented with 10 µM of radioactive 14C-Leu. Three microlitre of the soluble fraction of each reaction was loaded on a 4–12% NuPAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel in 3-(N-morpholino) propanesulfonic acid (MOPS) buffer (Invitrogen, Carlsbad, CA, USA) after heat-denaturation and reduction by 1,4-dithiothreitol (DTT). The gel was stained using InstantBlue Coomassie stain (Expedeon, San Diego, CA, USA) for 1 h, destained overnight, soaked in Gel Drying Solution (Bio-Rad, Hercules, CA, USA) for 30 min, fixed with cellophane films, dried overnight in GelAir Dryer (Bio-Rad, Hercules, CA, USA) and exposed for 72 h on Storage Phosphor Screen (GE Healthcare Biosciences, Pittsburgh, PA, USA). The autoradiogram was scanned using a Typhoon FLA7000 Imager (GE Healthcare Biosciences, Pittsburgh, PA, USA).
2.10 Cell viability test
Overnight culture of K361 strain grown in LB at 220 rpm at 37°C was diluted 100 times in fresh LB medium and kept incubating at 220 rpm at 37°C until OD600nm of 0.7–0.9. Cells were pelleted by centrifuging at 14 000×g at room temperature and washed twice with 0.85% NaCl solution and adjusted to OD600nm 0.01 (equivalent to 5.0 × 106 CFU/ml) or 1.0 (equivalent to 5.0 × 108 CFU/ml) with LB medium or 0.85% NaCl solution. Adjusted culture solution was aliquoted 1.2 ml into 14 ml culture tubes and incubated with the addition of cell-free products containing colicins (750 ng/ml) at 220 rpm at 37°C for 1 h. The samples were washed with 1 ml of 0.85% NaCl solution twice, diluted serially (101–106 dilution) and dropped 10 µl each on LB agar plate to quantify cell viability.
To determine the activity of each colicin, we treated cells at high initial population (5.0 × 108 CFU/ml) with different concentrations (2, 4, 8, 16, 32, 64, 128, 256, 512 and 1024 ng/ml) of each colicin for 3 min and 1 h. We quantified colicin cytotoxicity by calculating effective multiplicities (m). Effective multiplicity indicates the exponential loss of surviving cell populations (S) relative to populations of untreated control cells (S0) as a function of added colicin (i.e. S/S0 = e−m) (35).
2.11 Persister assay
Persister levels were determined by counting colony forming units grown on LB agar plates after exposing cells to antibiotics, washing and serial dilution (36). To determine the number of surviving persister cells, overnight culture of K361 strain grown in LB at 220 rpm at 37°C was diluted 100 times in fresh LB medium and incubated at 220 rpm at 37°C until OD600nm 0.7–0.9 for exponential phase or 3.0 for stationary phase. Cells were pelleted by centrifuging at 14 000×g at room temperature, washed twice with 0.85% NaCl solution and adjusted to OD600nm of 1.0 (equivalent to 5.0 × 108 CFU/ml) with LB medium containing 5 µg/ml of ciprofloxacin. Density-adjusted culture was aliquoted into 1.2 ml fractions in 14 ml culture tubes, exposed to 750 ng/ml of different colicins, and then incubated at 37°C at 220 rpm for 3 h. The samples were washed with 1 ml of 0.85% NaCl solution twice, diluted serially (101–104 dilution) and added 10 µl each onto LB agar plates to quantify persister cell viability.
For rifampin pretreatment, adjusted exponential phase cells were pretreated with 100 µg/ml of rifampin and washed twice with 0.85% NaCl solution. Cell culture was resuspended with LB medium containing 5 µg/ml of ciprofloxacin, exposed to 750 ng/ml of different colicins and incubated at 37°C at 220 rpm for 3 h. The samples were washed with 1 ml of 0.85% NaCl solution twice, diluted serially and added 10 µl each onto LB agar plate to quantify persister cell viability.
2.12 Statistical analysis
Statistical significance was assessed by using two-tailed t-tests between samples and no colicin controls. The null hypothesis stated that the means of the number of cells which survived colicin treatment are equal to the means of the number of untreated cells. If the P-value calculated from two-tailed t-tests is less than 0.05, then the null hypothesis has failed, indicating that the observed reduction in cell survival following colicin treatment is statistically significant. Statistical significance is indicated by asterisks in figures. All reported data are average ± standard deviation.
3. Results and discussions
3.1 Preparation of ‘cell-free’ extract
Cell lysate was prepared from a culture of E. coli BL21(DE3) Star strain using a sonication method (32). Although cell debris and most intact cells were removed via centrifugation during the extract preparation, 106 cells per ml of the extract remained. Since the presence of the unlysed cells may interfere with the assessment of colicin activity by cell viability, we eliminated the unlysed live cells from the extract by a syringe filtration and confirmed the complete removal of the cells in the extract (Supplementary Figure S1A). The ‘cell-free’ filtered extract exhibited similar protein synthesis activity compared to the unfiltered extract as assessed by the fluorescence intensity of green fluorescent protein (Supplementary Figure S1B). We used this filtered ‘cell-free’ extract to synthesize colicins.
3.2 Colicins synthesized via CFPS
We produced different types of colicins with various functions including peptidoglycan biosynthesis-inhibiting colicin M (7), pore-forming colicins Ia (5) and E1 (6) and nuclease colicin E2 (8). Although in vivo production requires co-production of immunity proteins to protect the host cells from colicin activity (1), CFPS is not limited by cell viability constraints (27, 28), allowing us to produce colicins at high titers without immunity proteins. Colicin genes were amplified using PCR from colicin plasmids. 5′- and 3′-untranslated regions that include T7 promoter and terminator sequences were added during the PCR amplification. We conducted the CFPS reactions at 30°C for 20 h by mixing PCR products with the ‘cell-free’ extract and CFPS reagents. Radioactive 14C-Leu incorporation showed that colicins Ia, E1 and E2 were almost 100% soluble with yields around 300 µg/ml (Figure 1A and Table 2). The yield of soluble colicins using CFPS was 10-fold higher compared to in vivo colicin production which is typically about 24–28 µg/ml (37). The majority of colicins produced using CFPS were full-length protein on the autoradiogram gel (Figure 1B). However, only 5% of the colicin M protein was soluble (Figure 1A and Table 2). Furthermore, only the soluble M was active in killing cells (Supplementary Figure S2). The observed low solubility of colicin M may explain the low yield of colicin M from living cells (5 mg/l) (7) compared to the other colicins (24–28 mg/l) (37), as only the soluble M might be purified from cells.
Figure 1.

Cell-free colicin production. (A) Total and soluble protein yield of cell-free produced colicins quantified by 14C-Leu scintillation counting. Error bars indicate standard deviations from three independent CFPS samples. (B) Autoradiogram gel of soluble colicins made in CFPS. (C) Time course of soluble colicin Ia production using PCR product (40 ng and 200 ng) and plasmid (200 ng) DNA templates.
Table 2.
Total and soluble colicin concentration
| Colicin | Total protein (µg/ml) | Soluble protein |
|---|---|---|
| M | 510 ± 70 | 30 ± 10 |
| Ia | 370 ± 10 | 350 ± 20 |
| E1 | 295 ± 8 | 300 ± 30 |
| E2 | 250 ± 20 | 260 ± 10 |
Total and soluble proteins of cell-free synthesized native and fusion colicins were quantified by radioactive 14C-Leu scintillation counting.
We chose to use linear DNA templates of colicin genes for more rapid colicin production compared to time-consuming cloning (26). To see if colicin production from the linear DNA template is comparable to colicin production from the plasmid template, we synthesized colicin Ia by adding plasmid and different amounts of linear DNA template in 15 µl cell-free reactions and then monitored colicin Ia production over time using 14C-Leu scintillation counting. Linear DNA template is susceptible to degradation during the cell-free reaction compared to the circular DNA template as reported previously (26). As expected, when the same copy number of templates was used [40 ng of Ia gene PCR product (2091 bp) compared to 200 ng of pCC1-cia plasmid (10 104 bp)], Ia production from the PCR template was only 75% of Ia yield from the plasmid template (Figure 1C). However, we could enhance the Ia yield to almost similar levels to the plasmid-templated Ia production by increasing the amount of PCR templates (200 ng of Ia gene PCR product compared to 200 ng of pCC1-cia plasmid) (Figure 1C). Moreover, we observed that most protein synthesis was achieved at early time points (3.5 h) and maintained over the time course (Figure 1C).
3.3 Nuclease colicin E2 is produced by CFPS without immunity protein
To avoid host-cell toxicity, a colicin must be blocked by the binding of a cognate immunity protein to the cytotoxicity domain (1). For example, colicin E2 functions as a DNase and the E2 immunity protein is coproduced in the same operon in vivo. We produced 260 ± 10 µg/ml of E2 using CFPS in the absence of E2 immunity protein (Figure 1A), but it is possible that E2 production might be inhibited by degrading its own DNA template during the cell-free reaction. To see if colicin E2 production is enhanced by the presence of an immunity protein, we added purified E2 immunity protein into the CFPS reaction so that the cytotoxicity domain of colicin E2 is immediately blocked by the E2 immunity protein upon synthesis, thereby protecting the E2 colicin DNA template. We confirmed that the purified E2 immunity protein was active by observing protection of the pSL101 plasmid DNA from degradation by colicin E2 (Figure 2A). Then, we added the purified E2 immunity protein at the start of the E2 colicin cell-free synthesis reaction. We added equimolar concentrations of the E2 immunity protein (10 kDa) to the anticipated yield of the E2 colicin (61 kDa) (Table 2) but did not observe an increase in cell-free E2 colicin production (Figure 2B). Further increase of E2 immunity protein (three times higher) adversely affected the yield of E2 colicin (Figure 2B). This result may be explained by an excess of DNA added as shown in Ia production (Figure 1C) or that transcription mediated by T7 polymerase may be fast enough to produce enough mRNA for translation (38) before the DNA template is degraded. These results show that nuclease E2 colicin can be produced using CFPS in the absence of the immunity protein without concern for DNA template degradation by the synthesized colicin E2.
Figure 2.

Effect of E2 immunity protein on DNA degradation and cell-free E2 production. (A) DNA degradation was assessed by mixing the plasmid DNA pSL101 (1000 ng) with cell-free product containing no colicin (lane 1), colicin E2 (lane 2) and colicin E2 and Immunity protein E2 (lane 3) after incubation at 37°C for 3 h. Purified DNA from these reactions was loaded on a 1% agarose gel. (B) Yield of cell-free synthesized colicin E2 with different molar ratios of E2 immunity protein (E2:Imm = 1:1 or 1:3).
3.4 Cell-free synthesized colicins are active in cell killing
We investigated the cell-killing activity of cell-free synthesized colicins without purification. First, we performed cell viability tests using various concentrations of colicin E1 to determine its optimal concentration. A culture of E. coli K361 strain was incubated to reach exponential growth phase with a turbidity (OD600nm) between 0.7 and 0.9, centrifuged, and resuspended in fresh LB to adjust cell populations to approximately 5.0 × 106 CFU/ml. The diluted cells were exposed to colicin E1 at 37°C for 1 h with shaking at 220 rpm. The surviving cells were quantified by serial dilution followed by plating on LB agar. We did not detect any surviving cells after exposure to colicin E1 at concentrations of 250 ng/ml or higher (Figure 3A). It was previously reported that colicins rapidly deactivate more than 99% of target cells within 1 min (39). Corroborating this previous study, we observed that cell-free synthesized E1 (750 ng/ml) inactivated cells over 99.9% with just a 1-min exposure (Figure 3B), and that the cell culture treated with E1 did not show growth but remained transparent as an evidence of killing (Figure 3C). However, the cultures without colicin treatment increased in cell population (Figure 3B) and the turbidity (Figure 3C). Along with the colicin E1 activity, we also observed substantial cell death in samples exposed to colicins Ia and E2 in LB medium (Figure 3D). These results indicate that cell-free synthesized colicins without purification are active and rapidly inactivate cells in a rich medium.
Figure 3.

Activity of cell-free synthesized colicins. K361 cells were exposed to cell-free synthesized colicins, no added colicin plasmid was assessed as a control (No Col). (A) K361 cells (initial cell density 5 × 106 CFU/ml) where treated with various concentrations of colicin E1 for 60 min at 37°C at 220 rpm. (B) Time course cell viability upon the addition of colicin E1 (750 ng/ml). (C) Cell growth of K361 culture (initial cell density 5 × 107 CFU/ml) with colicin E1 (750 ng/ml) treatment. The inset picture shows K361 culture after a 60 min exposure of cells to E1. Cell-killing activities of colicins Ia, E1 and E2 (750 ng/ml) at (D) low initial cell density (5 × 106 CFU/ml) and (E) high initial cell density (5 × 108 CFU/ml) during growth in LB medium and under a nutrient-free, non-growing condition in 0.85% NaCl solution. Error bars indicate standard deviations from two independent cultures with three plating replicates each. *Represents significant difference compared to No Col samples under the same media conditions with P-value <0.05. ND indicates ‘not detected’.
3.5 Comparison of activity of colicins in between nutrient-rich and nutrient-free conditions
Because mechanisms of colicin cell killing physically damage the inner membrane or cleave nucleic acids of target cells, we assessed the activities of colicins toward cells under non-growing, nutrient-free conditions to determine if colicins can provide an advantage over antibiotics, most of which can only kill growing cells. To inhibit cell growth, we washed and resuspended cells in a 0.85% NaCl solution. We confirmed that cells do not grow in 0.85% NaCl solution that does not contain nutrients (Supplementary Figure S3). Then, the cells were exposed to colicins under the same condition tested above except that LB medium was replaced with 0.85% NaCl. Contrary to its behavior in nutrient-rich LB medium, pore-forming colicin Ia barely reduced cell viability in 0.85% NaCl (∼3-fold) (Figure 3D). Interestingly, E1 (another pore-forming colicin) killed nearly all cells in the nutrient-free condition, similar to the E1 activity observed in the nutrient-rich condition (Figure 3D). Also, colicin E2 which has nuclease activity (8) killed nearly all of the cells in 0.85% NaCl solution (Figure 3D). Because we were unable to detect any surviving cells when colicins were added to K361 cultures with 5 × 106 CFU/ml, we were not able to compare the activity of colicins in the LB and 0.85% NaCl conditions. Therefore, we increased initial cell density to 5 × 108 CFU/ml and assessed the cell viability upon colicin treatment for 1 h in LB and 0.85% NaCl solution. We then calculated the effective multiplicity (m) which provides a quantitative measure of colicin activity using the relative ratio of surviving cells after colicin treatment to untreated cells (35). Contrary to the case of low initial cell density (Figure 3D), colicin Ia only slightly reduced cell viability with a multiplicity of 1.7 at 750 ng/ml (Table 3) in nutrient-rich LB medium and showed negligible inhibitory behavior (m = 1.1) under nutrient-free conditions at high initial cell density (Figure 3E). Colicins E1 and E2 decreased the cell viability by five orders of magnitude in LB (Figure 3E) with multiplicities of 13.1 and 12.2, respectively (Table 3). E1 and E2 did not show complete cell killing when the initial cell population was high, which is consistent with the previous observation that a small portion of the cells survived E1 treatment even at a very high concentration of this colicin (10 µg/ml) (40). In 0.85% NaCl solution, E1 resulted in a similar level of cell killing (m = 12.4) as in LB, but the E2 cell-killing activity was significantly attenuated by showing only 60-fold cell killing compared to the untreated cells (m = 4.0). We also observed a slight decrease in the cytotoxicity of E1 and Ia in 0.85% NaCl (Table 3, Figure 3E). The reduction of colicin cytotoxicity in 0.85% NaCl solution may be due to the lack of energy source in the solution because the initiation of colicin action requires energy (2). The amount of energy requirement may be different depending on the type of colicins and energy supply. For example, anaerobic conditions lower the cell-killing rate of colicins compared to aerobic conditions (41). Furthermore, starved cells reincubated in the presence of energy sources such as glucose and d-lactate initiated cell killing by colicins but exhibited different levels of cytotoxicity for colicins K, E2 and E3 (41). It was also reported that colicin E1 requires a small amount of cellular energy to initiate its transmission response (42). Our results confirm that colicin cytotoxicity is dependent on the metabolic state of the target cells, most likely because the uptake requires energy. Cell-killing activity may vary between types of colicins and may be reduced if target cells are not sufficiently energized by a nutrient supply.
Table 3.
Effective multiplicity of different colicins under growing (LB) and non-growing (0.85% NaCl) conditions
| Colicins | LB | 0.85% NaCl |
|---|---|---|
| No Col | 0 | 0 |
| Ia | 1.7 | 1.1 |
| E1 | 13.1 | 12.4 |
| E2 | 12.2 | 4.0 |
Effective multiplicity (m) was calculated based on the equation shown in Section 2 for quantitative measurement of colicin activity using cell-killing data shown in Figure 3E.
3.6 Comparison of cytotoxicity of different colicins at different concentrations
To compare cytotoxicity between cell-free synthesized colicins, we assessed cell viability upon colicin treatment with varying concentrations. First, we exposed K361 cells (5 × 108 CFU/ml) to colicins for 3 min (Figure 4A). We used a 3 min time-frame because colicins are known to rapidly deactivate the target cells (39) and previous studies have assessed the cytotoxicity of colicin E1 just 5 min after treating target cells (43). Greater colicin E1 concentrations led to increased cell killing until maximum cell killing was reached at concentrations above 128 ng/ml (Figure 4A) when multiplicity values of about 13 were observed (Supplementary Table S3). However, the multiplicity of Ia remained near 1, even at the highest concentration of 1024 ng/ml, indicating that the cell-free synthesized Ia is inefficient. The multiplicity of E2 was 4.3 at a concentration of 128 ng/ml and maintained similar multiplicity at higher concentrations (Supplementary Table S3). Because we previously observed active cell killing of both E1 and E2 after 1 h treatment (Figure 3E) but did not observe high cell killing by E2 in our 3 min cytotoxicity test (Figure 4A), we evaluated colicin cytotoxicity at various concentrations after a 1 h exposure (Figure 4B). Colicin E1 showed consistently high levels of cell killing after 1 h, confirming that E1 has high cytotoxicity. We found that the cytotoxicity of E2 was much higher after a 1 h treatment (m = 10.8 at 128 ng/ml) compared to a 3 min (m = 4.3 at 128 ng/ml) treatment (Supplementary Table S3). Since colicin E2 inactivates cells by degrading cellular DNA (8), E2 might require additional time to exhibit cytotoxicity compared to E1. A 1 h incubation was sufficient for E2 to effectively kill the target cells. On the other hand, the cytotoxicity of colicin Ia did not increase after a 1 h incubation (Figure 4B), further implying that the overall cytotoxicity of colicin Ia is substantially lower than E1 or E2. Our cytotoxicity experiment results confirm that the colicin concentration (750 ng/ml) that we used elsewhere in this study is well within the range of maximum cytotoxicity and that colicins E1 and E2 have high cytotoxicity, but Ia does not.
Figure 4.
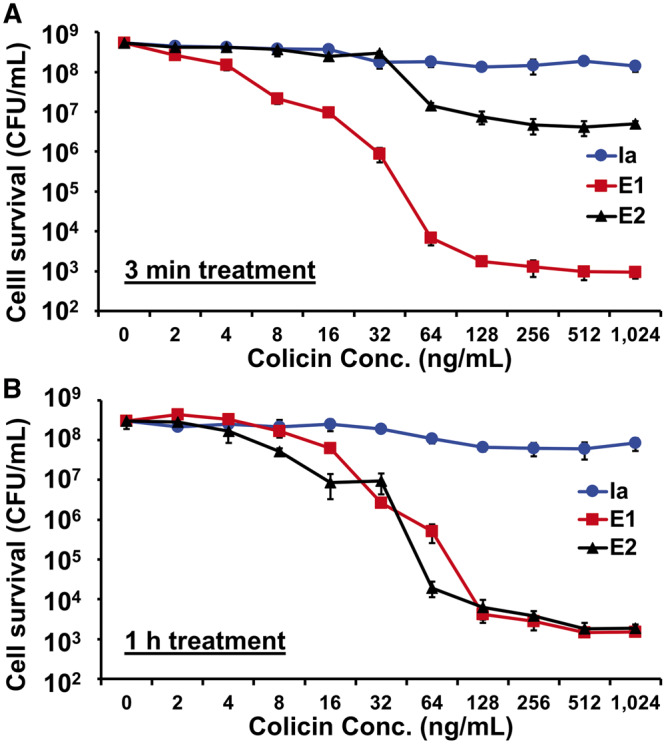
Cell-killing activity of colicins at different concentrations. K361 cells (initial cell density 5 × 108 CFU/ml) were treated with various concentrations of cell-free synthesized colicins for (A) 3 min and (B) 1 h at 37°C with shaking at 220 rpm. Cell-free reaction mixture without colicin production was added as a control in the 0 ng/mL condition. Error bars indicate standard deviation from two independent cultures with three plating replicates each. *Represents significant difference compared to 0 ng/mL samples under the same media conditions with P-value <0.05.
Previous studies reported that 200 ng/ml of colicin E1 produced in the cells had an effective multiplicity of 13 (44) and that 30 ng/ml of colicin E1 had a multiplicity of 6 after a 5 min exposure time (45). Corroborating these previous reports, we found that E1 exhibited a multiplicity of 12.9 at 256 ng/ml and 6.4 at 32 ng/ml after a 3 min treatment (Supplementary Table S3). These results indicate that the cytotoxicity of cell-free synthesized colicin E1 is comparable to that of in vivo produced colicin.
3.7 Colicins E1 and E2 kill persister cells
Since colicins E1 and E2 kill cells in 0.85% NaCl as well as LB (Figure 3D and E), we reasoned that these colicins may be effective in killing persister cells, which are dormant, non-metabolizing cell populations formed under stressed conditions (46). Because persister cells are not growing, they are insensitive to high concentrations of antibiotics, even without the acquisition of resistance mechanisms (46). As antibiotic levels drop, persister cells revive and repopulate (47). This resurgence is a serious problem in treating chronic infectious diseases (12). We performed a persister assay on E. coli K361 cells prepared from a culture grown to exponential growth phase then exposed to 5 µg/ml of ciprofloxacin for 3 h in LB. As expected, a small population (0.002%) of the cells were not killed by ciprofloxacin (Figure 5A), which is a characteristic of persister cells (48). When we added 750 ng/ml of each colicin, which is sufficient to maximize colicin cytotoxicity (Figure 4A and 4B), together with ciprofloxacin to the cell culture during persister cell formation, E1 and E2 killed nearly all remaining cells; however, Ia exhibited low cytotoxicity and barely decreased persister cell levels (Figure 5A). The enhanced cell killing achieved by adding E1 during persister cell formation was similar to the E1 activity toward cells under nutrient-free conditions (Figure 3E). However, treatment with E2 and ciprofloxacin was surprisingly effective at repressing persister levels given the lower cell-killing activity observed in 0.85% NaCl solution (Figure 3E). Contrary to the cells in the nutrient-free 0.85% NaCl solution, persister cells are still in nutrient-rich LB medium. However, persister cells do not utilize nutrients to maintain active cellular metabolism but become metabolically inactive to protect themselves from the antibiotics that typically target actively metabolizing cells. E2 might be effective in killing cells still in the process of entering dormant state in LB medium. When we tested persister cell formation by adding colicins (512 ng/ml Ia, 16 ng/ml E1 and 8 ng/ml E2) with the same multiplicity (∼1.6) (Supplementary Table S3), we did not observe a noticeable change in persister levels compared to the cells treated with ciprofloxacin only (Supplementary Figure S4).
Figure 5.
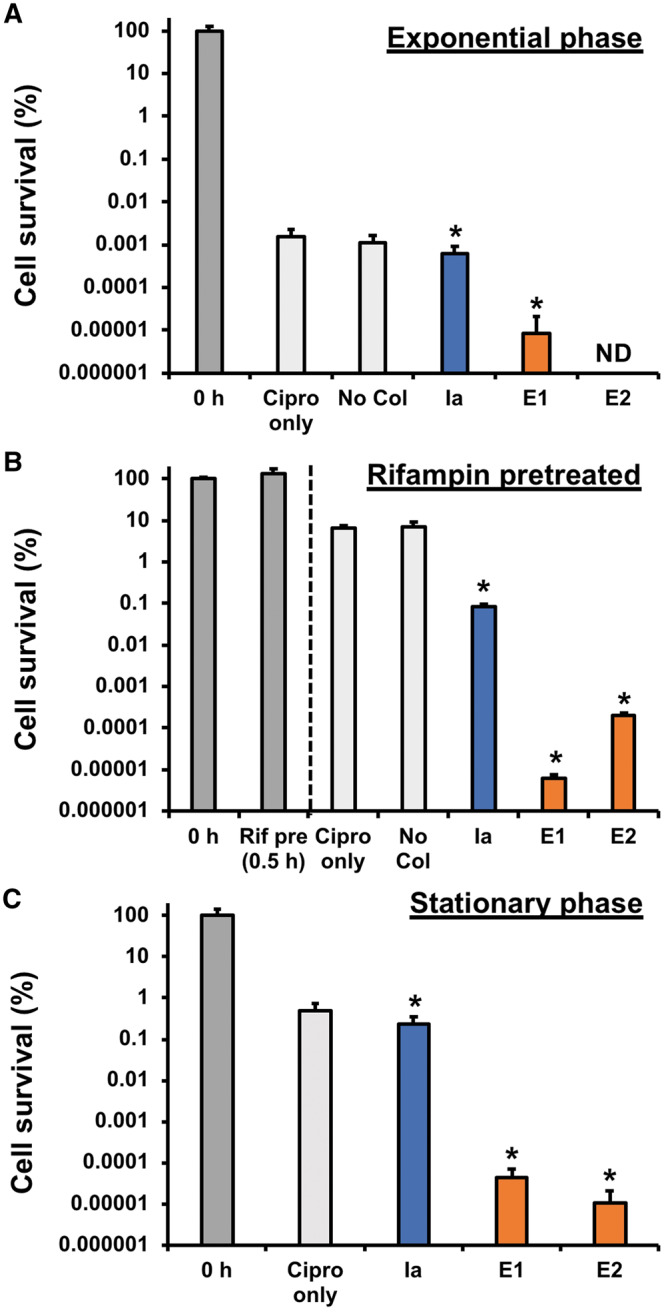
Eradication of persister cells by colicins. (A) K361 cells were grown to exponential phase, adjusted to OD600nm 1.0 with fresh LB, and exposed to ciprofloxacin (Cipro, 5 µg/ml) and each cell-free produced colicin (750 ng/ml) for 3 h with shaking at 220 rpm at 37°C. Cell viability was quantified by counting colony forming units (CFU). (B) Exponential phase cells were pretreated with rifampin (Rif pre, 100 µg/ml) for 30 min. Then, the Rif-pretreated cells were exposed to ciprofloxacin and different colicins for 3 h. (C) Stationary phase cells were exposed to ciprofloxacin and colicins for 3 h. Error bars indicate standard deviations from two independent cultures with three plating replicates each. *Represents significant difference compared to Cipro only with P-value <0.05. ND indicates ‘not detected’.
Next, we increased the level of persister cells formed at the start of our assay in two different ways by (i) treating cells with the antibiotic rifampin which halts transcription (49) and (ii) growing cells until stationary phase which makes cells grow more slowly (50). Rifampin pretreatment increased the fraction of persister cells remaining after ciprofloxacin treatment to 5% (Figure 5B), which was 1000-fold higher compared to no rifampin pretreatment (Figure 5A). Colicin E1 dramatically decreased persister populations by six orders of magnitude compared to the rifampin pretreated persister cells with ciprofloxacin only, and a similar result was obtained by E2 (Figure 5B). E1 and E2 also significantly decreased stationary phase persister cell population by four orders of magnitude compared to ciprofloxacin alone (Figure 5C). On the other hand, colicin Ia was less effective at killing persister cells, decreasing rifampin-pretreated persister cells by 80-fold (Figure 5B) and showing almost no effect to stationary phase cells (Figure 5C). Taken together, colicins can penetrate target cells in diverse persister states and physically damage inner membranes (by E1) or degrade DNA (by E2), which cannot be achieved by antibiotics. Unique cell-killing activities of colicins may lead us to the development of novel strategies to control non-growing persister cells.
The activity spectrum of native colicins is limited to E. coli and closely related bacterial species (51). However, the activity spectrum can be redirected by engineering colicin receptor binding domains or constructing chimeric colicins fused with other types of bacteriocins, as demonstrated previously by constructing pyocin–colicin chimeras to kill Pseudomonas with colicin activity or E. coli with pyocin activity (15). Activity domains of colicins with diverse modes of action can be fused with receptor binding and translocation domains of other bacteriocins to target specific types of cells and vice versa. We expect that the narrow spectrum of colicins will be a benefit for the development of target-specific antimicrobials that can selectively kill cells of interest without influencing other essential or beneficial bacteria. Understanding the interactions between colicin domains and the structural conformation changes would also aid in the construction of engineered or customized colicins as alternatives to antibiotics.
In summary, we showed that antimicrobial colicins can be produced using CFPS, and that cell-free synthesized colicins can effectively kill target cells without purification. CFPS enabled us to produce and characterize colicins rapidly from linear DNA templates and without the need for co-production of immunity proteins. We also found that colicins E1 and E2 exhibited cell-killing under nutrient-free conditions and eradicated persister cells. Effective killing of non-metabolizing and persister cells by colicins presents opportunities to use colicins as powerful new tools to address the growing problem of antimicrobial resistance. In addition, this study further demonstrates the advantages of CFPS for producing toxic proteins and expands the application of CFPS platforms for high-throughput protein synthesis and characterization.
Supplementary data
Supplementary Data are available at SYNBIO Online.
Supplementary Material
Acknowledgments
We thank Dr Karen S. Jakes at Albert Einstein College of Medicine for providing K361 strain and colicins E1 and E2 plasmids.
Funding
The Educational and Research Enhancement Fund at Illinois Institute of Technology. This material is based upon work supported by the National Science Foundation (Graduate Research Fellowship under Grant No. DGE-1324585).
Conflict of interest statement. None declared.
References
- 1. Cascales E., Buchanan S.K., Duché D., Kleanthous C., Lloubès R., Postle K., Riley M., Slatin S., Cavard D. (2007) Colicin biology. Microbiol. Mol. Biol. Rev., 71, 158–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jakes K.S., Cramer W.A. (2012) Border crossings: colicins and transporters. Annu. Rev. Genet., 46, 209–231. [DOI] [PubMed] [Google Scholar]
- 3. Kim Y.C., Tarr A.W., Penfold C.N. (2014) Colicin import into E. coli cells: a model system for insights into the import mechanisms of bacteriocins. Biochim. Biophys. Acta, 1843, 1717–1731. [DOI] [PubMed] [Google Scholar]
- 4. Kleanthous C., Walker D. (2001) Immunity proteins: enzyme inhibitors that avoid the active site. Trends Biochem. Sci., 26, 624–631. [DOI] [PubMed] [Google Scholar]
- 5. Jakes K.S., Finkelstein A. (2010) The colicin Ia receptor, Cir, is also the translocator for colicin Ia. Mol. Microbiol., 75, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jakes K.S. (2017) The colicin E1 TolC box: identification of a domain required for colicin E1 cytotoxicity and TolC binding. J. Bacteriol., 199, e00412-e00416.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. El Ghachi M., Bouhss A., Barreteau H., Touzé T., Auger G., Blanot D., Mengin-Lecreulx D. (2006) Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem., 281, 22761–22772. [DOI] [PubMed] [Google Scholar]
- 8. Sharma O., Yamashita E., Zhalnina M.V., Zakharov S.D., Datsenko K.A., Wanner B.L., Cramer W.A. (2007) Structure of the complex of the colicin E2 R-domain and its BtuB receptor. The outer membrane colicin translocon. J. Biol. Chem., 282, 23163–23170. [DOI] [PubMed] [Google Scholar]
- 9. Lancaster L.E., Savelsbergh A., Kleanthous C., Wintermeyer W., Rodnina M.V. (2008) Colicin E3 cleavage of 16S rRNA impairs decoding and accelerates tRNA translocation on Escherichia coli ribosomes. Mol. Microbiol., 69, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cotter P.D., Ross R.P., Hill C. (2013) Bacteriocins - a viable alternative to antibiotics? Nat. Rev. Microbiol., 11, 95–105. [DOI] [PubMed] [Google Scholar]
- 11. Walsh C. (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature, 406, 775–781. [DOI] [PubMed] [Google Scholar]
- 12. Fisher R.A., Gollan B., Helaine S. (2017) Persistent bacterial infections and persister cells. Nat. Rev. Microbiol., 15, 453–464. [DOI] [PubMed] [Google Scholar]
- 13. Murinda S.E., Rashid K.A., Roberts R.F. (2003) In vitro assessment of the cytotoxicity of nisin, pediocin, and selected colicins on simian virus 40-transfected human colon and Vero monkey kidney cells with trypan blue staining viability assays. J. Food Prot., 66, 847–853. [DOI] [PubMed] [Google Scholar]
- 14. Braun V., Frenz J., Hantke K., Schaller K. (1980) Penetration of colicin M into cells of Escherichia coli. J. Bacteriol., 142, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kageyama M., Kobayashi M., Sano Y., Masaki H. (1996) Construction and characterization of pyocin-colicin chimeric proteins. J. Bacteriol., 178, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Catherine C., Lee K.-H., Oh S.-J., Kim D.-M. (2013) Cell-free platforms for flexible expression and screening of enzymes. Biotechnol. Adv., 31, 797–803. [DOI] [PubMed] [Google Scholar]
- 17. Xie Q., Matsunaga S., Wen Z., Niimi S., Kumano M., Sakakibara Y., Machida S. (2006) In vitro system for high-throughput screening of random peptide libraries for antimicrobial peptides that recognize bacterial membranes. J. Pept. Sci., 12, 643–652. [DOI] [PubMed] [Google Scholar]
- 18. Cintas L.M., Casaus P., Holo H., Hernandez P.E., Nes I.F., Håvarstein L.S. (1998) Enterocins L50A and L50B, two novel bacteriocins from Enterococcus faecium L50, are related to staphylococcal hemolysins. J. Bacteriol., 180, 1988–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eichenlaub R. (1974) Synthesis of colicin E1 in a cell-free system. J. Bacteriol., 120, 1476–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ebina Y., Kishi F., Nakazawa T., Nakazawa A. (1979) Gene expression in vitro of colicin El plasmid. Nucleic Acids Res., 7, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salehi A.S.M., Smith M.T., Bennett A.M., Williams J.B., Pitt W.G., Bundy B.C. (2016) Cell-free protein synthesis of a cytotoxic cancer therapeutic: onconase production and a just-add-water cell-free system. Biotechnol J, 11, 274–281. [DOI] [PubMed] [Google Scholar]
- 22. Perez J.G., Stark J.C., Jewett M.C. (2016) Cell-free synthetic biology: engineering beyond the cell. Cold Spring Harb. Perspect. Biol., 8, a023853.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hodgman C.E., Jewett M.C. (2012) Cell-free synthetic biology: thinking outside the cell. Metab. Eng., 14, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon Y.C., Lee K.H., Kim H.C., Han K., Seo J.H., Kim B.G., Kim D.M. (2010) Cloning-independent expression and analysis of ω-transaminases by use of a cell-free protein synthesis system. Appl. Environ. Microbiol., 76, 6295–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahn J.-H., Chu H.-S., Kim T.-W., Oh I.-S., Choi C.-Y., Hahn G.-H., Park C.-G., Kim D.-M. (2005) Cell-free synthesis of recombinant proteins from PCR-amplified genes at a comparable productivity to that of plasmid-based reactions. Biochem. Biophys. Res. Commun., 338, 1346–1352. [DOI] [PubMed] [Google Scholar]
- 26. Schinn S.M., Broadbent A., Bradley W.T., Bundy B.C. (2016) Protein synthesis directly from PCR: progress and applications of cell-free protein synthesis with linear DNA. N. Biotechnol., 33, 480–487. [DOI] [PubMed] [Google Scholar]
- 27. Hong S.H., Kwon Y.-C., Jewett M.C. (2014) Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front. Chem., 2, 34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carlson E.D., Gan R., Hodgman C.E., Jewett M.C. (2012) Cell-free protein synthesis: applications come of age. Biotechnol. Adv., 30, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zemella A., Thoring L., Hoffmeister C., Kubick S. (2015) Cell-free protein synthesis: pros and cons of prokaryotic and eukaryotic systems. ChemBioChem, 16, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stech M., Kubick S. (2015) Cell-free synthesis meets antibody production: a review. Antibodies, 4, 12–33. [Google Scholar]
- 31. Jakes K.S. (2012) Translocation trumps receptor binding in colicin entry into Escherichia coli. Biochem. Soc. Trans., 40, 1443–1448. [DOI] [PubMed] [Google Scholar]
- 32. Kwon Y.-C., Jewett M.C. (2015) High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep., 5, 8663.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., Smith H.O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 34. Swartz J.R., Jewett M.C., Woodrow K.A. (2004) Cell-free protein synthesis with prokaryotic combined transcription-translation. Methods Mol. Biol., 267, 169–182. [DOI] [PubMed] [Google Scholar]
- 35. Sharma O., Cramer W.A. (2007) Minimum length requirement of the flexible N-terminal translocation subdomain of colicin E3. J. Bacteriol., 189, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hong S.H., Wang X., O’Connor H.F., Benedik M.J., Wood T.K. (2012) Bacterial persistence increases as environmental fitness decreases. Microb. Biotechnol., 5, 509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kienker P.K., Jakes K.S., Finkelstein A. (2008) Identification of channel-lining amino acid residues in the hydrophobic segment of colicin Ia. J. Gen. Physiol., 132, 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iost I., Guillerez J., Dreyfus M. (1992) Bacteriophage T7 RNA polymerase travels far ahead of ribosomes in vivo. J. Bacteriol., 174, 619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brunden K.R., Cramer W.A., Cohen F.S. (1984) Purification of a small receptor-binding peptide from the central region of the colicin E1 molecule. J. Biol. Chem., 259, 190–196. [PubMed] [Google Scholar]
- 40. Weber G., Helgerson S.L., Cramer W.A., Mitchell G.W. (1976) Changes in rotational motion of a cell-bound fluorophore caused by colicin E1: a study by fluorescence polarization and differential polarized phase fluorometry. Biochemistry, 15, 4429–4432. [DOI] [PubMed] [Google Scholar]
- 41. Jetten A.M., Jetten M.E. (1975) Energy requirement for the initiation of colicin action in Escherichia coli. Biochim. Biophys. Acta, 387, 12–22. [DOI] [PubMed] [Google Scholar]
- 42. Cramer W.A., Postma P.W., Helgerson S.L. (1976) An evaluation of N-phenyl-1-naphthylamine as a probe of membrane energy state in Escherichia coli. Biochim. Biophys. Acta, 449, 401–411. [DOI] [PubMed] [Google Scholar]
- 43. Shiver J.W., Cramer W.A., Cohen F.S., Bishop L.J., de Jong P.J. (1987) On the explanation of the acidic pH requirement for in vitro activity of colicin E1. J. Biol. Chem., 262, 14273–14281. [PubMed] [Google Scholar]
- 44. Shiver J.W., Cohen F.S., Merrill A.R., Cramer W.A. (1988) Site-directed mutagenesis of the charged residues near the carboxy terminus of the colicin E1 ion channel. Biochemistry, 27, 8421–8428. [DOI] [PubMed] [Google Scholar]
- 45. Schendel S.L., Click E.M., Webster R.E., Cramer W.A. (1997) The TolA protein interacts with colicin E1 differently than with other group A colicins. J. Bacteriol., 179, 3683–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wood T.K., Knabel S.J., Kwan B.W. (2013) Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol., 79, 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lewis K. (2010) Persister cells. Annu. Rev. Microbiol., 64, 357–372. [DOI] [PubMed] [Google Scholar]
- 48. Lewis K. (2007) Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol., 5, 48–56. [DOI] [PubMed] [Google Scholar]
- 49. Kwan B.W., Valenta J.A., Benedik M.J., Wood T.K. (2013) Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother., 57, 1468–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Keren I., Kaldalu N., Spoering A., Wang Y., Lewis K. (2004) Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett., 230, 13–18. [DOI] [PubMed] [Google Scholar]
- 51. Schulz S., Stephan A., Hahn S., Bortesi L., Jarczowski F., Bettmann U., Paschke A.-K., Tusé D., Stahl C.H., Giritch A.. et al. (2015) Broad and efficient control of major foodborne pathogenic strains of Escherichia coli by mixtures of plant-produced colicins. Proc. Natl. Acad. Sci. USA, 112, E5454–E5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Levengood-Freyermuth S.K., Click E.M., Webster R.E. (1993) Role of the carboxyl-terminal domain of TolA in protein import and integrity of the outer membrane. J. Bacteriol., 175, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hong S.H., Ntai I., Haimovich A.D., Kelleher N.L., Isaacs F.J., Jewett M.C. (2014) Cell-free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site-specific non-standard amino acid incorporation. ACS Synth. Biol., 3, 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. La Rosa S.L., Diep D.B., Nes I.F., Brede D.A. (2012) Construction and application of a luxABCDE reporter system for real-time monitoring of Enterococcus faecalis gene expression and growth. Appl. Environ. Microbiol., 78, 7003–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.