

Abstract
Gene therapy for auditory diseases is gradually maturing. Recent progress in gene therapy treatments for genetic and acquired hearing loss has demonstrated the feasibility in animal models. However, a number of hurdles, such as lack of safe viral vector with high efficiency and specificity, robust deafness large animal models, translating animal studies to clinic etc., still remain to be solved. It is necessary to overcome these challenges in order to effectively recover auditory function in human patients. Here, we review the progress made in our group, especially our efforts to make more effective and cell type-specific viral vectors for targeting cochlea cells.
Subject terms: Targeted gene repair, DNA damage and repair
Introduction
Hearing loss is a common neurological disorder. Both genetic causes and environmental factors, such as ototoxic chemicals, chronic ear infections, large noise, and aging, can lead to hearing loss and deafness. In China, around 2 of every 1000 children are born with a clinically significant hearing loss in one or both ears [1] and about 60,000 babies born in China each year have the hearing loss syndrome and about half of them has a genetic etiology.
Current therapies for hearing loss include hearing aids, middle ear prostheses/active implants to amplify the sound signal, or cochlear implants to directly stimulate spiral neurons [2]. These approaches enable patients to hear the outside sound to some degree, but the therapeutic result remains far from effective in restoring natural hearing, especially in case that patients have deficiencies in frequency sensitivity, natural sound perception, and speech discrimination in noisy environments [2]. Therefore, more effective methods are still urgently required for treating hearing loss.
The inner ear system contains three major types of functional cells: hair cells (HCs), supporting cells (SCs), and spiral ganglion neurons (SGNs), all of which play an important role in the process of hearing production and perception. There are two types of sensory HCs in the cochlea: the outer HCs (OHCs) and the inner HCs (IHCs). OHCs amplify sound signals, while IHCs convert the mechanical information carried by sound waves into electrical signals that are transmitted to the neurons [3].
Genes required for cochlea function and hearing in cochlea cells
Many genes in both HCs and SCs play essential roles in the development or maintenance of cochlea and thus are involved in regulating the cochlea function and hearing. Here we provide a very brief information in this area. For more details, please refer other elegant review articles [4–6].
Transmembrane channel-like 1 (TMC1) is a pore-forming component of channels that participate in mechanoelectrical transduction of sound in cochlear and vestibular HCs [7]. Otoferlin (OTOF) is an essential protein in HC ribbon synapses [8], and was previously found playing an important role in regulating the mode of exocytosis in IHCs [9]. Mutations on these genes as well as others, like MYO7A, PCDH15, and POU4F3, cause the dysfunction of cochlea and lead to deafness [10–14].
Besides hair-cell gene mutations, SC gene mutations have also been linked to deafness. SCs are located at the bottom of the inner and OHCs, anchoring the sensory epithelium to the basilar membrane, thus playing a mechanical role in protecting and maintaining the surrounding environment for HCs. Some key deafness genes mainly express and have functions in SCs, such as GJB2, which affects the SC’s gap junction and is the most common hereditary deafness gene [1, 6, 15, 16]. In mammals, hair-cell loss due to environmental and genetic stress is thought to be permanent [17]. However, recent studies suggest that SCs are potential inner ear progenitor cells from which HCs can be regenerated [18, 19]. Therefore, SCs are a potential target for gene therapy, not only to correct inherited hearing defects, but also for hair-cell regeneration.
SGNs, located in a bony channel (Rosenthal’s canal) that spirals around axis of the cochlea (modiolus), are primary neurons of the auditory system. The HCs release glutamate neurotransmitters upon sound stimulation to bind NMDAR2 and mGluRIs on the SGN membrane to produce excitatory electricals. SGNs transmit these electrical signals to the auditory cortex through the eighth cranial nerve, enabling us to hear outside sound. Thus, SGNs, as the bridge between HCs and brain, are required for normal hearing. Unfortunately, noise exposure, ototoxic drugs, and genetic factors can cause the irreversible SGN damage or death, and thus the communication between HCs and brain is disturbed, leading to sensorineural hearing loss [17].
A brief gene therapy history in auditory disease
In recent years, gene therapy has emerged as an important method to treat inherited diseases (Table 1). Although 140 deafness-associated alleles have been identified, few treatments are available to slow or reverse genetic deafness. Clearly, there is an urgent need to develop biotherapies for restoring auditory function. Among them, the gene therapy has become the most promising therapy for hereditary deafness [5]. The inner ear is an ideal target for gene therapy, and many viral and nonviral vectors have been developed for the transmission of genetic material in the cochlea [20]. The adeno-associated virus (AAV) is widely used in gene therapy due to its high infection efficiency, low pathogenicity and toxicity, sustained expression of the carried genes, as well as its simple, cheap, and fast production [21–24]. Several studies have achieved good results by AAV vector-mediated gene therapy using animal models with different mutated genes in several types of cells in cochlea [2, 21, 25–30].
Table 1.
The brief history of gene therapy in hearing diseases.
| Animal model | Treatment reagent | Injection time and delivery method | Ave. ABR improvement (best freq.) and treatment efficacy | Targeted cells and major morphological improvement |
|---|---|---|---|---|
| Vglut3−/− mice [2] | AAV1-Vglut3 |
P1–3 and P10 Route: AC and RWM |
~50 dB (90 dB of control) Lasted for 3–6 months |
IHCs/Improve the morphology of partial afferent IHC ribbon synapses. |
| Kcnq1−/− mice [54] | AAV1-Kcnq1 |
P0–2 Route: Scala media |
~45 dB (90 dB of control) Lasted for 4–6 months |
SV marginal cells/rescue the collapse of Reissner’s membrane death of HCs and cells in the SG. |
| MsrB3−/− mice [55] | AAV2/1-MsrB3 |
E12.5 Route: in utero |
~40–50 dB | IHCs and OHCs/recovery hearing and the morphology of the stereociliary bundles. |
| Slc26a4−/− and Slc26a4tm1Dontuh/ tm1Dontuh mice [56] | rAAV2/1-Slc26a4 |
E12.5 Route: in utero |
~20–40 dB(8–12 weeks) | IHCs, OHCs and stria vascularis/restored hearing phenotypes included normal hearing and progressive hearing loss. |
| Gjb2cKO mice Cx26fl/flP0-Cre [57] | AAV5-Cx26 |
P0 and P42 Route: RWM |
~20 dB in P0 (100 dB of control mice) ~0 dB in P42 |
IHC, OHCs, and SCs/rescue the formation of organ of Corti and HCs. No morphology change in P42. |
| Whrnwi/wi mice [58] | AAV2/8-whirilin |
P1–5 Route: PSC |
~20 dB Lasted for 4 months Rescue the vestibular function |
IHCs/rescue the morphology and function of stereociliary bundles and temporarily the death of IHCs. |
| Clrn1ex4/– mice [28] | AAV2/8-Clrn1 |
P1–P3 Route: RWM |
~30–40 dB | IHCs and OHCs/restored hearing phenotypes included normal hearing as well as the synaptic ribbons. |
| Clrn1–/–-TgAC1 mice [59] | AAV2/8-Clrn1 | P1–P3 Route: RWM |
~30–40 dB Lasted for 5 months |
IHCs and OHCs/restored the hair bundle structure and hearing. |
| Usher1c (c.216G>A) [29] | AAV2-harmonin |
P0–1 and P10–12 Route: RWM |
~50–60 dB (110 dB of control) Lasted for 6 months |
IHCs and OHCs/rescue the function of stereociliary bundles and death of HCs. |
| Otof−/− mice [21] | Dual AAV |
P10 and P17 and P30 Route: RWM |
~30–40 dB Lasted for 5–6 months |
IHCs/restore the number of ribbons by promoting their production. |
| Otof−/− mice [60] | Dual AAV2/6half-vector |
P6–7 Route: RWM |
~50–60 dB(110 dB of control) | IHCs/restore the exocytosis function of IHC and partially the number of ribbons. |
| TMC−/− mice [61] | AAV2/1-Cba-Tmc |
P0–2 Route: RWM |
~20–30 dB (110 dB of control) | IHCs and OHCs/restore the sensory transduction current of HCs, SCs, and SGs. |
| TMC−/− mice [62] | sAAV-Tmc1 |
P0–2 Route: RWM |
~50–60 dB (110 dB of control) Lasted for 3 months Rescue the vestibular function |
IHCs and OHCs/rescue the function of stereociliary bundles, sensory transduction current, and death of HCs. |
| Tmc1Bth/+ mice [63] | Cas9: gRNA |
P1 Route:Scala media |
~20–30 dB | IHCs and OHCs/rescue the death of HCs. |
| Tmc1Bth/+ mice [31] | rAAV2/9miTmc1 |
P0–2 Route: RWM |
~30–40 dB | IHCs and OHCs/rescue the number of IHCs and OHCs partially. |
| Tmc1Bth/+ mice [27] | AAV-SaCas9-KKH |
P1 Route: Scala media |
~30–40 dB | IHCs and OHCs/rescue the morphology of stereociliary bundles and the death of IHCs. |
| Tmc1Bth/+ mice3 | AAV9.miTmc1 |
P15–16 and P56–60 and P84–90 Route: RWM + SF |
~30–40 dB No difference in P84–90 |
IHCs, OHCs, and SV/rescue the morphology of stereociliary bundles and temporarily the death of IHCs. |
PSC posterior semicircular canal, AC apical cochleostomy, SF semicircular fenestration.
Akil et al. loaded the VGLUT3 gene with AAV1 vectors to VGLUT3 knockout neonatal mice that displayed deafness by round window membrane (RWM) injection [2]. VGLUT3 gene was strongly expressed in whole cochlea. In addition, acoustic brainstem response (ABR) experiments showed that VGLUT3 overexpression with AAV vector successfully rescued the hearing phenotype in VGLUT3 knockout mice. Murine Beethoven (Bth) mutation (Tmc1 c.1235T>A [p.Met412Lys]) leads to the autosomal-dominant hearing loss. Shibata et al. used rAAV2/9 as the viral vector to deliver designed artificial microRNAs to rescue the progressive hearing loss [31]. In their study, rAAV2/9 predominantly localized to IHCs with about 74% efficiency of infection and the hearing function get some recovery as tested by ABR and distortion product otoacoustic emissions. Notably, many conventional AAV serotypes can transduce IHCs with high efficiency, but still exhibit no or very low transducing efficiency in OHCs. In 2017, a breakthrough study carried by Landegger et al.’s group demonstrated that Anc80L65 transduced both IHCs and OHCs in mice with very high efficiency, a substantial improvement over conventional AAV vectors [32]. Anc80L65 successfully delivered wild-type Ush1c into the inner ears of the neonatal Ush1c c.216G>A mice model [29]. Taking the advantage of Anc80L65 transducing HCs, Pan et al. showed the most complete recovery of auditory and vestibular function with gene therapy approach with AAVs [29].
The challenges in the cochlea gene therapy
Despite of many exciting advances in gene therapy for deafness in animal models, there is still a long way to go before it can be applied to deafness in humans. There are currently more than 20 clinical trials for hearing loss therapies in the United States with six potential therapeutic molecules. Intriguingly, there is one clinical trial involving gene therapy for auditory diseases [33]. Moreover, a number of AAV-related gene therapy drugs have been approved by the U.S. FDA, which fully proves the clinical potential of AAV. In the field of hearing, however, there is no clinical drug based on AAV. As mentioned above, there is some success in gene therapy animal studies with AAV delivery. However, the specificity and efficiency of these viruses remain weak and may cause unwanted side effects by expressing genes in other untargeted cells.
It is feasible to systematically characterize the specificity of different AAVs to transduce the different cell types in the cochlea. An elegant study showed that AAVs specifically transduced different types of retina in both mice and nonhuman primate under the control of different gene modulator components [34]. In China, Li et al.’s group at Fudan University screened the available AAV variants to target the SCs in cochlea, which are essential for the function of both HCs and SGNs and have the potential to transdifferentiate to hair-cell-like cells. They found that AAV9-PHP.eB showed relatively high transduction efficacy in both OHCs and IHCs, and this is consistent with the results from a recent study performed at Lee et al.’s group [35–37]. They found that AAV-DJ had relatively high efficiency in SCs, surpassing what has been reported previously [35, 38]. However, the existing AAV variants do not transduce the cochlea cells in an efficient way and especially SCs are not sufficiently targeted by these AAVs [37–39]. Thus, in order to make the gene therapy with AAVs, it is necessary to generate new AAV variants which should have two properties, high transducing efficiency and specificity.
The development of AAV-ie
To achieve this, we employed a strategy similar to a previous study [22] and aim to discover AAV variants with high transducing efficiency by inserting select peptides into an AAV vector and tested the transducing efficiency in the in vitro cell culture and in vivo animals [40]. AAV vectors can be successfully delivered to the inner ear to transduce cochlea cells by injection through RWM [41]. Thus, the AAV needs to cross a mesothelial cell layer to infect HCs and SCs. We reasoned that novel AAV variants with the ability to cross the mesothelial cell layer may increase gene transfer efficiency [40]. Since an earlier study demonstrated that the insertion of a peptide (DGTLAVPFK) helped the new AAV vector cross the blood–brain barrier [22], we inserted the DGTLAVPFK peptide into the VP1 capsid of AAV-DJ and found that the new AAV variant, which they named as AAV-ie (inner ear), dramatically increase the transducing rate to 80% of SCs in cochlea [40].
Cochlear SCs contain different cell types: Hensen’s cells, Deiters cells, pillar cells, inner phalangeal cells, and inner border cells. We found that high-dose AAV-ie infected all cell types of SCs with high efficiency without obvious toxicity to the cochlea function and auditory behaviors. Manipulation of signaling pathways and transcription factors such as gene Atoh1 can lead to transdifferentiation of SC into HCs [42]. To assess the potential of the AAV-ie vector for HC regeneration, we used AAV-ie-Atoh1-NLS-mNeonGreen (AAV-ie-Atoh1) to deliver mouse Atoh1 into the cochlea. New hair-cell-like cells were generated in the AAV-ie-Atoh1 group as unambiguously demonstrated by the immunofluorescence labeling and SEM experiments (Fig. 1). The HC regeneration by the Atoh1 overexpression with AAV-ie is comparable with a previous genetic study that used Foxg1-Cre-mediated Atoh1 overexpression mice, indicating AAV-ie is a powerful tool to deliver genes into SCs and could represent a potential tool to be used as HC regeneration. Indeed, we further demonstrated the newly generated hair-cell-like cells displayed excitable membrane properties relatively similar to the electrophysiological properties of HCs [40]. Using ex vivo human samples taken from ear surgery, we further demonstrated that AAV-ie can transduce the SCs in human utricle SCs. Recent collaborative experiments show that AAV-ie can transdifferentiate human utricle SCs to hair-cell-like cells in in vitro culture (data not shown). To our knowledge, this is the first study to use AAV as the deliver tool to show the unambiguous hair-cell-like cell regeneration in both rodent animal cochlea and culture human utricle cultures. Thus, AAV-ie may hold the potential for correcting genetic hearing impairment of SCs and also for HC regeneration to treat environmental and age-induced hearing loss or genetic auditory diseases given that in general AAVs have the lowest toxicity as viral vectors.
Fig. 1. Adeno-associated virus-inner ear-Atoh1 (AAV-ie-Atoh1) induces new hair cells (HCs) in vivo with stereocilia.

a Representative confocal projection image of control and AAV-ie-Atoh1 cochlea. Scale bar, 10 µm. b Scanning electron microscopy (SEM) images of AAV-ie- and AAV-ie-Atoh1-injected cochlea at apical regions. Regenerated HC-like cells were artificially colored magenta.
We reported that AAV-ie not only transduced SCs but also HCs in both animal models and human utricle samples. The nonspecific transducing properties of AAV-ie may limit it as an appropriate vector to deliver genes to SCs to treat either genetic or acquired hearing loss. Thus, further optimization of AAV variants to increase the transducing efficiency and specificity as gene transfer vectors for clinical use is much needed. We will discuss our current efforts to achieve the above goal.
Improving the AAV efficiency
The existing AAV variants did not evolve for the purposes of highly transduce the cochlea cells, especially SCs [35, 37, 39]. Modification of these AAV variants to improve their efficacy and specificity of their potential use in inner ear gene therapy is much needed. There are many strategies to increase the transducing efficiency of AAV variants as illustrated in Fig. 2. Rational design of point mutations may increase the chance of AAV variants trafficking to the nucleus by the lack of AAV capsid ubiquitination [43–46]. Another strategy is to randomly fragment and reassemble the capsid genome of wild-type AAV serotypes 1–13 by PCR to generate a chimeric capsid library. Newly generated capsids may give the synthetic AAV different properties, such as tissue tropism and transducing efficiency [47–50]. In addition to the above two methods, peptides can be inserted into specific regions of AAV capsids to change their properties and several AAV variants are found to be highly efficient to transduce cells in central nervous system and in cochlea [22, 23, 40]. These efforts to optimize the capsids have led to the development of new AAV variants that are capable of high efficiency transduction at lower doses, and this increases the chance of their use in human gene therapy.
Fig. 2.

The strategies used to change the capsid to increase the transducing efficiency of AAV variants.
Achieving the AAV specificity
AAV variants displaying high transducing efficiency often lack specificity and may bring severe side effects. To circumvent this shortcoming, efforts are needed to make the expression of interested genes in specific types of cells. The production of cell-targeted AAV can be achieved by selecting cell-specific promoters [22]. To achieve the specificity of AAV variants, we searched the literature for genes specifically expressed in the different types of cochlea cells, HCs, SCs, and SGNs (Table 2). Our single cell sequencing data are consistent with this information (data not shown).
Table 2.
Genes are specifically expressed in cochlea cells.
| HCs | SCs | SGNs |
|---|---|---|
| Ocm [64] | Gjb2 [30] | Syn [65] |
| Slc26a5 [66] | Lgr5 [67, 68] | NeuN [69] |
| Otof [70] | GFAP [71] | Map2 [72] |
| Atp2a3 [70, 73] | Fgfr3 [74] | Tuj1 [75] |
| Tpbgl [70] | Tak1 [76] | Calb1 [77] |
| Dnajc5b [70] | Sox21 [78] | Calb2 [79] |
| Myo7a [80] | Sox2 [81, 82] | Nos1 [83] |
| Myo6 [84] | PLP1 [68] | Runx1 |
| Zfp [85] | CD44 [86] | Prph [87] |
| Tmc1 [73, 88] | Prox1 [89, 90] | Cacna1h [91] |
| Tmc2 [88] | CX30 [92] | Slc6a4 [83] |
| Cabp2 [93] | Aquaporin4 [94] | Grm8 [95] |
| Brip1 [96] | Brn3a [97] | |
| Zmat3 [98] | Trim54 | |
| Strip2 [96] |
The promoter sequences of specific expression genes are chosen using four different methods (Fig. 3). At the 5′ end of the gene specifically expressed in inner ear cells, the region between 500 and 3500 bp was selected to intercept the gene sequence as the synthesis promoter. The 5′ end sequence (synthetic promoter) is constructed using four different strategies [34]. ProA contains a sequence upstream of the initiation codon of a cell-specific gene in the inner ear of the mouse cochlea, the bases of the sequence at both ends of −1500 to 500 and −3000 to −1000 extracted from the 5′ −3000 to 500 bp of the gene specifically expressed in the inner ear cell. ProB is an ordered assembly of systemically inherited and conserved DNA elements identified in nucleotide sequences prior to at least two hair-cell-specific gene transcription initiation sites. The conserved genetic sites were predicted by the database of the University of California Santa Crus and National Center for Biotechnology. ProC is composed of multiple inner ear cell-specific repeat sequences of transcription factor binding sites (TFBS) and random sequence crossover. TFBS can be predicted by searching the literature and JASPER database [51]. ProD was determined based on the combination of epigenetics and transcriptome analysis. The hypomethylation sequence of cis-acting elements specifically expressed by inner ear cells could be predicted by MethPrimer and other databases. This part of hypomethylated cis-acting elements could be amplified from the genome as the synthesis promoter of ProD. ProC and ProD also contain the minimal TATA box synthetic promoter (minP) element. These studies are intended to obtain synthetic promoters of genes specifically expressed in inner ear cells, and prepare for the next step of in vivo screening.
Fig. 3. Different strategies are used in constructing the synthetic promoter.
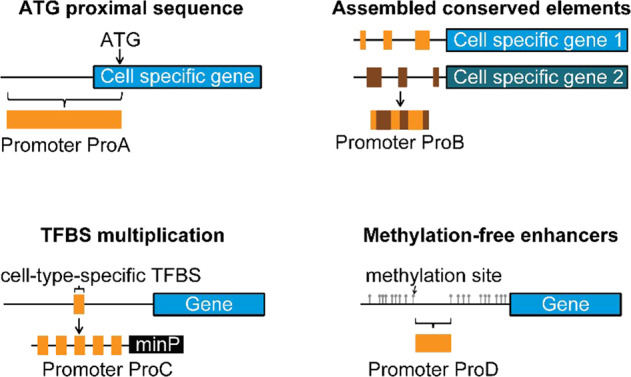
ProA: A 5′ end of a specific type of inner ear cell-specific expressed gene −3000 to 500 bp. ATG is the translation start site. ProB: A phylogenetic conserved sequence before the transcription initiation site of a gene specifically expressed by at least two specific types of inner ear cells. ProC: Transcription factor binding site (TFBS) repeats for multiple specific types of inner ear cell-specific transcription factors. ProD: Hypomethylated sequences of cis-acting elements of genes specifically expressed by specific types of inner ear cells.
By using these strategies to choose the promotor sequence of specific genes in cochlea cells, we are able to generate the highly transducing AAV variants in HCs, SCs, or SGNs (data not shown). It is a substantial amount of work to generate these AAV variants and screen for their transducing efficiency and specificity. We will continue these efforts to optimize AAV variants, which can transduce the cochlea cells in adult mice and other large animal models, such as pigs and nonhuman primates.
Large animal models for hearing research
While most work related to inner ear gene therapy is conducted in rodents, larger animals, such as pigs or nonhuman primates, which have ears that are closer to those of humans, are better animal models for evaluating the efficiency/specificity and toxicity of AAV variants. Thus, these large animal models may extend translational proof-of-principle studies. China has established the largest pool of pig models and a large amount of mutations have been generated [52, 53]. Interestingly, the mutation of SOX10 (R109W) in pigs by N-ethyl-N-nitrosourea mutagenesis causes inner ear malfunctions and hearing loss [52], and might represent a good model to test the gene therapy approach for hearing loss. Right now, we are conducting collaborative experiments to evaluate the efficiency/specificity and toxicity of AAV variants in pigs and expect some AAV variants may present as potential options to be used in clinical trials.
It is an exciting time for inner ear gene therapy. With the advent of new AAV variants displaying high efficiency and specificity in transducing cochlea cells and with the establishment of large animal models, such as pigs and nonhuman primates, we would expect the rapid translation from basic research to clinical trials is feasible. There are more than 100 different genes causing genetic hearing loss, yet the mechanisms underlying hearing dysfunction by distinct gene mutations are different and need to be fully investigated before developing the gene therapy strategy for each hearing deaf gene. Despite many challenges, there are reasons for optimism as new AAV variants, which specifically and efficiently target different cochlea cells, are developed and more collaborative projects, from both basic scientists and clinical doctors, are conducted to develop feasible gene therapy strategies for hearing loss.
Acknowledgements
This work was supported by the National Natural Science Foundation of China, 81970878 and 31771130 (GZ), Shanghai Municipal Government, and ShanghaiTech University.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
2/11/2021
A Correction to this paper has been published: 10.1038/s41434-020-00186-x
References
- 1.Dai P, Huang LH, Wang GJ, Gao X, Qu CY, Chen XW, et al. Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am J Hum Genet. 2019;105:803–12. doi: 10.1016/j.ajhg.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akil O, Seal RP, Burke K, Wang C, Alemi A, During M, et al. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron. 2012;75:283–93. doi: 10.1016/j.neuron.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LeMasurier M, Gillespie PG. Hair-cell mechanotransduction and cochlear amplification. Neuron. 2005;48:403–15. doi: 10.1016/j.neuron.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed H, Shubina-Oleinik O, Holt JR. Emerging gene therapies for genetic hearing loss. J Assoc Res Otolaryngol. 2017;18:649–70. doi: 10.1007/s10162-017-0634-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Géléoc GSG, Holt JR. Sound strategies for hearing restoration. Science. 2014;344:1241062. doi: 10.1126/science.1241062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang W, Kim SM, Wang W, Cai C, Feng Y, Kong W, et al. Cochlear gene therapy for sensorineural hearing loss: current status and major remaining hurdles for translational success. Front Mol Neurosci. 2018;11:221. doi: 10.3389/fnmol.2018.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan B, Akyuz N, Liu X-P, Asai Y, Nist-Lund C, Kurima K, et al. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. 2018;99:736–53.e6. doi: 10.1016/j.neuron.2018.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–89. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 9.Takago H, Oshima-Takago T, Moser T. Disruption of otoferlin alters the mode of exocytosis at the mouse inner hair cell ribbon synapse. Front Mol Neurosci. 2019;11:492–492. doi: 10.3389/fnmol.2018.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16:188–90. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science. 1998;279:1950–4. doi: 10.1126/science.279.5358.1950. [DOI] [PubMed] [Google Scholar]
- 13.Xiang M, Maklad A, Pirvola U, Fritzsch B. Brn3c null mutant mice show long-term, incomplete retention of some afferent inner ear innervation. BMC Neurosci. 2003;4:2. doi: 10.1186/1471-2202-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Li B, Apisa L, Yu H, Entenman S, Xu M, et al. ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23(erl/ erl) mutant mice. Cell Death Dis. 2016;7:e2485–e2485. doi: 10.1038/cddis.2016.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang YP, Tang WX, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. P Natl Acad Sci USA. 2005;102:15201–6. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park H-J, Houn Hahn S, Chun Y-M, Park K, Kim H-N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110:1535–8. doi: 10.1097/00005537-200009000-00023. [DOI] [PubMed] [Google Scholar]
- 17.Wagner EL, Shin J-B. Mechanisms of hair cell damage and repair. Trends Neurosci. 2019;42:414–24. doi: 10.1016/j.tins.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franco B, Malgrange B. Concise review: regeneration in mammalian cochlea hair cells: help from supporting cells transdifferentiation. Stem Cells. 2017;35:551–6. doi: 10.1002/stem.2554. [DOI] [PubMed] [Google Scholar]
- 19.Shu Y, Li W, Huang M, Quan YZ, Scheffer D, Tian C, et al. Renewed proliferation in adult mouse cochlea and regeneration of hair cells. Nat Commun. 2019;10:5530. doi: 10.1038/s41467-019-13157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sacheli R, Delacroix L, Vandenackerveken P, Nguyen L, Malgrange B. Gene transfer in inner ear cells: a challenging race. Gene Ther. 2013;20:237–47. doi: 10.1038/gt.2012.51. [DOI] [PubMed] [Google Scholar]
- 21.Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci USA. 2019;116:4496–501. doi: 10.1073/pnas.1817537116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–9. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34:204–9. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki J, Hashimoto K, Xiao R, Vandenberghe LH, Liberman MC. Cochlear gene therapy with ancestral AAV in adult mice: complete transduction of inner hair cells without cochlear dysfunction. Sci Rep. 2017;7:45524. doi: 10.1038/srep45524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshimura H, Shibata SB, Ranum PT, Moteki H, Smith RJH. Targeted allele suppression prevents progressive hearing loss in the mature murine model of human TMC1 deafness. Mol Ther. 2019;27:681–90. doi: 10.1016/j.ymthe.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isgrig K, McDougald DS, Zhu J, Wang HJ, Bennett J, Chien WW. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat Commun. 2019;10:427–427. doi: 10.1038/s41467-018-08243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.György B, Meijer EJ, Ivanchenko MV, Tenneson K, Emond F, Hanlon KS, et al. Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of Usher syndrome 3A and transduces hair cells in a non-human primate. Mol Ther. 2019;13:1–13. doi: 10.1016/j.omtm.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dulon D, Papal S, Patni P, Cortese M, Vincent PF, Tertrais M, et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J Clin Investig. 2018;128:3382–401. doi: 10.1172/JCI94351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol. 2017;35:264–72. doi: 10.1038/nbt.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Q, Wang Y, Chang Q, Wang J, Gong S, Li H, et al. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014;21:71–80. doi: 10.1038/gt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibata SB, Ranum PT, Moteki H, Pan B, Goodwin AT, Goodman SS, et al. RNA interference prevents autosomal-dominant hearing loss. Am J Hum Genet. 2016;98:1101–13. doi: 10.1016/j.ajhg.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol. 2017;35:280–4. doi: 10.1038/nbt.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren Y, Landegger LD, Stankovic KM. Gene therapy for human sensorineural hearing loss. Front Cell Neurosci. 2019;13:323–323. doi: 10.3389/fncel.2019.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jüttner J, Szabo A, Gross-Scherf B, Morikawa RK, Rompani SB, Hantz P, et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nat Neurosci. 2019;22:1345–56. doi: 10.1038/s41593-019-0431-2. [DOI] [PubMed] [Google Scholar]
- 35.Hu X, Wang J, Yao X, Xiao Q, Xue Y, Wang S, et al. Screened AAV variants permit efficient transduction access to supporting cells and hair cells. Cell Discov. 2019;5:49. doi: 10.1038/s41421-019-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J, Nist-Lund C, Solanes P, Goldberg H, Wu J, Pan B, et al. Efficient viral transduction in mouse inner ear hair cells with utricle injection and AAV9-PHP.B. Hear Res. 2020;107882. [DOI] [PubMed]
- 37.Shu Y, Tao Y, Wang Z, Tang Y, Li H, Dai P, et al. Identification of adeno-associated viral vectors that target neonatal and adult mammalian inner ear cell subtypes. Hum Gene Ther. 2016;27:687–99. doi: 10.1089/hum.2016.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu X, Chai R, Guo L, Dong B, Li W, Shu Y, et al. Transduction of adeno-associated virus vectors targeting hair cells and supporting cells in the neonatal mouse cochlea. Front Cell Neurosci. 2019;13:8. doi: 10.3389/fncel.2019.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao Y, Huang M, Shu Y, Ruprecht A, Wang H, Tang Y, et al. Delivery of adeno-associated virus vectors in adult mammalian inner-ear cell subtypes without auditory dysfunction. Hum Gene Ther. 2018;29:492–506. doi: 10.1089/hum.2017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Qi J, Chen X, Tang M, Chu C, Zhu W, et al. Critical role of spectrin in hearing development and deafness. Sci Adv. 2019;5:eaav7803. doi: 10.1126/sciadv.aav7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chien WW, McDougald DS, Roy S, Fitzgerald TS, Cunningham LL. Cochlear gene transfer mediated by adeno-associated virus: comparison of two surgical approaches. Laryngoscope. 2015;125:2557–64. doi: 10.1002/lary.25317. [DOI] [PubMed] [Google Scholar]
- 42.Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, et al. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–41. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- 43.Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105:7827–32. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berns KI, Srivastava A. Next generation of adeno-associated virus vectors for gene therapy for human liver diseases. Gastroenterol Clin North Am. 2019;48:319–30. doi: 10.1016/j.gtc.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buning H, Srivastava A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol Ther Methods Clin Dev. 2019;12:248–65. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Lieshout LP, Domm JM, Rindler TN, Frost KL, Sorensen DL, Medina SJ, et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Mol Ther Methods Clin Dev. 2018;9:323–9. doi: 10.1016/j.omtm.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16:1252–60. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 48.Koerber JT, Jang JH, Schaffer DV. DNA shuffling of adenoassociated virus yields functionally diverse viral progeny. Mol Ther. 2008;16:1703–9. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82:5887–911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choudhury SR, Fitzpatrick Z, Harris AF, Maitland SA, Ferreira JS, Zhang Y, et al. In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol Ther. 2016;24:1247–57. doi: 10.1038/mt.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mathelier A, Fornes O, Arenillas DJ, Chen C-y, Denay G, Lee J, et al. JASPAR 2016: a major expansion and update of the openaccess database of transcription factor binding profiles. Nucleic Acids Res. 2015;44:D110–5. doi: 10.1093/nar/gkv1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hai T, Cao C, Shang H, Guo W, Mu Y, Yang S, et al. Pilot study of large-scale production of mutant pigs by ENU mutagenesis. Elife. 2017;6:e26248. doi: 10.7554/eLife.26248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hao QQ, Li L, Chen W, Jiang QQ, Ji F, Sun W, et al. Key genes and pathways associated with inner ear malformation in SOX10 (p.R109W) mutation pigs. Front Mol Neurosci. 2018;11:181. doi: 10.3389/fnmol.2018.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang Q, Wang J, Li Q, Kim Y, Zhou B, Wang Y, et al. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol Med. 2015;7:1077–86. doi: 10.15252/emmm.201404929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim MA, Cho HJ, Bae SH, Lee B, Oh SK, Kwon TJ, et al. Methionine sulfoxide reductase B3-targeted In utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid Redox Signal. 2016;24:590–602. doi: 10.1089/ars.2015.6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim MA, Kim SH, Ryu N, Ma JH, Kim YR, Jung J, et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics. 2019;9:7184–99. doi: 10.7150/thno.38032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iizuka T, Kamiya K, Gotoh S, Sugitani Y, Suzuki M, Noda T, et al. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum Mol Genet. 2015;13:13. doi: 10.1093/hmg/ddv109. [DOI] [PubMed] [Google Scholar]
- 58.Isgrig K, Shteamer JW, Belyantseva IA, Drummond MC, Fitzgerald TS, Vijayakumar S, et al. Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol Ther. 2017;25:780–91. doi: 10.1016/j.ymthe.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geng R, Omar A, Gopal RS, Chen H-CD, Stepanyan R, Basch LM, et al. Modeling and preventing progressive hearing loss in Usher syndrome III. Sci Rep. 2017;7:13480. doi: 10.1038/s41598-017-13620-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Moyed H, Cepeda AP, Jung S, Moser T, Kügler S, Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. 2019;11:e9396. doi: 10.15252/emmm.201809396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Askew C, Rochat C, Pan B, Asai Y, Ahmed H, Child E, et al. Tmc gene therapy restores auditory function in deaf mice. Sci Transl Med. 2015;7:295ra108. doi: 10.1126/scitranslmed.aab1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nist-Lund CA, Pan B, Patterson A, Asai Y, Chen T, Zhou W, et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat Commun. 2019;10:236. doi: 10.1038/s41467-018-08264-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao X, Tao Y, Lamas V, Huang M, YehW-H, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553:217–21. doi: 10.1038/nature25164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simmons DD, Tong B, Schrader AD, Hornak AJ. Oncomodulin identifies different hair cell types in the mammalian inner ear. J Comp Neurol. 2010;518:3785–802. doi: 10.1002/cne.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maison S, Liberman LD, Liberman MC Type II cochlear ganglion neurons do not drive the olivocochlear reflex: re-examination of the cochlear phenotype in peripherin knock-out mice. eNeuro. 2016;3:ENEURO.0207–16.2016. [DOI] [PMC free article] [PubMed]
- 66.Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–55. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]
- 67.McLean WJ, Yin X, Lu L, Lenz DR, McLean D, Langer R, et al. Clonal expansion of Lgr5-positive cells from mammalian cochlea and high-purity generation of sensory hair cells. Cell Rep. 2017;18:1917–29. doi: 10.1016/j.celrep.2017.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bermingham-McDonogh O, Oesterle EC, Stone JS, Hume CR, Huynh HM, Hayashi T. Expression of Prox1 during mouse cochlear development. J Comp Neurol. 2006;496:172–86. doi: 10.1002/cne.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boström M, Anderson M, Lindholm D, Park K-H, Schrott-Fischer A, Pfaller K, et al. Neural network and “ganglion” formations in vitro: a video microscopy and scanning electron microscopy study on adult cultured spiral ganglion cells. Otol Neurotol. 2007;28:1109–19. doi: 10.1097/MAO.0b013e318159e710. [DOI] [PubMed] [Google Scholar]
- 70.Ranum P, Goodwin A, Yoshimura H, Kolbe D, Walls W, Koh JY, et al. Insights into the biology of hearing and deafness revealed by single-cell RNA sequencing. Cell Rep. 2019;26:3160–71. doi: 10.1016/j.celrep.2019.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rio C, Dikkes P, Liberman MC, Corfas G. Glial fibrillary acidic protein expression and promoter activity in the inner ear of developing and adult mice. J Comp Neurol. 2002;442:156–62. doi: 10.1002/cne.10085. [DOI] [PubMed] [Google Scholar]
- 72.Ladrech S, Lenoir M, Ruel J, Puel J-L. Microtubule-associated protein 2 (MAP2) expression during synaptic plasticity in the guinea pig cochlea. Hear Res. 2003;186:85–90. doi: 10.1016/s0378-5955(03)00302-2. [DOI] [PubMed] [Google Scholar]
- 73.Liu H, Pecka JL, Zhang Q, Soukup GA, Beisel KW, He DZZ. Characterization of transcriptomes of cochlear inner and outer hair. Cells. 2014;34:11085–95. doi: 10.1523/JNEUROSCI.1690-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pannier S, Couloigner V, Messaddeq N, Elmaleh-Berges M, Munnich A, Romand R, et al. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim Biophys Acta. 2009;1792:140–7. doi: 10.1016/j.bbadis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 75.Huang X, Liu J, Wu W, Hu P, Wang Q. Taurine enhances mouse cochlear neural stem cell transplantation via the cochlear lateral wall for replacement of degenerated spiral ganglion neurons via sonic hedgehog signaling pathway. Cell Tissue Res. 2019;378:49–57. doi: 10.1007/s00441-019-03018-6. [DOI] [PubMed] [Google Scholar]
- 76.Parker MA, Jiang K, Kempfle JS, Mizutari K, Simmons CL, Bieber R, et al. TAK1 expression in the cochlea: a specific marker for adult supporting cells. J Assoc Res Otolaryngol. 2011;12:471–83. doi: 10.1007/s10162-011-0265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spencer RF, Shaia WT, Gleason AT, Sismanis A, Shapiro SM. Changes in calcium-binding protein expression in the auditory brainstem nuclei of the jaundiced Gunn rat. Hear Res. 2002;171:129–41. doi: 10.1016/s0378-5955(02)00494-x. [DOI] [PubMed] [Google Scholar]
- 78.Hosoya M, Fujioka M, Matsuda S, Ohba H, Shibata S, Nakagawa F, et al. Expression and function of Sox21 during mouse cochlea development. Neurochem Res. 2011;36:1261–9. doi: 10.1007/s11064-011-0416-3. [DOI] [PubMed] [Google Scholar]
- 79.Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV. Sensory neuron diversity in the inner ear is shaped by activity. Cell. 2018;174:1229–46.e17. doi: 10.1016/j.cell.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li S, Mecca A, Kim J, Caprara GA, Wagner EL, Du T-T, et al. Myosin-VIIa is expressed in multiple isoforms and essential for tensioning the hair cell mechanotransduction complex. Nature Communications. 2020;11:2066. doi: 10.1038/s41467-020-15936-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR. Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol. 2008;9:65–89. doi: 10.1007/s10162-007-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steevens AR, Glatzer JC, Kellogg CC, Low WC, Santi PA, Kiernan AE. SOX2 is required for inner ear growth and cochlear nonsensory formation before sensory development. Development. 2019;146:dev170522. doi: 10.1242/dev.170522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vyas P, Wu JS, Jimenez A, Glowatzki E, Fuchs PA. Characterization of transgenic mouse lines for labeling type I and type II afferent neurons in the cochlea. Sci Rep. 2019;9:5549. doi: 10.1038/s41598-019-41770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roux I, Hosie S, Johnson SL, Bahloul A, Cayet N, Nouaille S, et al. Myosin VI is required for the proper maturation and function of inner hair cell ribbon synapses. Hum Mol Genet. 2009;18:4615–28. doi: 10.1093/hmg/ddp429. [DOI] [PubMed] [Google Scholar]
- 85.Nagy I, Bodmer M, Schmid S, Bodmer D. Promyelocytic leukemia zinc finger protein localizes to the cochlear outer hair cells and interacts with prestin, the outer hair cell motor protein. Hear Res. 2005;204:216–22. doi: 10.1016/j.heares.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 86.Hertzano R, Puligilla C, Chan SL, Timothy C, Depireux DA, Ahmed Z, et al. CD44 is a marker for the outer pillar cells in the early postnatal mouse inner ear. J Assoc Res Otolaryngol. 2010;11:407–18. doi: 10.1007/s10162-010-0211-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Froud KE, Wong ACY, Cederholm JME, Klugmann M, Sandow SL, Julien J-P, et al. Type II spiral ganglion afferent neurons drive medial olivocochlear reflex suppression of the cochlear amplifier. Nat Commun. 2015;6:7115. doi: 10.1038/ncomms8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan B, Géléoc Gwenaelle S, Asai Y, Horwitz Geoffrey C, Kurima K, Ishikawa K, et al. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron. 2013;79:504–15. doi: 10.1016/j.neuron.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bermingham-McDonogh O, Oesterle EC, Stone JS, Hume CR, Huynh HM, Hayashi T. Expression of Prox1 during mouse cochlear development. J Comp Neurol. 2006;496:172–86. doi: 10.1002/cne.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu S, Wang Y, Lu Y, Li W, Liu W, Ma J, et al. The key transcription factor expression in the developing vestibular and auditory sensory organs: a comprehensive comparison of spatial and temporal patterns. Neural Plast. 2018;2018:7513258. doi: 10.1155/2018/7513258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie D, Hu P, Xiao ZA, Wu W, Chen Y, Xia K. Subunits of voltage-gated calcium channels in murine spiral ganglion cells. Acta oto-laryngologica. 2007;127:8–12. doi: 10.1080/00016480600627927. [DOI] [PubMed] [Google Scholar]
- 92.Zhao HB, Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J Comp Neurol. 2006;499:506–18. doi: 10.1002/cne.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang T, Scholl ES, Pan N, Fritzsch B, Haeseleer F, Lee A. Expression and localization of CaBP Ca2+ binding proteins in the mouse cochlea. PLoS ONE. 2016;11:e0147495. doi: 10.1371/journal.pone.0147495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J, Verkman AS. Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem. 2001;276:31233–7. doi: 10.1074/jbc.M104368200. [DOI] [PubMed] [Google Scholar]
- 95.Girotto G, Vuckovic D, Buniello A, Lorente-Cánovas B, Lewis M, Gasparini P, et al. Expression and replication studies to identify new candidate genes involved in normal hearing function. PLoS ONE. 2014;9:e85352–e85352. doi: 10.1371/journal.pone.0085352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scheffer DI, Shen J, Corey DP, Chen ZY. Gene expression by mouse inner ear hair cells during development. J Neurosci. 2015;35:6366–80. doi: 10.1523/JNEUROSCI.5126-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang EJ, Liu W, Fritzsch B, Bianchi LM, Reichardt LF, Xiang M. Brn3a is a transcriptional regulator of soma size, target field innervation and axon pathfinding of inner ear sensory neurons. Development. 2001;128:2421–32. doi: 10.1242/dev.128.13.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hickox AE, Wong ACY, Pak K, Strojny C, Ramirez M, Yates JR, et al. Global analysis of protein expression of inner ear hair. Cells. 2017;37:1320–39. doi: 10.1523/JNEUROSCI.2267-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]