

Abstract
Increasing evidence suggests that overt oxidative stress within the retina plays an important role in the progression of age-related retinal decline, and in particular, in the disease age-related macular degeneration (AMD). Nuclear factor erythroid 2-like 2 (Nrf2) is a master transcription factor that upregulates numerous of antioxidant/detoxification genes. Nrf2−/− mice develop progressive retinal degeneration that includes the formation of drusen-like deposits, lipofuscin, and sub-retinal pigment epithelium (RPE) deposition of inflammatory proteins. Furthermore, strategies that promote Nrf2 activation have shown promise for the treatment of cone/rod dystrophies and other forms of retinal degeneration. Herein we explored whether utilizing a small molecule-inducible version of Nrf2 confers additional protection against oxidative stresses when compared to a constitutively expressed version of Nrf2. Stable populations of human ARPE-19 cells were generated that express either constitutive FLAG-tagged (FT) Nrf2 (FT cNrf2) or doxycycline (dox)-inducible FT Nrf2 (FT iNrf2) at low levels (~4.5 fold vs. endogenous). Expression of either FT cNRF2 or FT iNrf2 upregulated canonical antioxidant genes (e.g., NQO1, GCLC). Both FT cNrf2 and FT iNrf2 ARPE-19 cells were protected from cigarette smoke extract-induced nitric oxide generation to similar extents. However, only FT iNrf2 cells demonstrated enhanced resistance to doxorubicin and cumene hydroperoxide-mediated increases in mitochondrial superoxide and lipid peroxidation, respectively, and did so in a dox-dependent manner. These results suggest that therapeutic approaches which conditionally control Nrf2 activity may provide additional protection against acute oxidative stresses when compared to constitutively expressed Nrf2 strategies.
The generation of reactive oxygen/nitrogen species (ROS/RNS) due to a number of cellular processes including inflammation (Thelen et al., 1993), oxidative phosphorylation (Wei, 1998), and metal-induced autoxidation (Kasprzak, 1995), is a normal aspect of cellular physiology. Thus, cells are constantly challenged by endogenous factors as well as environmental stressors (e.g., xenobiotics, UV light, pro-oxidant chemicals, etc.) that generate oxidative stress (i.e., accumulation of highly reactive oxygen-based species capable of detrimentally modifying critical cellular components such as proteins, lipids, and DNA). The retina is a tissue within the body that is particularly susceptible to such forms of oxidative stress. It is among the highest oxygen-consuming tissues (Yu and Cringle, 2005), constantly exposed to light, and also contains high levels of easily-oxidized polyunsaturated fatty acids (Jeffrey et al., 2001).
Several lines of evidence indicate that oxidative stress in the retina plays a role in the onset and progression of eye diseases such as age-related macular degeneration (AMD) (Beatty et al., 2000; Jarrett and Boulton, 2012; Winkler et al., 1999). For example, the Age-Related Eye Disease Study (AREDS) found that administering high levels of antioxidants to patients with intermediate dry AMD reduced their risk of progression to advanced dry AMD by ~25% (Age-Related Eye Disease Study Research, 2001). In addition, cigarette smoke, which contains high levels of ROS and RNS, is the primary modifiable risk factor for the development of geographic atrophy and choroidal neovascularization (Smith et al., 2001; Tomany et al., 2004). Furthermore, two independent human association studies linked polymorphisms in superoxide dismutase 2 (SOD2), an antioxidant gene, to the progression of exudative AMD (Kimura et al., 2000; Kowalski et al., 2010), although these results have been challenged by other groups (Esfandiary et al., 2005; Kondo et al., 2009). Nonetheless, a number of transgenic mice deficient in, or lacking, key antioxidant enzymes develop retinal abnormalities reminiscent of pathology observed in AMD. For example, superoxide dismutase 1−/− (SOD1−/−) mice develop drusen-like deposits, retinal pigment epithelium (RPE) dysfunction (junction protein disruption), and choroidal neovascularization (Imamura et al., 2006). SOD2 deficient mice demonstrate increased autofluoresecence including higher levels of N-retinylidene-N-retinyl-ethanolamine (A2E) and iso-A2E bis-retinoids (Biswal et al., 2016; Justilien et al., 2007). DJ-1 null mice are characterized by RPE degeneration (vacuolization), retinal thinning, and accelerated RPE atrophy after an oxidative challenge (Bonilha et al., 2015, 2017). And lastly, nuclear factor erythroid 2-like 2 (Nrf2) null mice show age-dependent drusen-like protein deposition, RPE degeneration (vacuolization), and sporadic choroidal neovascularization (Zhao et al., 2011).
Given the strong epidemiologic and empirical support for the involvement of oxidative stress in the initiation and/or progression of retinal diseases, targeting oxidative stress as a therapy for diseases such as AMD has received increased attention. Specifically, the ubiquitously expressed basic leucine zipper transcription factor, Nrf2, is consistently proposed to confer potential wide-ranging antioxidant and detoxification benefits in the retina and in RPE cells (Barnett and Handa, 2013; Cano et al., 2010; Lambros and Plafker, 2016; Liu et al., 2016a, 2016b). Under basal, non-oxidizing conditions, Nrf2 is sequestered in the cytoplasm and constitutively degraded by an inhibitory ubiquitin E3 ligase complex composed of Kelch-like ECH-associated protein 1 (KEAP1), cullin-3 (CUL3), and RING-box 1 (RBX1) (Taguchi et al., 2011; Villeneuve et al., 2010). Upon modification of KEAP1 with electrophiles such as ROS/RNS, Nrf2 is released and translocates to the nucleus where, along with small Maf protein cofactors, it upregulates the transcription of phase II antioxidant and detoxification genes. Once Nrf2 is translocated, a series of autoregulatory feedback loops are initiated which ultimately limit the extent and duration of Nrf2 activation, including transcriptional upregulation of KEAP1/CUL3/RBX1 (Kaspar and Jaiswal, 2010; Lee et al., 2007) and the phosphorylation of Nrf2 by glycogen synthase kinase 3β (GSK-3β), which promotes its nuclear export (Jain and Jaiswal, 2006), guiding the cell back towards homeostasis. Thus, under physiologic conditions, levels of nuclear Nrf2 typically proceed in a sinusoidal manner, peaking after oxidative stress, and returning to baseline after the stress is remedied.
It is generally accepted that the ability of many cells within the body (including retinal cells) to mount an effective antioxidant response declines with age (Bruns et al., 2015; Sachdeva et al., 2014; Suh et al., 2004). Thus, from an eye disease perspective, strategies have been developed to counteract this potential decline in Nrf2 signaling in the retina through direct over-expression of Nrf2 (Xiong et al., 2015), or through disrupting the inhibitory KEAP1-Nrf2 interaction (Ildefonso et al., 2016). While these strategies protected against photoreceptor/retinal ganglion cell loss and NaIO3-induced retinal degeneration, respectively, these approaches, in theory, would likely force constitutive Nrf2 activation in a non-physiological manner that may lead to unanticipated adverse effects. Due to the potential for autoregulatory feedback of Nrf2, and possible complications due to constitutive Nrf2 overexpression, we decided to explore whether there was any difference in signaling and/or protection against oxidative insults conferred by constitutive vs. inducible expression of Nrf2 in RPE cells. To achieve this, we generated two sets of ARPE-19-based cell lines (ATCC#: CRL-2302, routinely mycoplasma tested, STR verified, University of Arizona Genomics Core, Tucson, AZ) by infecting them with VSV-G-pseudotyped lentivirus and selecting for stable populations using puromycin. The first set of cells were infected with virus packaged with either empty pLenti CMV/TO Puro DEST (Addgene plasmid# 17293, gift from Eric Campeau), or pLenti CMV/TO Puro containing an N-terminally FLAG-tagged (FT), full length Nrf2. Since ARPE-19 cells lack endogenous tet-repressor, use of this Nrf2 construct results in constitutive expression (referred to as FT cNrf2 here onward). The second set of cells we generated were used for dox-inducible expression. For this set of cells, we infected ARPE-19 cells with virus generated using either empty vector (pCW57-MCS1-2A-MCS2, Addgene plasmid # 71782, gift from Adam Karpf), or pCW57 containing FT Nrf2. pCW57 is an ‘all-in-one’ tet-on vector that is the sole vector necessary for generating a dox-inducible system. Use of this Nrf2 construct results in inducible Nrf2 expression (referred to as FT iNrf2 here onward). After the selection process with puromycin (for ≥ 2 weeks), both sets of these cells, along with their respective control cells, were characterized by quantitative PCR (qPCR) and western blotting to assess for upregulation of Nrf2 as well as classic phase II antioxidant genes downstream of Nrf2. Cells were plated at near confluence, allowed to attach overnight, and maintained in culture for an additional 96 h [without dox for FT cNrf2 and control cells, with or without dox (100 ng/mL) for FT iNrf2 and control cells]. Extraction of total mRNA (Aurum, BioRad, Hercules, CA) followed by cDNA synthesis (qScript cDNA Supermix, Quanta Bio, Beverly, MA) and qPCR (Quant Studio 6, Life Technologies, Carlsbad, CA) demonstrated low levels of FT cNrf2 overexpression (4.6 ± 0.5 vs. endogenous, p < 0.01, TaqMan assay Hs00975960_m1, Thermo Fisher Scientific, Waltham, MA) in the constitutive cells compared to empty vector control cells (Fig. 1A) after normalizing to β-actin (TaqMan assay Hs03023880_g1). At these low levels of Nrf2 overexpression, we did not observe substantial increases in the transcription of downstream phase II genes glutamate cysteine ligase catalytic unit (GCLC, 1.4 ± 0.2, TaqMan assay Hs00155249_m1), heme oxygenase 1 (HO1, 1.2 ± 0.1, TaqMan assay Hs01110250_m1), or SOD1 (0.9 ± 0.08, TaqMan assay Hs00533490_m1). However, NADPH quinone reductase 1 (NQO1) was significantly upregulated (3.6 ± 0.8, p < 0.05, TaqMan assay Hs00168547_m1). qPCR results were corroborated by western blotting using whole cell lysates (WCL, lysis buffer: [50 mM HEPES (pH 8), 10 mM KCl, 2 mM MgCl2, 1% SDS], ~15–20 μg/sample loaded, Fig. 1B) run under reducing and denaturing conditions. FT cNrf2 cells demonstrated slight increases in GCLC, as well as NQO1 protein levels, but not in HO1 protein levels. Nrf2 nuclear translocation in FT cNrf2 cells was confirmed after cell fractionation using NE-PER (Thermo Fisher Scientific, 15–25 μg loaded for cytosolic fractions, 1.5–20 μg loaded for nuclear fractions, Fig. 1B). ARPE-19 cells inducibly expressing FT iNrf2 at similar levels to FT cNrf2 cells (4.6 ± 1.1 vs. endogenous, p < 0.05, Fig. 1C) demonstrated a similar phase II gene expression profile; GCLC (1.6 ± 0.6), HO1 (1.3 ± 0.3), SOD1 (1.3 ± 0.3), and NQO1 (5.1 ± 1.8, p < 0.05, Fig. 1C). Elevation of protein levels of GCLC and NQO1 was confirmed using western blotting of WCLs (Fig. 1D). Lack of significant upregulation of canonical Nrf2 downstream genes such as HO1 and SOD1 could be due to the timing (96 h) of Nrf2 induction for these experiments, and might be observable at earlier timepoints, or by using higher levels of over-expression. Nuclear fractionation again confirmed translocation of FT iNrf2 (Fig. 1D). Thus, within this limited assessment of Nrf2-dependent phase II genes, FT cNrf2 and FT iNrf2 appear to have a similar ability to activate downstream genes, yet we did note that iNrf2 consistently had higher nuclear Nrf2 levels than cNrf2. (cf. Fig. 1B nuclear to Fig. 1D nuclear). We subsequently assessed whether transcript or protein levels of KEAP1, a negative regulator of Nrf2, were altered differently in cNrf2 vs. iNrf2 cells in a compensatory autoregulatory mechanism (Lee et al., 2007). We found no changes to either KEAP1 protein levels or transcript levels (TaqMan assay Hs00202227_m1), nor did we observe any changes in transcripts of other members (i.e., CUL3, RBX1, TaqMan assays Hs00180183_m1 and Hs00360274_m1, respectfully) of the E3 ubiquitin ligase complex (Fig. 1E and F).
Fig. 1.
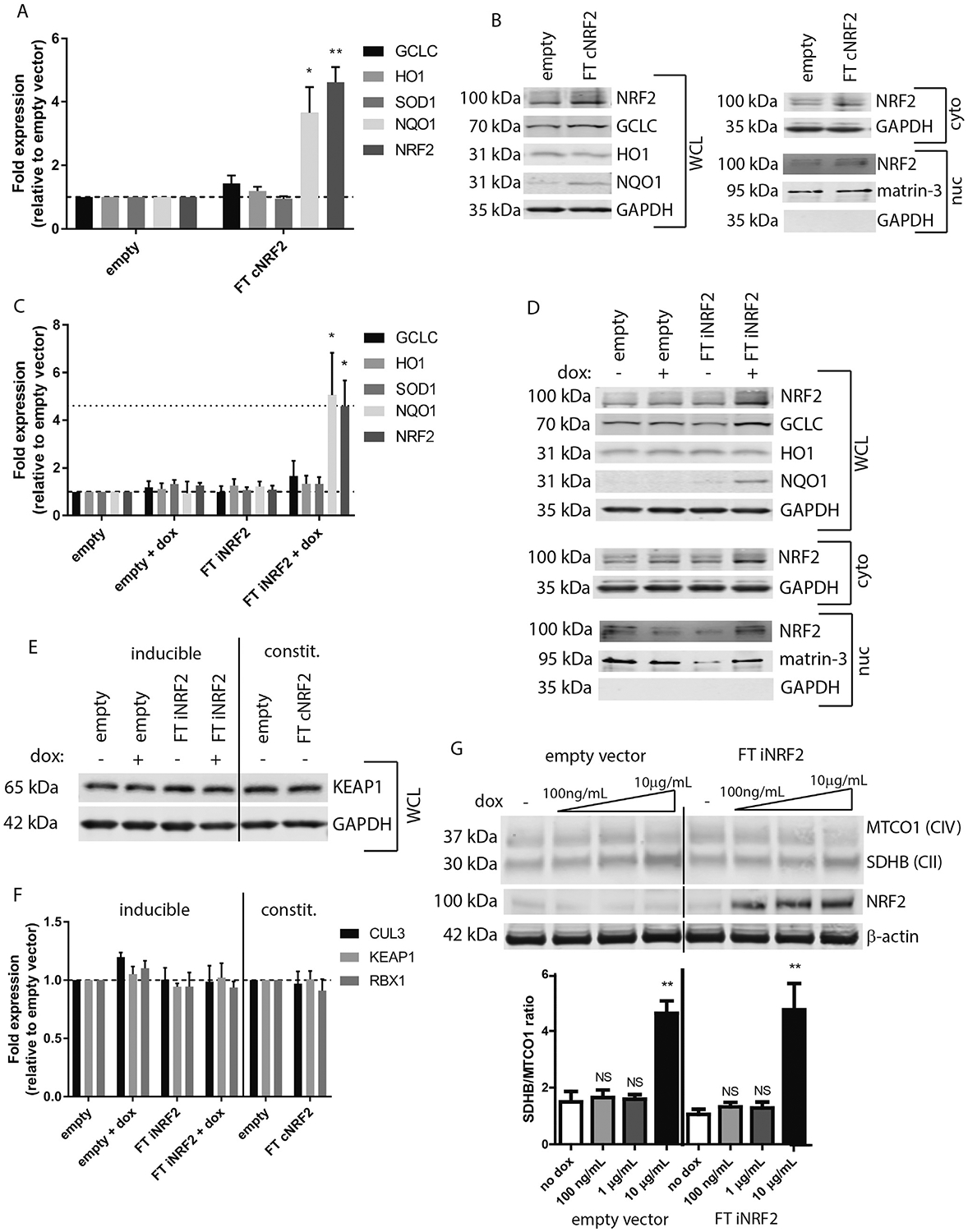
Constitutive and inducible Nrf2 expression results in increased phase II and other oxidative stress response genes. (A, B) ARPE-19 cells constitutively expressing FLAG-tagged (FT) Nrf2 (FT cNrf2) were assessed for canonical downstream Nrf2 genes using TaqMan probes (A) or western blotting (B). Primary antibodies were as follows: Nrf2 (Proteintech, Chicago, IL), NQO1 (Santa Cruz, Dallas, TX), HO1 (Bethyl Laboratories, Montgomery, TX), GCLC (Proteintech), GAPDH (Santa Cruz), and matrin-3 (Bethyl Laboratories). Nrf2 and KEAP1 (see panel ‘E’) were imaged on a LI-COR Odyssey Fc (LI-COR, Lincoln, NE) using either SuperSignal West Femto substrate (Thermo Fisher Scientific) or an IRDye secondary antibody (LI-COR, Lincoln, NE), while remaining proteins were detected solely using IRDye secondary antibodies and imaged on a LI-COR CLx (LI-COR). (C, D) ARPE-19 cells expressing inducible FT Nrf2 (FT iNrf2) were also assayed by qPCR (C) and western blotting (D). Dotted line in panel ‘C’ represents FT cNrf2 levels, as shown in panel ‘A’. (E, F) KEAP1 protein levels and KEAP1/CUL3/RBX1 ubiquitin ligase complex transcripts are not changed in FT cNrf2 or FT iNrf2 samples. KEAP1 primary antibody was from Santa Cruz. (G) Low levels of dox (100 ng/mL-1 μg/mL) for 96 h do not perturb mitonuclear protein balance. OXPHOS antibody cocktail (Abcam, Cambridge, MA), β-actin (LI-COR)). For qPCR experiments, n = 3, mean ± S.D., *p < 0.05, **p < 0.01, one sample t-test against a hypothetical mean of 1 (i.e., unchanged). Representative data of ≥3 independent experiments for all western blots, mean ± S.D. **p < 0.01, unpaired Student’s t-test assuming equal variance amongst samples.
Many estimates suggest that the majority of endogenously produced ROS originate from the normal activity of the electron transport chain in the mitochondria. Since mitochondria are most likely descendants of ancient proteobacteria (Gray et al., 1999), and antibiotics such as tetracyclines (e.g., dox) target conserved aspects of bacterial protein synthesis (Schnappinger and Hillen, 1996), use of tetracyclines to control gene expression in mammalian cells has drawn scrutiny (Chatzispyrou et al., 2015; Moullan et al., 2015). Thus, we purposely used low levels of dox (100 ng/mL) in our ARPE-19 culture system for 96 h and confirmed that these levels did not disrupt mitonuclear protein imbalance as described previously using higher levels of dox (Fig. 1G, (Moullan et al., 2015)). Furthermore, we did not observe any consistent dox-dependent increase in nitric oxide, mitochondrial superoxide, or lipid peroxidation (Fig. 2B, D, F, respectively) that would indicate that the levels of dox we used significantly altered the redox state of ARPE-19 cells.
Fig. 2.
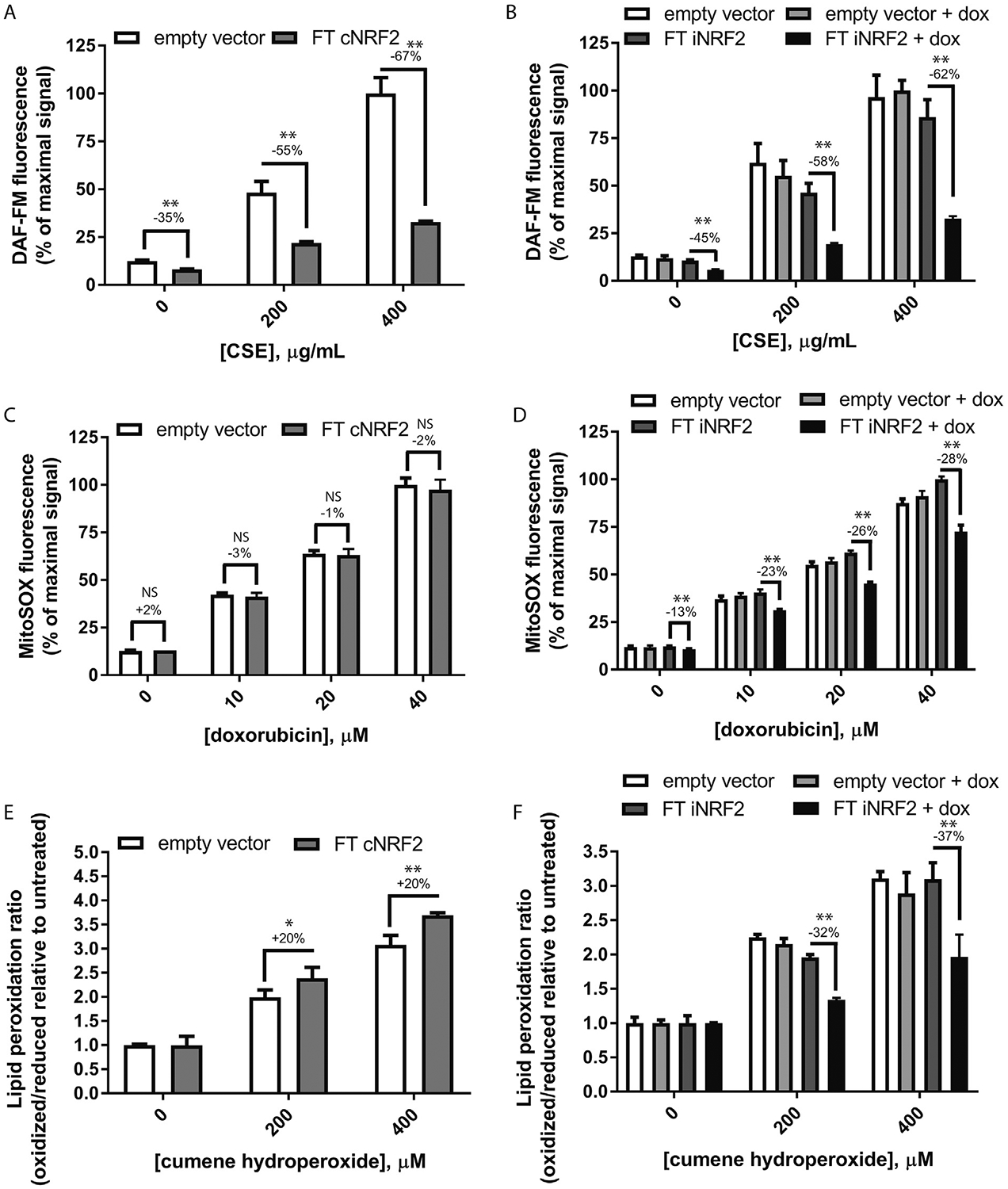
Dox-mediated induction of Nrf2 confers enhanced protection against three different acute oxidative stresses. Sets of constitutive or inducible Nrf2 cells were treated with either (A, B) CSE and evaluated for NO levels, (C, D) doxorubicin and evaluated for mitochondrial superoxide, or (E, F) cumene hydroperoxide and assessed for changes to the lipid peroxidation profile. Representative data ≥3 independent experiments performed using biological triplicates/quadruplicates for each data point, mean ± S.D. * = p < 0.05, **p < 0.01, unpaired Student’s t-test assuming equal variance amongst samples.
Next, we assessed the potential functional consequences of FT cNrf2 or FT iNrf2 expression by assaying the cells for antioxidant protection using three acute oxidative stresses. Control and FT cNrf2 or FT iNrf2 (induced or uninduced) ARPE-19 cells were labeled with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM, Thermo Fisher Scientific), a nitric oxide (NO)-sensitive dye, followed by treatment with cigarette smoke extract (CSE, Murty Pharmaceuticals, Lexington, KY) for 1 h. Cells were then washed, trypsinized, neutralized, and analyzed on an Attune NxT Flow Cytometer (Thermo Fisher Scientific) using an autosampler and the BL1 channel. Cells expressing either FT cNrf2 or FT iNrf2 were significantly protected from NO production after CSE treatment (Fig. 2A and B). Constitutive expression of FT Nrf2 resulted in a reduction of 55–67% in the DAF-FM signal after CSE treatment, whereas inducible Nrf2 expression yielded a comparable 58e62% reduction in signal (Fig. 2A and B). Notably, basal NO levels in vehicle-treated constitutive and inducible cells were also significantly downregulated by 35 and 45%, respectively (Fig. 2A and B). These data demonstrate that both approaches for inducing Nrf2 expression in ARPE-19 cells are able to significantly prevent NO formation and do so to similar extents.
Given the observation of polymorphisms in SOD2 associated with AMD (Kimura et al., 2000; Kowalski et al., 2010) and the observations that mice deficient in SOD1/2 suffer from various phenotypes associated with retinal degeneration (Biswal et al., 2016; Imamura et al., 2006; Justilien et al., 2007), we next assessed whether ARPE-19 cells expressing Nrf2 would be protected from superoxide formation. FT cNrf2 or FT iNrf2 ARPE-19 cells were treated with doxorubicin for 1 h followed by cell labeling with MitoSOX Red (Thermo Fisher Scientific) for 30 min. Cells were processed as described above for flow cytometery using the BL2/BL3 channel. We were surprised to find that cells constitutively expressing Nrf2 were not protected to any degree against doxorubicin-mediated superoxide formation (Fig. 2C). In contrast, however, dox-dependent induction of FT iNrf2 significantly protected ARPE-19 cells from superoxide formation at levels ranging from 13 to 28% (Fig. 2D), demonstrating enhanced protection against acute oxidative stress using an inducible form of Nrf2.
Finally, we measured the extent of cumene hydroperoxide-induced lipid peroxidation between constitutive and inducible Nrf2 cells. Lipid peroxidation has been demonstrated to increase with age in the rodent retina (Castorina et al., 1992), and lipid peroxidation products [such as malondialdehyde (MDA) or 4-hydroxynonenal (4-HNE)] can wreak havoc on a number of critical RPE pathways including lysosomal degradation (Kaemmerer et al., 2007) and inflammasome activation (Kauppinen et al., 2012). ARPE-19 cells were treated with cumene hydroperoxide for 1 h followed by labeling with Image-iT Lipid Peroxidation Sensor (Thermo Fisher Scientific). Cells were then processed as described above and lipid peroxidation was measured using flow cytometry (BL1 for oxidized lipid/YL1 for reduced lipid (Fig. 2E and F)). We were surprised to find that cells expressing FT cNrf2 actually demonstrated a 20% higher ratio of oxidized/reduced lipid peroxidation than empty vector control cells (Fig. 2E). However, consistent with our previous observations monitoring NO or superoxide, dox-treated FT iNrf2 cells demonstrated significant protection (by 32–37%) against acute lipid peroxidation generated by cumene hydroperoxide (Fig. 2F).
To the best of our knowledge, this is the first proof-of-principle demonstration that cells stably encoding a regulatable Nrf2 transgene are protected from oxidative insults in a small molecule-dependent manner. In fact, our results in a simple, cultured, RPE system indicate that use of a dox-inducible form of Nrf2 bestows enhanced antioxidant properties when compared to a constitutively expressed form of Nrf2. However, a simple analysis of classic Nrf2 downstream genes, or prominent repressive mechanisms (e.g., KEAP1/CUL3/RBX1 complex) in FT cNrf2 vs. FT iNrf2 cells showed no clear differences between these two strategies, signaling-wise. These observations suggest that measurement of a few downstream genes cannot be used to definitively predict a functional outcome in Nrf2 signaling, and that actual protection from certain acute oxidative stresses is likely a culmination of global changes to antioxidant signaling pathways and/or repressive mechanisms.
Importantly, our data demonstrate that inducible Nrf2 expression could serve as an effective gene therapy approach for the treatment of oxidative stress-associated retinal degeneration since it is regulatable, and it can protect against multiple unique, acute stresses with distinct mechanisms of action. It remains to be determined how cells with constitutive vs. inducible Nrf2 overexpression would fare when exposed to more chronic oxidative stress, as is likely to happen in vivo. Caution must be taken before moving this strategy forward to animal models since it would require either systemic administration of dox, or repeated ocular injections. This type of approach may cause unwanted off-target effects of dox in susceptible ocular or extra-ocular tissues. However, a recent study used transient (2 days on/5 days off) and cyclic (up to 35 cycles) treatment of genetically engineered mice with dox without any observation of deleterious effects (Ocampo et al., 2016). In fact, dox administration in the Ocampo et al. study was used to reduce age-associated hallmarks by partial reprogramming of stem cells. Alternatively, these potentially confounding issues with using dox in vivo could be circumvented by the use of alternative inducible systems such as the cumate switch system (Mullick et al., 2006), or even a light-induced strategy (Wang et al., 2012), although the latter may come with its own challenges (Organisciak and Vaughan, 2010). Nonetheless, our results have important implications for gene therapy approaches that utilize activation of the Nrf2 pathway for protection of the retina and suggest that an inducible strategy may yield effective protection against a number of oxidative stresses.
Acknowledgements
This work was funded in part by an endowment from the Roger and Dorothy Hirl Research Fund (JDH), a vision research grant from the Karl Kirchgessner Foundation (JDH), a National Eye Institute Visual Science Core Grant (EY020799), an unrestricted grant from Research to Prevent Blindness (RPB), a Career Development Award from RPB (JDH), and a Medical Student Research Fellowship from RPB (KTV).
References
- Age-Related Eye Disease Study Research, G., 2001. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol 119, 1417–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett BP, Handa JT, 2013. Retinal microenvironment imbalance in dry age-related macular degeneration: a mini-review. Gerontology 59, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty S, Koh H, Phil M, Henson D, Boulton M, 2000. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol 45, 115–134. [DOI] [PubMed] [Google Scholar]
- Biswal MR, Ildefonso CJ, Mao H, Seo SJ, Wang Z, Li H, Le YZ, Lewin AS, 2016. Conditional induction of oxidative stress in RPE: a mouse model of progressive retinal degeneration. Adv. Exp. Med. Biol 854, 31–37. [DOI] [PubMed] [Google Scholar]
- Bonilha VL, Bell BA, Rayborn ME, Samuels IS, King A, Hollyfield JG, Xie C, Cai H, 2017. Absence of DJ-1 causes age-related retinal abnormalities in association with increased oxidative stress. Free Radic. Biol. Med 104, 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilha VL, Bell BA, Rayborn ME, Yang X, Kaul C, Grossman GH, Samuels IS, Hollyfield JG, Xie C, Cai H, Shadrach KG, 2015. Loss of DJ-1 elicits retinal abnormalities, visual dysfunction, and increased oxidative stress in mice. Exp. Eye Res 139, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns DR, Drake JC, Biela LM, Peelor FF 3rd, Miller BF, Hamilton KL, 2015. Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models. Oxidative Med. Cell. Longev 2015, 732596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano M, Thimmalappula R, Fujihara M, Nagai N, Sporn M, Wang AL, Neufeld AH, Biswal S, Handa JT, 2010. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res 50, 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castorina C, Campisi A, Di Giacomo C, Sorrenti V, Russo A, Vanella A, 1992. Lipid peroxidation and antioxidant enzymatic systems in rat retina as a function of age. Neurochem. Res 17, 599–604. [DOI] [PubMed] [Google Scholar]
- Chatzispyrou IA, Held NM, Mouchiroud L, Auwerx J, Houtkooper RH, 2015. Tetracycline antibiotics impair mitochondrial function and its experimental use confounds research. Cancer Res. 75, 4446–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfandiary H, Chakravarthy U, Patterson C, Young I, Hughes AE, 2005. Association study of detoxification genes in age related macular degeneration. Br. J. Ophthalmol 89, 470–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF, 1999. Mitochondrial evolution. Sci. (New York, N.Y) 283, 1476–1481. [DOI] [PubMed] [Google Scholar]
- Ildefonso CJ, Jaime H, Brown EE, Iwata RL, Ahmed CM, Massengill MT, Biswal MR, Boye SE, Hauswirth WW, Ash JD, Li Q, Lewin AS, 2016. Targeting the Nrf2 signaling pathway in the retina with a gene-delivered secretable and cell-penetrating peptide. Investig. Ophthalmol. Vis. Sci 57, 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura Y, Noda S, Hashizume K, Shinoda K, Yamaguchi M, Uchiyama S, Shimizu T, Mizushima Y, Shirasawa T, Tsubota K, 2006. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A 103, 11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain AK, Jaiswal AK, 2006. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem 281, 12132–12142. [DOI] [PubMed] [Google Scholar]
- Jarrett SG, Boulton ME, 2012. Consequences of oxidative stress in age-related macular degeneration. Mol. Aspect. Med 33, 399–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey BG, Weisinger HS, Neuringer M, Mitchell DC, 2001. The role of docosahexaenoic acid in retinal function. Lipids 36, 859–871. [DOI] [PubMed] [Google Scholar]
- Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, Kim SR, Sparrow JR, Hauswirth WW, Lewin AS, 2007. SOD2 knockdown mouse model of early AMD. Investig. Ophthalmol. Vis. Sci 48, 4407–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaemmerer E, Schutt F, Krohne TU, Holz FG, Kopitz J, 2007. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Investig. Ophthalmol. Vis. Sci 48, 1342–1347. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Jaiswal AK, 2010. An autoregulatory loop between Nrf2 and Cul3-Rbx1 controls their cellular abundance. J. Biol. Chem 285, 21349–21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzak KS, 1995. Possible role of oxidative damage in metal-induced carcinogenesis. Cancer Investig. 13, 411–430. [DOI] [PubMed] [Google Scholar]
- Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K, 2012. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells-implications for age-related macular degeneration (AMD). Immunol. Lett 147, 29–33. [DOI] [PubMed] [Google Scholar]
- Kimura K, Isashiki Y, Sonoda S, Kakiuchi-Matsumoto T, Ohba N, 2000. Genetic association of manganese superoxide dismutase with exudative age-related macular degeneration. Am. J. Ophthalmol 130, 769–773. [DOI] [PubMed] [Google Scholar]
- Kondo N, Bessho H, Honda S, Negi A, 2009. SOD2 gene polymorphisms in neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Mol. Vis 15, 1819–1826. [PMC free article] [PubMed] [Google Scholar]
- Kowalski M, Bielecka-Kowalska A, Oszajca K, Eusebio M, Jaworski P, Bartkowiak J, Szemraj J, 2010. Manganese superoxide dismutase (MnSOD) gene (Ala-9Val, Ile58Thr) polymorphism in patients with age-related macular degeneration (AMD). Med. Sci. Mon. Int. Med. J. Exp. Clin. Res 16, CR190–196. [PubMed] [Google Scholar]
- Lambros ML, Plafker SM, 2016. Oxidative stress and the Nrf2 anti-oxidant transcription factor in age-related macular degeneration. Adv. Exp. Med. Biol 854, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee OH, Jain AK, Papusha V, Jaiswal AK, 2007. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J. Biol. Chem 282, 36412–36420. [DOI] [PubMed] [Google Scholar]
- Liu X, Ward K, Xavier C, Jann J, Clark AF, Pang IH, Wu H, 2016a. The novel triterpenoid RTA 408 protects human retinal pigment epithelial cells against H2O2-induced cell injury via NF-E2-related factor 2 (Nrf2) activation. Redox Biol. 8, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xavier C, Jann J, Wu H, 2016b. Salvianolic acid B (sal B) protects retinal pigment epithelial cells from oxidative stress-induced cell death by activating glutaredoxin 1 (Grx1). Int. J. Mol. Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, Auwerx J, 2015. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: a Call for Caution in Biomedical Research. Cell reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullick A, Xu Y, Warren R, Koutroumanis M, Guilbault C, Broussau S, Malenfant F, Bourget L, Lamoureux L, Lo R, Caron AW, Pilotte A, Massie B, 2006. The cumate gene-switch: a system for regulated expression in mammalian cells. BMC Biotechnol. 6, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, Araoka T, Vazquez-Ferrer E, Donoso D, Roman JL, Xu J, Rodriguez Esteban C, Nunez G, Nunez Delicado E, Campistol JM, Guillen I, Guillen P, Izpisua Belmonte JC, 2016. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733 e1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organisciak DT, Vaughan DK, 2010. Retinal light damage: mechanisms and protection. Prog. Retin. Eye Res 29, 113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva MM, Cano M, Handa JT, 2014. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res 119, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, Hillen W, 1996. Tetracyclines: antibiotic action, uptake, and resistance mechanisms. Arch. Microbiol 165, 359–369. [DOI] [PubMed] [Google Scholar]
- Smith W, Assink J, Klein R, Mitchell P, Klaver CC, Klein BE, Hofman A, Jensen S, Wang JJ, de Jong PT, 2001. Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology 108, 697–704. [DOI] [PubMed] [Google Scholar]
- Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM, 2004. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. U. S. A 101, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Motohashi H, Yamamoto M, 2011. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells Devoted Mol. Cell. Mech 16, 123–140. [DOI] [PubMed] [Google Scholar]
- Thelen M, Dewald B, Baggiolini M, 1993. Neutrophil signal transduction and activation of the respiratory burst. Physiol. Rev 73, 797–821. [DOI] [PubMed] [Google Scholar]
- Tomany SC, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, Vingerling JR, Klein BE, Smith W, De Jong PT, 2004. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology 111, 1280–1287. [DOI] [PubMed] [Google Scholar]
- Villeneuve NF, Lau A, Zhang DD, 2010. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxidants Redox Signal. 13, 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen X, Yang Y, 2012. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Meth 9, 266–269. [DOI] [PubMed] [Google Scholar]
- Wei YH, 1998. Oxidative stress and mitochondrial DNA mutations in human aging. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med 217, 53–63. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Boulton ME, Gottsch JD, Sternberg P, 1999. Oxidative damage and age-related macular degeneration. Mol. Vis 5, 32. [PMC free article] [PubMed] [Google Scholar]
- Xiong W, MacColl Garfinkel AE, Li Y, Benowitz LI, Cepko CL, 2015. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig 125, 1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, 2005. Retinal degeneration and local oxygen metabolism. Exp. Eye Res 80, 745–751. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J, 2011. Age-related retinopathy in NRF2-deficient mice. PLoS One 6, e19456. [DOI] [PMC free article] [PubMed] [Google Scholar]