

Abstract
Genome-wide association studies have identified approximately 170 loci associated with Crohn’s disease (CD), and defining which genes drive these association signals is a major challenge. The primary aim of this study was to define which CD-locus genes are most likely to be disease-related. We developed a gene prioritization regression model (GPRM) by integrating complementary mRNA expression datasets, including bulk RNA-Seq from the terminal ileum of 302 newly-diagnosed, untreated CD patients and controls and in stimulated monocytes. Transcriptome-wide association and co-expression network analyses were performed on the ileal RNA-Seq datasets, identifying forty genome-wide significant genes. Co-expression network analysis identified a single gene module which was substantially enriched for CD-locus genes and most highly expressed in monocytes. By including expression-based and epigenetic information, we refined likely CD genes to 2.5 prioritized genes per locus from an average of 7.8 total genes. We validated our model structure using cross-validation and our prioritization results by protein-association network analyses, which demonstrated significantly higher CD gene interactions for prioritized compared to non-prioritized genes. Although individual datasets cannot convey all of the information relevant to a disease, combining data from multiple relevant expression-based datasets improves prediction of disease genes and helps to further understanding of disease pathogenesis.
Introduction
Crohn’s disease (CD) is an inflammatory bowel disease (IBD) that most commonly affects the terminal ileum. A series of genome wide association studies (GWAS) have identified 170 loci1, 2 which are associated with CD risk, but determination of causal genes remains challenging. Focus on protein altering alleles has been a strong focus of functional studies; such studies have led to identification and characterization of many specific mutations in genes that overlap strong CD association signals, including Arg381Gln in IL23R3–5, Ala300Thr in ATG16L16, 7, NOD28–10, Ile231Leu GPR656, Ile254Vale in LACC111 and autosomal recessive loss of function variants in the IL10 pathway12. Furthermore, a large international fine-mapping effort identified 45 independent signals for which the credible SNP list could be resolved to single variant resolution with greater than 50% confidence13. For 13 of these, the implicated single variant involved a protein-altering allele, strongly implicating that gene in CD pathogenesis. However, this leaves over one hundred CD signals for which fine mapping was not possible.
For the large majority of CD loci, non-coding alleles, presumably altering regulation of gene expression, drive the association signal. Specifically mapping association signals to altered gene expression has proved challenging. Early enthusiasm that GWAS signals would be enriched in expression quantitative trait loci (eQTL) has subsequently been tempered by 1) the lack of significant enrichment observed for precisely defined, credible trait-associated SNP sets with specific gene-SNP eQTL pairs13 and 2) the observation that the large majority of transcripts contain eQTL in some biologic context14–16 The ubiquity of eQTL across genes observed with large, cell-specific in vitro culture studies, often enhanced with stimulation (e.g. with cytokines or microbial products), may or may not reflect disease-driving regulation of gene expression14; it may be reasoned that examining the affected disease tissue, directly ex-vivo, may provide important complementary insight. Since treatment of CD patients can strongly influence cytokine expression and modify cell proportionality17–20, examination of tissue from newly-diagnosed, untreated patients provides the most relevant view of expression changes that are disease related. Another challenge is the establishment of a connection between gene expression changes and association signals. Two approaches to this include single-point, colocalization of eQTL and association signals and consideration of all of the CD associated SNPs which modify gene expression transcriptome-wide. Colocalization of signal is best able to identify overlap in cases where a single strong eQTL and association signal exist for a locus, while a multipoint transcriptome wide association study is able to integrate many SNPs to account for loci with multiple or weaker signals.
In this study, we developed a gene prioritization regression model (GPRM) that integrates expression, association, and epigenetic information to identify which genes in CD loci are the most likely to be related to pathogenesis. Validation of the model was performed by comparing the number of protein interactions between prioritized and non-prioritized genes in CD loci, and results were found to be robust to modification of the list of CD reference genes. To improve the flexibility of the GPRM and account for future improvements to CD knowledge, we also developed an app with the ability to integrate changes in CD loci definition, dataset inclusion, and reference gene list.
Results
Single-point mapping of disease trait with expression quantitative trait loci
In order to decouple disease and treatment effects on gene expression, we examined an expression dataset21, 22 of newly diagnosed, untreated children with CD. We first tested for the colocalization of trait association and eQTL signals23, 24. Cis eQTL were identified using RNA-Seq and Illumina 2.5 Omni genotype data from 302 untreated individuals of European descent in the RISK cohort21, 22. We performed quality control and imputation25–27, including 25 expression covariates determined using probabilistic estimation of expression residuals (PEER)28, 29 and the top 5 genotypic principal components as covariates. We observed significant associations to 12 506 genes, (Supplementary Figure S1), corresponding to 1 274 331 gene-eSNP pairs. Next, we used coloc23, 24 to determine whether the eSNPs coincided with trait association signals. Of the 159 CD loci that contained an eSNP (Figure 1a), only 10 had a posterior probability > 0.75 that the CD association and altered expression were the result of the same SNP, including ATG16L1 (Figure 1b). The co-localization of ATG16L1 was unexpected, because the association signals include a protein altering allele, Thr300Ala, where the alanine risk allele increases proteolytic cleavage by caspase-37, 30, thereby decreasing ATG16L1 protein levels. In contrast, 17 had a posterior probability > 0.75 that the two signals were the result of distinct SNPs (Figure 1a & 1c). The large majority of loci do not show strong evidence for shared or distinct signals since they have only weak evidence for a single signal in either the eQTL or association data, commonly resulting from multiple signals in the same region, showing that it is important to use a complementary approach able to consider overlap between multiple SNPs in a locus.
Figure 1:
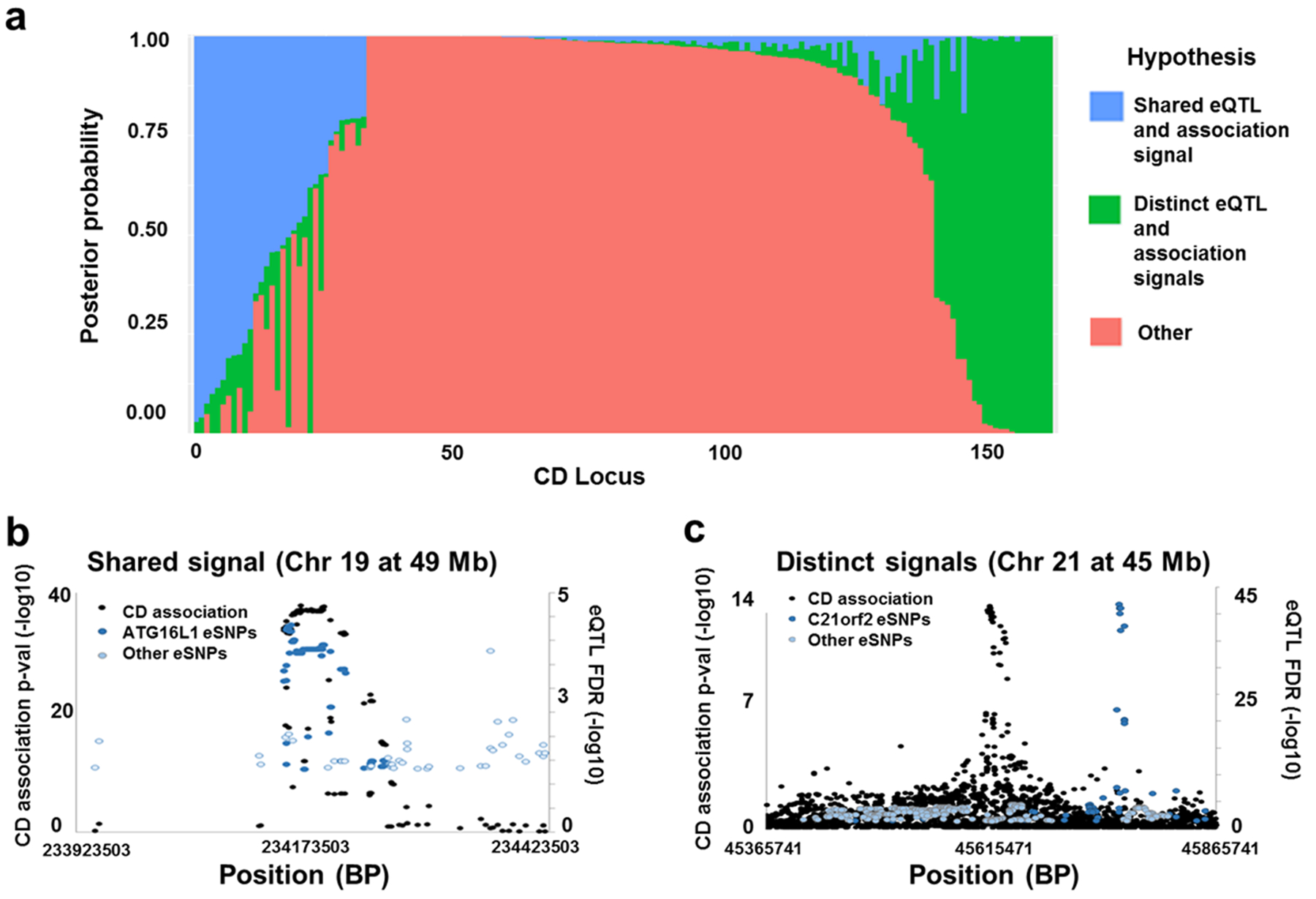
Measurement of colocalization between CD associations and single-point eQTL in each CD locus. a. Distribution of posterior probability values reported by Coloc across CD loci show that a subset of loci have overlapping signal. Each bar represents a CD locus, while coloration represents the posterior probabilities reported. b. and c. Plots comparing eSNP and CD association p-value peaks in representative loci with high probability of shared signal (b) and distinct signals (c).
Transcriptome-wide association studies
Recently developed transcriptome-wide association approaches (TWAS)31, 32 were used to integrate the identified CD-association expression changes into a gene prioritization model, as it can include multiple SNPs to return gene-level information on CD-associated expression changes. It does so by measuring expression modification in a cohort for which both genotype and expression data is available, then imputing expression levels in a much larger dataset with only genotype and phenotype data to improve power to test for association between expression levels and CD status. First, a restricted maximum likelihood approach33 was used to identify a total of 6 029 genes predicted to be heritable within the RISK dataset21, 32, 34. Each heritable gene was then tested for TWAS significance, resulting in 40 significant genes (Supplementary Table S2). Twenty of these TWAS significant genes reside within 15 distinct CD loci defined by a 500 kb window (Table 1, ordered by TWAS significance)2, 35. The top TWAS signal, CDC42SE2 (CDC42 small effector 2), is a strong candidate due to its actin cytoskeletal localization and role in early contractile events in macrophage phagocytosis36. Regions with very strong and broad disease association signals and/or high linkage disequilibrium such as the IBD5 locus37, the major histocompatibility complex (MHC) region and the chromosome 3p region at 49.7 Mb contain multiple TWAS significant genes. Of note, TWAS newly implicates three genes (C9orf173, FAM166A, and C9orf139), all contained within a 226 kb region on chromosome 9, which is not near a disease-associated genome-wide significant locus. Finally, six TWAS significant genes correspond to coloc-defined signals (Table 1, loci marked with asterisks), while the remaining four loci coloc predicted to have shared signal contain eQTL related to genes with nominally significant (P-values < 0.05) TWAS association signals.
Table 1:
Top TWAS results in IBD loci.
| Chr | base pairs | Phenotype | Significant genes in locus | Significant genes (P) |
|---|---|---|---|---|
| 5 | 130613600 | IBD | 1 | CDC42SE2 (2.3 × 10−12) |
| 6 | 167373547* | IBD | 1 | RNASET2 (2.6 × 10−12) |
| 5 | 150277909 | IBD | 1 | ZNF300P1 (5.8 × 10−11) |
| 3 | 49721532 | IBD | 2 | APEH (6.9 × 10−11), CAMKV (3.6 × 10−8) |
| 9 | 139266405 | IBD | 1 | SEC16A (1.8 × 10−10) |
| 5 | 131778452 | IBD | 4 | PDLIM4 (3.9 × 10−9), P4HA2 (9.2 × 10−9), LOC553103 (5.9 × 10−8), SLC22A5 (5.7 × 10−7) |
| 5 | 96252803* | IBD | 1 | ERAP2 (5.1 × 10−10) |
| 2 | 234173503* | CD | 1 | ATG16L1 (3.2 × 10−9) |
| 16 | 28517709* | IBD | 2 | APOBR (8.6 × 10−9), CCDC101 (1.0 × 10−7) |
| 10 | 35295431 | IBD | 1 | CUL2 (2.0 × 10−7) |
| 22 | 39659773 | IBD | 1 | PDGFB (3.7 × 10−7) |
| 1 | 155878732* | IBD | 1 | RIT1 (4.2 × 10−7) |
| 17 | 37912377* | IBD | 1 | GSDMB (5.6 × 10−7) |
| 1 | 159799910 | CD | 1 | KCNJ10 (5.9 × 10−6) |
| 1 | 160856964 | IBD | 1 | SLAMF7 (6.7 × 10−6) |
, locus also implicated in coloc analysis
Weighted gene co-expression network analysis (WGCNA) demonstrates a striking enrichment of CD-loci genes in an immune module enriched for monocyte genes based on single cell sequence data
We next sought to examine the effects of gene coexpression networks in CD ileal tissue. We performed weighted gene coexpression network analysis (WGCNA)38 and observed 29 modules, 11 of which contained more than 500 genes. One module, the black module containing 691 genes, demonstrated uniquely strong enrichment of CD locus genes (Table 2, 13.7% of black module genes are within CD loci compared to the genome wide total of 6.7%, enrichment P = 6.49 × 10−10). Unsurprisingly, GO enrichment analysis of the black module showed enrichment for immune, defense and inflammatory genes39 (Supplementary Table S3), generally implicating leukocytes. To further refine leukocyte gene expression contributing to the black module gene enrichment, we analyzed a peripheral blood mononuclear cell (PBMC) dataset in which single cell mRNA sequencing (scRNASeq) was performed using the 10X Genomics Chromium system40. We performed unsupervised clustering (Methods), and identified 29 cell clusters which mapped extremely well with general leukocyte classification markers for B cells/plasmablasts (CD19), T cells (CD3E), NK cells (NCAM1), dendritic cells (CD1C) and monocytes (CD14) (Figure 2a). As expected, the black module genes within CD loci demonstrated broad gene expression across PBMC cell clusters generally (Figure 2a). However, when we summed the unique molecular identifiers (UMIs) for all black module genes for each cell, we observed a marked enrichment of black module gene expression within classical (CD14++CD16-) and non-classical (CD14+CD16++) monocytes41, 42 (P < 1 × 10−200, Supplementary Table S4) compared to other leukocyte clusters (Figure 2b).
Table 2:
WGCNA coexpression modules
| Module | Gene number | CD gene number | CD % | CD enrichment P | Top significant GO term |
|---|---|---|---|---|---|
| All genes | 18308 | 1225 | 6.7 | NA | NA |
| black | 691 | 95 | 13.8 | 6.5 × 10−10 | immune system process |
| blue | 2066 | 171 | 8.3 | 0.0054 | developmental process |
| yellow | 1315 | 105 | 8.0 | 0.078 | No GO enrichment |
| green | 1220 | 91 | 7.5 | 0.29 | translation |
| brown | 1563 | 110 | 7.0 | 0.60 | small molecule metabolic process |
| red | 1212 | 82 | 6.8 | 0.91 | No GO enrichment |
| pink | 580 | 39 | 6.7 | 0.93 | respiratory electron transport chain |
| magenta | 546 | 32 | 5.9 | 1 | cell cycle process |
| turquoise | 5336 | 308 | 5.8 | 1 | nucleic acid metabolic process |
| purple | 508 | 23 | 4.5 | 1 | bicarbonate transport |
| grey | 1290 | 43 | 3.3 | 1 | nucleosome assembly |
Figure 2:
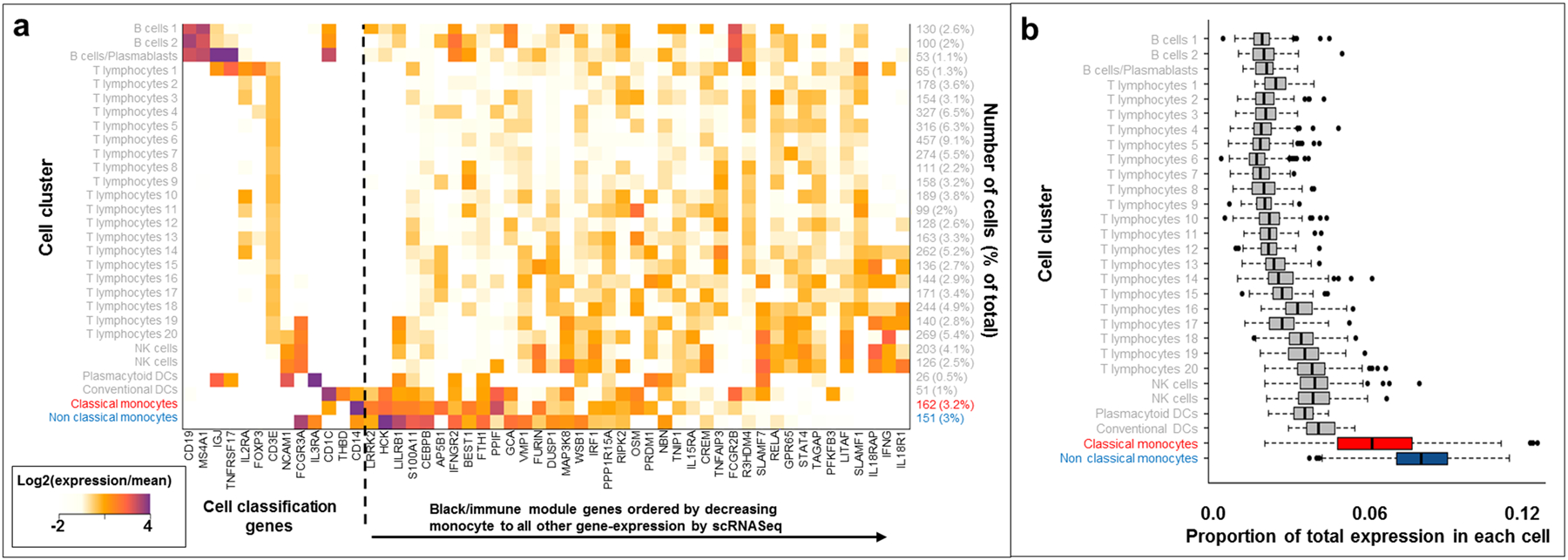
Single cell mRNA-Seq analyses of black module genes. a. Heatmap showing relative expression values for cell classification genes and black module genes in 5000 PBMC cells. b. Distribution of total UMI proportion corresponding to black module gene expression across cell types.
Development and validation of a gene prioritization regression model
We used a set of curated CD reference genes (n=96) prioritized from four association and fine-mapping studies1, 2, 13, 35 to prioritize all CD locus genes (Supplementary Table S5). Two logistic regression models were used; the first model included only expression and epigenetic trait information, then used backward stepwise regression in order to determine relative contribution values for each dataset independent of trait association signals (Supplementary Table S6). Predictive mRNA expression data included multiple WGCNA modules, genes differentially expressed between M1 versus M2 macrophage subsets43 and genes significant by TWAS; non-predictive gene mRNA expression lists included differentially expressed genes in CD compared to control terminal ileum, and genes demonstrating ileal expression greater than the median (FPKM greater than 3.4). Predictive epigenetic datasets included open chromatin from CD4+ Th17 cells, but not the intestinal mucosa, nor monocytes44. With these significantly predictive expression and epigenetic traits, the second gene prioritization regression model (GPRM) also included gene-level CD association as predictors (Table 3, Supplementary Table S7) to generate prioritization scores for likely CD genes, the full list of which is listed in Supplementary Table S8. Other than overlap with open chromatin in Th17 cells (p-value = 0.058) the same predictors were significant in the gene prediction model: TWAS significance (p-value < 1.9E-4), differential expression between M1 proinflammatory and M2 wound-healing macrophages (p-value = 2.7E-11), and coexpression module categorization (p-value <1.7E-14). Pairwise comparisons between GPRM scores and their associated standard errors were performed to compare genes within each of the 162 CD loci that contained genes (Figure 3a). For each locus, genes having significantly higher GPRM scores (red) than other genes in the locus (blue) were defined using Welch’s t-test. Among the 162 CD loci, an average of 7.8 genes are present within the 500 kb windows, which is refined to 2.5 prioritized genes across loci (red, Figure 3a) by regression analyses.
Table 3:
Gene Prioritization Regression Model (GPRM) results
| Estimate | Standard Error | t value | P | |
|---|---|---|---|---|
| Intercept | 4.7 × 10−5 | 0.0011 | 0.042 | 0.97 |
| Gene Level CD association (p < 3 × 10−6) | 0.13 | 0.0049 | 27.2 | < 2.0 × 10−16 |
| Gene Level CD association (3 × 10−6 < p <0.05) | 0.027 | 0.0018 | 15.2 | < 2.0 × 10−16 |
| WGCNA module (black) | 0.023 | 0.0030 | 7.7 | 1.7 × 10−14 |
| WGCNA module (green) | −0.0069 | 0.0024 | −2.9 | 0.0041 |
| WGCNA module (greenyellow) | −0.010 | 0.0040 | −2.6 | 0.0097 |
| WGCNA module (yellow) | −0.0054 | 0.0024 | −2.3 | 0.021 |
| Differential expression M1/M2 | 0.012 | 0.0018 | 6.7 | 2.7 × 10−11 |
| TWAS (8.3 × 10−6 < P < 0.05) | 0.041 | 0.011 | 3.7 | 1.9 × 10−4 |
| TWAS (P < 8.3 × 10−6) | 0.0088 | 0.0046 | 1.9 | 0.057 |
| Open chromatin Th17 cells | 0.0022 | 0.0012 | 1.9 | 0.058 |
M1/M2, genes with higher expression in M1 compared to M2 in vitro differentiated macrophages
Figure 3:
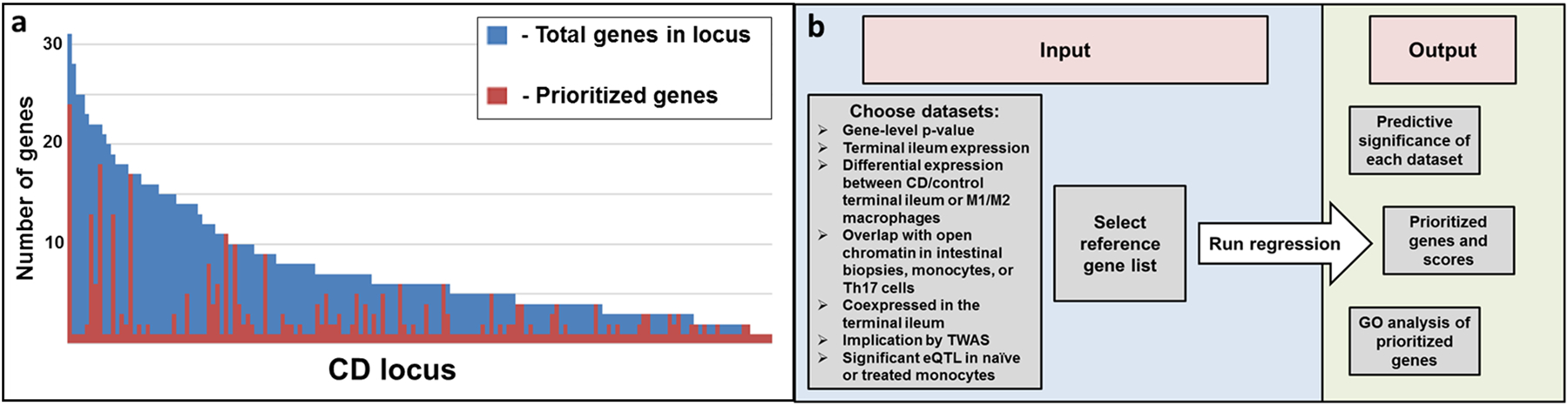
Prioritized genes across CD loci and schematic of prioritization app. a. Top prioritized genes per locus defined according to significant difference in standard error of prioritization scores when compared to other genes in the same locus. Each bar represents a CD-locus, with coloration representing prioritization status of the genes within the locus. The average number of genes per locus was 7.8 (shown in blue), while the average number of prioritized genes per locus was 2.5 (shown in red). b. Schematic of CD gene prioritization app. Datasets to be included and a list of reference genes can be submitted by the user with outputs including significant estimates of datasets, gene prioritization and GO analysis of prioritized genes.
To validate the prioritized gene lists, we generated a protein-protein interaction network45 of all CD-locus genes (n=1264), then calculated the average number of connections for each gene, counting interactions with CD reference genes and interactions with all CD-locus genes. The 398 prioritized genes compared to the 866 non-prioritized genes had significantly more reference gene interactions (average of 2.7 vs. 1.5, P = 1.1 × 10−4) and total interactions counting all 1264 CD-locus genes (average of 11.1 compared to 7.5, P value = 1.4 × 10−4). To test the sensitivity of these results to the reference gene list, we removed the 36 genes which had previously been prioritized by DAPPLE (protein-protein interactions)1, resulting in a reduced 60 gene reference list. After re-running the regression model, the number of prioritized genes increased to 478, and only 7 genes from the original 398 gene list were not present in the new prioritized list. Furthermore, the STRING analysis45 comparing the new 478 prioritized and 786 non-prioritized gene lists showed similar numbers of reference gene (0.74 versus 0.47, P = 9.8 × 10−4) and total (10.7 versus 7.4, P = 1.4 × 10−4) interactions. To ensure that there was value being added and that already validated genes were not driving our validation results we reran the STRING analyses but excluded all reference genes, and then compared interactions between both the newly prioritized genes and non-prioritized genes with CD reference and CD locus genes. With all reference genes removed the newly prioritized genes still had significantly more interactions with both CD-locus genes (8.60 vs 6.69, p-value = 0.008) and CD reference genes (1.65 vs 1.09, p-value = 0.009).
Taken together, the GPRM gene results are substantively robust to the reference gene list. We expect that with increasing functional studies of CD-associated candidate genes and with more sequencing studies across populations that may identify rare protein-coding variants, the reference gene list will continually be improved. Anticipating this, we have developed a web-based app where user-defined inputs such as reference gene lists may be applied to continually improve prioritization of CD genes (Figure 3b, https://rcg.bsd.uchicago.edu/ibdgc/cdprioritization/).
Discussion
In this study, we have developed a gene prioritization regression model based on a curated CD reference gene list that integrates transcriptome-wide association results and co-expression analyses from ileal mucosal biopsies from a pediatric IBD case and control cohort. We show using a single cell RNA sequence dataset40 that transcripts within the intestinal tissue-based WGCNA module most enriched for CD locus genes are highly expressed in classical and non-classical monocytes. Given this enrichment, our GPRM model also includes differential expression of in vitro differentiated monocytes, as well as Th17 (T lymphocytes) DNase-Seq peaks.
The marked enrichment of genes within the CD-gene enriched WGCNA-defined immune module from bulk ileal RNA-Seq data within monocytes compared to dendritic cells by scRNA-Seq analysis was unexpected (Figure 3b), due to the highly overlapping gene expression between these closely related innate immune cell subtypes. This difference could reflect the greater abundance of monocytes/macrophages within intestinal tissues46 and thus greater impact on tissue-based networks.
Alternatively, a unique feature of the genetic architecture of European ancestry CD is the absence, compared to most chronic inflammatory diseases, of dominant MHC associations1, which implicate antigen-specific epitope presentation, most prominently by dendritic cells, to lymphocytes. In its stead, the presence of high effect risk alleles of innate immunity in European ancestry CD (e.g. NOD2, LRRK2, CSF2RB47, 48 implicates altered first-line innate immune defense against microbes generally, as opposed to in an antigen-specific manner. Similar to European ancestry CD, a recent African-American GWAS reported a top association to the PTGER4 gene region49. This cross-population association, combined with PTGER4’s top score in our prioritization model (Supplementary Table S8), further highlight the role of lipid mediators and monocyte immunity in CD pathogenesis.
Because of the polygenic nature of CD, some degree of uncertainty in defining likely contributing genes within genome-wide significant loci will always be present. Using purely statistical, fine-mapping approaches, progressively greater uncertainty is observed with loci of smaller effects. Even when using the largest available case-control cohorts, only 45 independent signals could be fine-mapped to a single variant with greater than 50% certainty(13). For these reasons, utilizing complementary expression datasets as applied in this study provides substantively improved prioritization capacity. Importantly, our prioritized gene lists were extremely robust to our reference gene list. Developing web-based tools (https://rcg.bsd.uchicago.edu/ibdgc/cdprioritization/) that empower the broader community to examine and adjust assumptions (e.g. through continually improving reference gene lists) and integrate large datasets will accelerate biologic understanding of genetic association results.
Integration of TWAS results, coexpression network information, open chromatin overlap, and differential expression results using stepwise regression has shown that many expression and epigenetic datasets each have unique value, and together can best predict which genes are most likely to be disease related. This makes sense for study of complex disease behavior, since a combination of cell types in different conditions contribute to pathogenesis.
While CD GWAS signals are most strongly enriched in T cells44 by epigenetic marks, the presented mRNA-based analyses consisting of bulk RNA-Seq from terminal ileum (leveraging both eQTL/TWAS and WGCNA analyses), scRNA-Seq from PBMCs, and in vitro studies of purified cell populations stimulated with cytokines and microbial products all independently implicate monocytes in CD pathogenesis. Our findings suggest potential pathogenic roles for both classical (CD14++CD16-) and non-classical (CD14+CD16++) monocytes; non-classical monocytes have been implicated to play an important role in in vivo patrolling behavior50 and are expanded in infectious and inflammatory conditions41, 51.
Materials/Subjects and Methods
RNA-Seq data
Full biopsies from the terminal ileum were collected from 302 newly diagnosed individuals under age 17 from the RISK cohort21, 34, 52 (GEO accession GSE57945, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57945), including 213 CD patients, 50 UC patients, 4 unspecified IBD patients, and 35 healthy controls. Samples were barcoded up to 12 per lane and sequenced using the Illumina HiSeq 2000. RNA-seq reads were mapped using TopHat253 (to the human reference genome version 19. Initial RNA-seq mapping results indicate that approximately 20 million reads were successfully mapped for each individual. Following RNA-seq mapping, expression levels at the gene and isoform levels was determined and expression quantified using Cufflinks54, 55 to generate FPKM estimates and HTseq56 to generate raw read counts. In the regression model, genes were coded 0 or 1 for terminal ileum expression according to whether their average expression levels were above or below the median of the average FPKM values (FPKM>=3.4) across all genes.
Genotype data
Genotype data was collected from 302 newly diagnosed individuals under age 17 from the RISK cohort21, 34, 52 using the Illumina Omni 2.5M kit, then imputed using SHAPEIT26, 27 and IMPUTE225. Plink 1.957, 58 was used to remove SNPs which had a call rate < 90%, failed Hardy-Weinberg equilibrium, or had a minor allele frequency < 1% in the full population and to remove individuals with a missing SNP rate > 0.02%.
eQTL generation
Genotype and ileal RNA-seq expression data from the RISK cohort was used for eQTL generation. Genes with low expression (< 0.1 FPKM) were removed, along with SNPs with a minor allele frequency low enough that fewer than 10 copies were represented in the population tested. After normalization of expression values, RNA-seq expression and genotyping results were combined to identify eQTL using the program Matrix eQTL59. The first five principle components measured in the genotype data, first 25 PEER components28, 29 measured in the RNA-seq data, disease state, gender, and the lane number each RNA-seq sample was run in were included as covariates in the eQTL analysis.
Definition of IBD loci
Each locus was defined to be a 500kb window surrounding the 140 CD SNPs in Supplementary Table 2 of Jostins et al.1 and the 30 newly defined CD loci in Supplementary Table 3 of Liu et al.2 to give a total of 170 CD loci.
Colocalization of eQTL with CD associations
Association results from imputed CD association data and eQTL results from this project were used to determine colocalization of signals. Overlapping SNP within 500kb of the IBD loci defined in Jostins et al.1 and Liu et al.2 were used as input for the R package coloc23, 24, which returned posterior probabilities for each of the following hypothesis tests:
No single causal variant in either dataset
A causal variant only in the association data
A causal variant only in the eQTL data
Distinct causal variants in each dataset
A shared causal variant
The full set of posterior probabilities reported for each CD locus are reported in Supplementary Table S1.
Heritability testing and TWAS
TWAS was run using the RISK expression and genotype data to impute expression values for the 6,299 CD cases and 15,148 controls used in the association study described in Jostins et al(1). GCTA60 was first used to perform restricted maximum likelihood (REML) testing on variant stabilizing transformation-normalized read counts per gene and each SNP within 100kb of the gene region. Based on REML results, 6,029 genes were heritable (P < 0.01) and were included for analysis using TWAS31, 32. TWAS was run using VST-normalized read counts per gene and each SNP within 1Mb of the gene region.
Coexpression analysis
Input FPKM-normalized expression data from the 302 individuals of European ancestry to generate coexpression modules in WGCNA using the preset standard parameters38. The CD and eGene enrichment tests reported in Supplementary Table S3 were performed in R using Fisher’s exact test.
Testing for differential expression between CD case and control terminal ileum samples
Included 213 CD cases and 35 controls of European descent from the RISK cohort, and used the R package DESeq261 to determine significance of differential expression according to disease status. Genes were coded 0 or 1 in the prioritization model, according to whether or not they had an FDR significance <= 0.1 in the DESeq2 output.
Logistic regression
Stepwise logistic regression models were developed in R both to determine how well each of the expression and epigenetic traits included in the study were able to predict a curated list of high-confidence CD genes and then to use the predictive datasets to generate scores which could suggest which genes are more likely to be associated with CD. The first model included only expression trait and epigenetic information and used backward stepwise regression in order to determine relative contribution values for each expression dataset independent of pure association signal (though TWAS integrated association information and showed some informational overlap with association scores), while the second included the predictive datasets from the initial model along with gene-level CD association in order to generate a prioritized list of CD genes. Data included in the first model included TWAS significance (genes coded 0 if p > 0.05, 1 if 0.05 >= p > 8.29E-6, and 2 if p <= 8.29E-6), high expression in intestine (coded 1 if expression was greater than the median across all genes or 0 otherwise), differential expression between CD cases and controls (coded 1 if p < 0.01 or 0 otherwise), overlap with open chromatin region in Th17 cells, monocytes, and intestinal biopsy (coded 1 if overlap occurred or 0 otherwise), differential expression between M1 proinflammatory and M2 wound-healing macrophages (coded 1 if FDR p-value < 0.01 at 6 or 24 hours post differentiation or 0 otherwise), identification of eQTL in monocytes in Fairfax et al.(14), and inclusion in 29 coexpression modules as potential predictors (coded network as an indicator variable with separate levels for each module). After backward stepwise regression, significant predictors in the expression model included TWAS significance (p-value < 2E-16), overlap with open chromatin region in Th17 cells (p-value = 0.00375) (GEO accession GSM1014541, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1014541), differential expression between M1 proinflammatory and M2 wound-healing macrophages (p-value = 1.64E-13), and coexpression module categorization (p-value <2E-16) (Supplemental Table S6). Each of these significantly predictive datasets were then used for the prioritization model, along with gene-level p-values. Most of the same predictors were significant in the gene prediction model (TWAS significance (p-value < 1.9*10−4), differential expression between M1 proinflammatory and M2 wound-healing macrophages (p-value = 2.7*10−11), and coexpression module categorization (p-value <1.7*10−14)), though overlap with open chromatin in Th17 cells lost predictive power (p-value = 0.058). Cross-validation was then performed using the DAAG package in R, and mean squared error values were compared between the full prediction model and models with each variable removed using 10 folds (2 000 genes each) to make sure that overfitting was not occurring. The full model did not have a higher mean squared error than any model with removed variables (0.00452 for the full model compared to 0.00472, 0.00453, 0.00452, 0.00454, and 0.00452 for models with each variable removed).
Code availability
Prioritization app and data descriptions available at: https://rcg.bsd.uchicago.edu/ibdgc/cdprioritization/
Gene prioritization within CD loci
Prioritization scores and standard error values from the regression model were used to compare genes within 500kb of each of the CD or IBD loci defined by Jostins et al1 and Liu et al.2. Pairwise comparisons between prioritization scores and their associated error values were made using Welch’s t-test for each of the genes in each locus by taking the gene with the highest prioritization rank and comparing it to see if the score was significantly higher than the next highest gene in each locus until a significant different was noted. The top prioritized gene or genes within each locus were then defined to be those which were most highly prioritized and which did not differ significantly in score from one another.
Selection of CD reference genes
To train the GPRM to identify CD genes a total of 96 reference genes were selected based on the results of four recent CD-association manuscripts. Genes were selected from the top association results in Jostins et al.1 if they had strong evidence for CD involvement based on multiple bioinformatics approaches implemented in the manuscript, demonstrated evidence of Mendelian inheritance of IBD, or contained an excess of rare variants in cases or controls in deeply sequenced individuals. Genes from Liu et al.2 were selected based on both GRAIL and DAPPLE significance, genes from Huang et al.13 were selected on the condition that a coding SNP for the gene was present in one of the credible sets of causal SNPs annotated in Supplementary Table 1 of their manuscript, and genes were selected from the loci identified in de Lange et al.35 based on reported disease implication. A full list of CD reference genes and their sources are included in Supplementary Table S5.
Gene level CD association p-values
Gene-level CD association p-values were calculated using association results from a Hapmap3-imputed dataset containing 6,333 CD and 15,056 control samples62. SNP-level p-values for 1,235,490 SNPs were first transformed so that the genomic inflation factor was 163, then VEGAS64 was used to calculate gene-level p-values for 17,031 genes (taking into account the p-value of the most significant SNP and linkage disequilibrium structure within ±50 kb of the gene). Next, 1,000,000 simulations were run to obtain a p-value for each gene, allowing for a maximal significance of 1E-6. Within the prioritization model, gene-level p-values were coded into 3 groups (coded as 2 if p < 3E-6, 1 if 3E-6 < p < 0.05, and 0 if p > 0.05). The threshold of 3E-6 was based on Bonferroni correction of a p-value of 0.05 after testing the 17,214 genes which had SNP-level CD-association data.
DNase-seq open chromatin data
DNase-seq results stored in narrowPeak files for Th17 cells, monocytes, and intestinal samples were downloaded from the ENCODE database44. Genes were defined to overlap open chromatin if the gene region or 2kb upstream region overlapped to any degree with open chromatin regions in each cell type.
M1-M2 cell culture and differential expression analysis
Peripheral blood mononuclear cells were isolated from individuals with informed consent as approved by the Yale University Institutional Review Board using the Ficoll-Hypaque gradient. Monocyte-derived-macrophages were generated as in Pena et al.65 with slight modifications. Specifically, after PBMC isolation, 5 × 10^6 cells in serum-free RPMI 1640 were seeded in each well of a 6-well plate and incubated at 37°C for an hour. The media was then replaced with fresh complete RPMI media containing M-CSF (10ng/ml) (Shenandoah Biotechnology, Warwick, PA). Cells were cultured for 7 days and media were changed every two days. On day 7, 5 × 10^6 cells from one well were harvested in QIAzol (Qiagen) and designated M0 cells (cells before macrophage polarization). The remaining cells were stimulated in complete RPMI with 100 ng/mL LPS (Sigma-Aldrich, St. Louis, MO) and 20 ng/mL interferon-γ (R&D Systems, Minneapolis, MN) for M1 polarization or 20 ng/mL IL-4 (R&D Systems) for M2 polarization. Cells were harvested in QIAzol after 6 and 24 hours post-stimulation. Total RNA was extracted using the Qiagen miRNAeasy mini kit (Qiagen) according to the manufacturer’s protocol. Microarrays were performed using Illumina HumanHT-12 v3 Expression BeadChips (Illumina, San Diego, CA). In total, 7 healthy controls and 6 CD cases were included. The microarray expression data were analyzed by the R bio-conductor packages lumi66 and gplots. Genes were considered to be differentially expressed in the prioritization model if they had a FDR < 0.01 at either the 6 hour or 24 hour time points.
Single cell analysis
Single cell data included a randomly selected subset of 5k out of the 68k total PBMCs collected by Zheng et al.40). UMI were quantified and clustered into cell types using unsupervised EM-like clustering optimizing the likelihood of the data to a multinomial mixture model67, then all of the transcripts within each of the modules defined by WGCNA were summed. UMI counts per module were then normalized by dividing by the total counts per cell to determine the fraction of total UMI per cell that corresponded to each module. A linear regression model was then used to determine which modules had expression values that were most strongly correlated with each cell type (Supplementary Table 3).
Protein-protein interaction analyses
The number of interactions between all of the genes in the 170 CD loci were calculated based on a STRING45 network generated using all 1,264 CD locus genes. The average number of total interactions and interactions with genes used as a reference in the prioritization model were averaged for the both the set of 398 genes which were most highly prioritized by the model and the 866 remaining CD-locus genes. Genes which were prioritized had significantly more connections overall compared to non-prioritized genes (mean of 11.1/standard deviation of 17.03 vs a mean of 7.5/standard deviation of 13.41, Welch’s t-test p-value = 1.4 × 10−4) and when comparing only connections to reference genes (mean of 2.7/standard deviation of 5.92 vs. a mean of 1.5/standard deviation of 4.31, Welch’s t-test p-value = 1.1 × 10−4). Results were also tested with reference genes excluded from the prioritized gene list to ensure they were not the source of the observed enrichment, and both IBD locus gene connections and reference gene connections were still higher among prioritized genes compared to non-prioritized genes (mean of 8.6 vs a mean of 6.7, Welch’s t-test p-value = 8.2 × 10−3 for all IBD gene connections and a mean of 1.6 vs a mean of 1.1, Welch’s t-test p-value = 8.9 × 10−3 for reference gene interactions).
Supplementary Material
Acknowledgements
This work was supported by the HPC facilities operated by, and the staffs of, the Yale Center for Research Computing and the Yale Center for Genome Analysis, as well as NIH grant 1S10OD018521-01, which helped fund the cluster.
Funding
NIH research grants (U01 DK62429, U01 DK62422, R01 DK106593, and P30 DK078392) as well as the Crohn’s and Colitis Foundation and the Sanford Grossman Charitable Trust
Footnotes
Conflict of interest:
The authors declare that they have no competing interests
References
- 1.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491(7422): 119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nature genetics 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Meglio P, Di Cesare A, Laggner U, Chu CC, Napolitano L, Villanova F et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PloS one 2011; 6(2): e17160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pidasheva S, Trifari S, Phillips A, Hackney JA, Ma Y, Smith A et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PloS one 2011; 6(10): e25038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proceedings of the National Academy of Sciences of the United States of America 2011; 108(23): 9560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lassen KG, McKenzie CI, Mari M, Murano T, Begun J, Baxt LA et al. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity 2016; 44(6): 1392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014; 506(7489): 456–62. [DOI] [PubMed] [Google Scholar]
- 8.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001; 411(6837): 603–6. [DOI] [PubMed] [Google Scholar]
- 9.Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 2003; 124(1): 140–6. [DOI] [PubMed] [Google Scholar]
- 10.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet 2005; 365(9473): 1794–6. [DOI] [PubMed] [Google Scholar]
- 11.Lahiri A, Hedl M, Yan J, Abraham C. Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nature communications 2017; 8: 15614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. The New England journal of medicine 2009; 361(21): 2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H, Fang M, Jostins L, Umicevic Mirkov M, Boucher G, Anderson CA et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017; 547(7662): 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014; 343(6175): 1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348(6235): 648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierson E, Consortium GT, Koller D, Battle A, Mostafavi S, Ardlie KG et al. Sharing and Specificity of Co-expression Networks across 35 Human Tissues. PLoS computational biology 2015; 11(5): e1004220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holtta V, Sipponen T, Westerholm-Ormio M, Salo HM, Kolho KL, Farkkila M et al. In Crohn’s Disease, Anti-TNF-alpha Treatment Changes the Balance between Mucosal IL-17, FOXP3, and CD4 Cells. ISRN Gastroenterol 2012; 2012: 505432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eriksson C, Rantapaa-Dahlqvist S, Sundqvist KG. Changes in chemokines and their receptors in blood during treatment with the TNF inhibitor infliximab in patients with rheumatoid arthritis. Scand J Rheumatol 2013; 42(4): 260–5. [DOI] [PubMed] [Google Scholar]
- 19.Eder P, Lykowska-Szuber L, Krela-Kazmierczak I, Stawczyk-Eder K, Zabel M, Linke K. The influence of infliximab and adalimumab on the expression of apoptosis-related proteins in lamina propria mononuclear cells and enterocytes in Crohn’s disease - an immunohistochemical study. Journal of Crohn’s & colitis 2013; 7(9): 706–16. [DOI] [PubMed] [Google Scholar]
- 20.Hvas CL, Kelsen J, Agnholt J, Dige A, Christensen LA, Dahlerup JF. Discrete changes in circulating regulatory T cells during infliximab treatment of Crohn’s disease. Autoimmunity 2010; 43(4): 325–33. [DOI] [PubMed] [Google Scholar]
- 21.Haberman Y, Tickle TL, Dexheimer PJ, Kim MO, Tang D, Karns R et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. The Journal of clinical investigation 2014; 124(8): 3617–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kugathasan S, Denson LA, Walters TD, Kim MO, Marigorta UM, Schirmer M et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS genetics 2014; 10(5): e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plagnol V, Smyth DJ, Todd JA, Clayton DG. Statistical independence of the colocalized association signals for type 1 diabetes and RPS26 gene expression on chromosome 12q13. Biostatistics 2009; 10(2): 327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics 2009; 5(6): e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nature methods 2013; 10(1): 5–6. [DOI] [PubMed] [Google Scholar]
- 27.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nature methods 2011; 9(2): 179–81. [DOI] [PubMed] [Google Scholar]
- 28.Parts L, Stegle O, Winn J, Durbin R. Joint genetic analysis of gene expression data with inferred cellular phenotypes. PLoS genetics 2011; 7(1): e1001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS computational biology 2010; 6(5): e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proceedings of the National Academy of Sciences of the United States of America 2014; 111(21): 7741–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature genetics 2016; 48(3): 245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasaniuc B, Zaitlen N, Shi H, Bhatia G, Gusev A, Pickrell J et al. Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics (Oxford, England) 2014; 30(20): 2906–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR et al. Common SNPs explain a large proportion of the heritability for human height. Nature genetics 2010; 42(7): 565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cutler DJ, Zwick ME, Okou DT, Prahalad S, Walters T, Guthery SL et al. Dissecting Allele Architecture of Early Onset IBD Using High-Density Genotyping. PloS one 2015; 10(6): e0128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nature genetics 2017; 49(2): 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ching KH, Kisailus AE, Burbelo PD. Biochemical characterization of distinct regions of SPEC molecules and their role in phagocytosis. Exp Cell Res 2007; 313(1): 10–21. [DOI] [PubMed] [Google Scholar]
- 37.Rioux JD, Daly MJ, Silverberg MS, Lindblad K, Steinhart H, Cohen Z et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nature genetics 2001; 29(2): 223–8. [DOI] [PubMed] [Google Scholar]
- 38.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009; 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R et al. Massively parallel digital transcriptional profiling of single cells. Nature communications 2017; 8: 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011; 118(5): e16–31. [DOI] [PubMed] [Google Scholar]
- 42.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017; 356(6335). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ning K, Gettler K, Zhang W, Ng SM, Bowen BM, Hyams J et al. Improved integrative framework combining association data with gene expression features to prioritize Crohn’s disease genes. Human molecular genetics 2015; 24(14): 4147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015; 518(7539): 337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic acids research 2017; 45(D1): D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunological reviews 2014; 260(1): 102–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chuang LS, Villaverde N, Hui KY, Mortha A, Rahman A, Levine AP et al. A Frameshift in CSF2RB Predominant Among Ashkenazi Jews Increases Risk for Crohn’s Disease and Reduces Monocyte Signaling via GM-CSF. Gastroenterology 2016; 151(4): 710–723 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine AP, Pontikos N, Schiff ER, Jostins L, Speed D, Consortium NIBDG et al. Genetic Complexity of Crohn’s Disease in Two Large Ashkenazi Jewish Families. Gastroenterology 2016; 151(4): 698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brant SR, Okou DT, Simpson CL, Cutler DJ, Haritunians T, Bradfield JP et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans With Inflammatory Bowel Disease. Gastroenterology 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010; 33(3): 375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ziegler-Heitbrock L The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol 2007; 81(3): 584–92. [DOI] [PubMed] [Google Scholar]
- 52.Rosen MJ, Karns R, Vallance JE, Bezold R, Waddell A, Collins MH et al. Mucosal Expression of Type 2 and Type 17 Immune Response Genes Distinguishes Ulcerative Colitis From Colon-Only Crohn’s Disease in Treatment-Naive Pediatric Patients. Gastroenterology 2017; 152(6): 1345–1357 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics (Oxford, England) 2009; 25(9): 1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 2012; 7(3): 562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome biology 2011; 12(3): R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 2015; 31(2): 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 2007; 81(3): 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015; 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics (Oxford, England) 2012; 28(10): 1353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. American journal of human genetics 2011; 88(1): 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014; 15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 2010; 42(12): 1118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Devlin B, Roeder K. Genomic control for association studies. Biometrics 1999; 55(4): 997–1004. [DOI] [PubMed] [Google Scholar]
- 64.Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet 2010; 87(1): 139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. Journal of immunology 2011; 186(12): 7243–54. [DOI] [PubMed] [Google Scholar]
- 66.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics (Oxford, England) 2008; 24(13): 1547–8. [DOI] [PubMed] [Google Scholar]
- 67.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014; 343(6172): 776–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.