

Abstract
A copper catalyst in combination with N-fluorobenzenesulfonimide (NFSI) has been reported to functionalize benzylic C–H bonds to the corresponding benzylic sulfonimides via C–N coupling. Here, we reported a closely related Cu-catalyzed method with NFSI that instead leads to C–F coupling. This switch in selectivity arises from changes to the reaction conditions (Cu:ligand ratio, temperature, addition of base) and further benefits from inclusion of MeB(OH)2 in the reaction. MeB(OH)2 is shown to serve as a “redox buffer” in the reaction, responsible for rescuing inactive Cu(II) for continued promotion of fluorination reactivity.
Graphical Abstract:
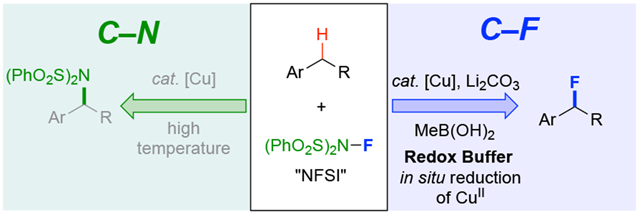
N–Fluorobenzenesulfonimide (NFSI) is a widely used reagent in organic synthesis. It is commonly used as a terminal oxidant in transition metal-catalyzed oxidations1 and as a group-transfer reagent for sulfonylation, fluorination, and sulfonimidation of organic molecules.2,3 The majority of these methods take advantage of the reactive N–F bond. For example, NFSI is commonly featured in electrophilic and radical fluorination of carbanions, carbon-centered radicals, and acidic C–H bonds,4 while complementary methods and mechanisms have been identified for C–N bond formation with the sulfonimide group.5 This bifurcation in reactivity between C–F and C–N bond formation is well documented in C(sp2)–H functionalization of (hetero)arenes with NFSI (Scheme 1A).3 We and others have recently been exploring Cu catalyzed methods for site-selective functionalization of benzylic C(sp3)–H bonds.6,7 These reactions employ NFSI in a radical-relay mechanism that enables oxidative C–H coupling with diverse nucleophilic partners, including cyanide, azide, trifluoromethyl, alcohols, and arylboronic acids (Scheme 1B). The earliest example of this reactivity featured direct transfer of the sulfonimide group from NFSI to the benzylic position (Scheme 1C, top).6 Here, we show that variation of the Cu/NFSI C–H sulfonimidation reaction conditions leads to C–F rather than C– N bond formation, complementing other benzylic C–H fluorination methods in the literature.8 Mechanistic studies provide insights into the origin of this switch in selectivity, highlighting the role of MeB(OH)2 as a “redox buffer” and Li2CO3 as a Brønsted base in the reaction. Additional studies reveal the intrinsic lability of secondary benzylic fluorides, making them susceptible to nucleophilic substitution. The latter reactivity is noted here, but provides basis for a complementary effort, leading to a versatile new class of benzylic C–H cross-coupling reactions.9
Scheme 1.
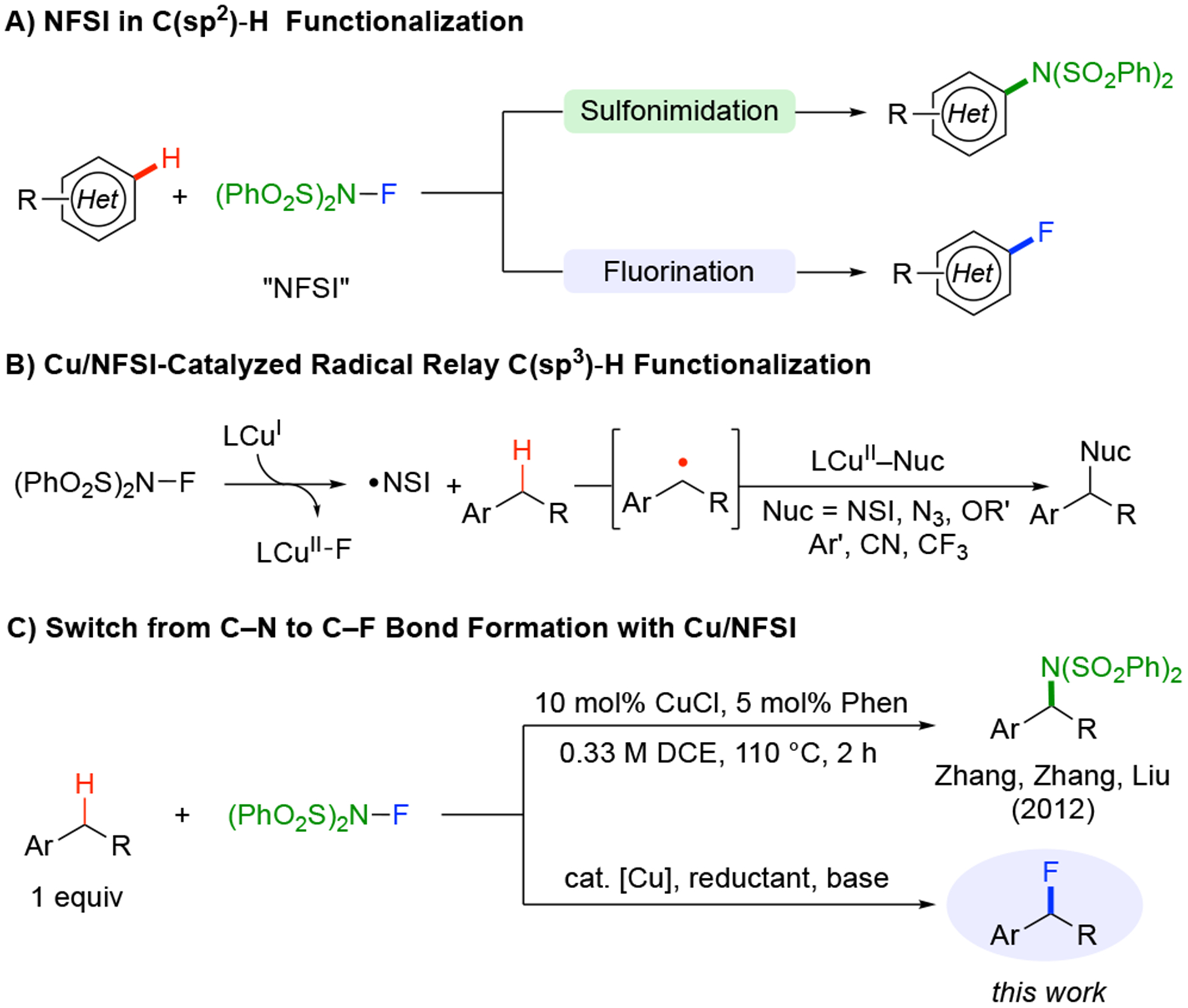
C–N to C–F Bond Formation when Using NFSI in C(sp2) and C(sp3) C–H Functionalization
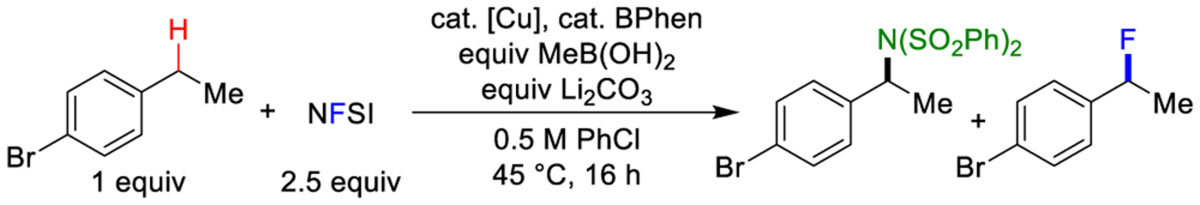
The present study was initiated by investigating the original Cu/NFSI-catalyzed sulfonimidation reaction (Scheme 1C).6 This reaction takes place at much higher temperatures than more recent Cu/NFSI reactions, which often proceed near room temperature.7 Attempting the sulfonimidation of p-bromoethylbenzene at lower temperature led to low conversion, with a preference for C–N over C–F bond formation (Table 1, entry 2). The formation of a C–F bond could arise from reaction of an intermediate benzylic radical with a CuII–F species10 or via a Cu-promoted radical-chain process involving NFSI.8g, 11 Addition of MeB(OH)2 as an in situ reductant for the Cu catalyst7e (see further discussion below) led to complete substrate conversion, but only moderate yield of the C–N product was observed, with no C–F product (Table 1, entry 3). Further variation of the conditions, however, including addition of Li2CO3 as a base, using PhCl as the solvent, and lowering the catalyst loading, led to C–H fluorination in good yield (81%, Table 1, entry 7), with no C–N product formation (see additional screening data in the Supporting Information). Difluorination of the benzylic position is the primary side reaction.
Table 1.
Fluorination Reaction Optimizations
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | [Cu] (mol%) | BPhen (mol%) | MeB(OH)2 (equiv) | Li2CO3 (equiv) | Conv (%) | C–N (%) | C–F (CF2)a (%) |
| 1b,c | CuCl (10) | see Scheme 1A for conditions | nd | 76 | nd | ||
| 2c | CuCl (10) | 5 | - | - | 24 | 19 | 4 |
| 3c | CuCl (10) | 5 | 2 | - | 100 | 42 | - |
| 4c | CuCl (10) | 5 | 2 | 3 | 100 | 44 | 21 (4) |
| 5 | CuCl (10) | 5 | 2 | 3 | 100 | 25 | 40 (6) |
| 6 | CuCl (2) | 1 | 2 | 3 | 86 | 4 | 74 (4) |
| 7 | CuOAc (2) | 2.4 | 2 | 3 | 100 | - | 81 (11) |
0.3 mmol scale. 1H NMR yields; int. std. = CH2Br2.
Reported results with ethylbenzene (ref. 6).
1,2-dichloroethane (DCE) used as the solvent.
Several fundamental studies were undertaken to gain insights into the observed reactivity and the role of MeB(OH)2 and other reaction components. Copper(I) is proposed to react with NFSI, forming a CuII–F species and an imidyl radical, •NSI (Scheme 1B). The imidyl radical can promote hydrogen atom transfer from the benzylic C–H bond, but it can react even more rapidly with another CuI center,7e,12 generating a second equivalent of CuII and halting catalysis. To probe Cu/NFSI reactivity, NFSI was titrated into a solution of BPhenCuI(OAc) in PhCl (Bphen = bathophenanthroline). Nearly isosbestic behavior was observed by UV-visible spectroscopy, corresponding to oxidation of CuI to CuII species by NFSI (Figure 1A, step 1). Complete consumption of CuI was observed upon addition of 0.5 equiv of oxidant. This oxidation is rapid at room temperature, occurring on the timescale of mixing. Addition of NFSI beyond 0.5 equiv has no effect on the UV-visible spectrum, suggesting that CuII does not react further with NFSI.
Figure 1. Fundamental insights into Cu/NFSI-catalyzed fluorination reactions.
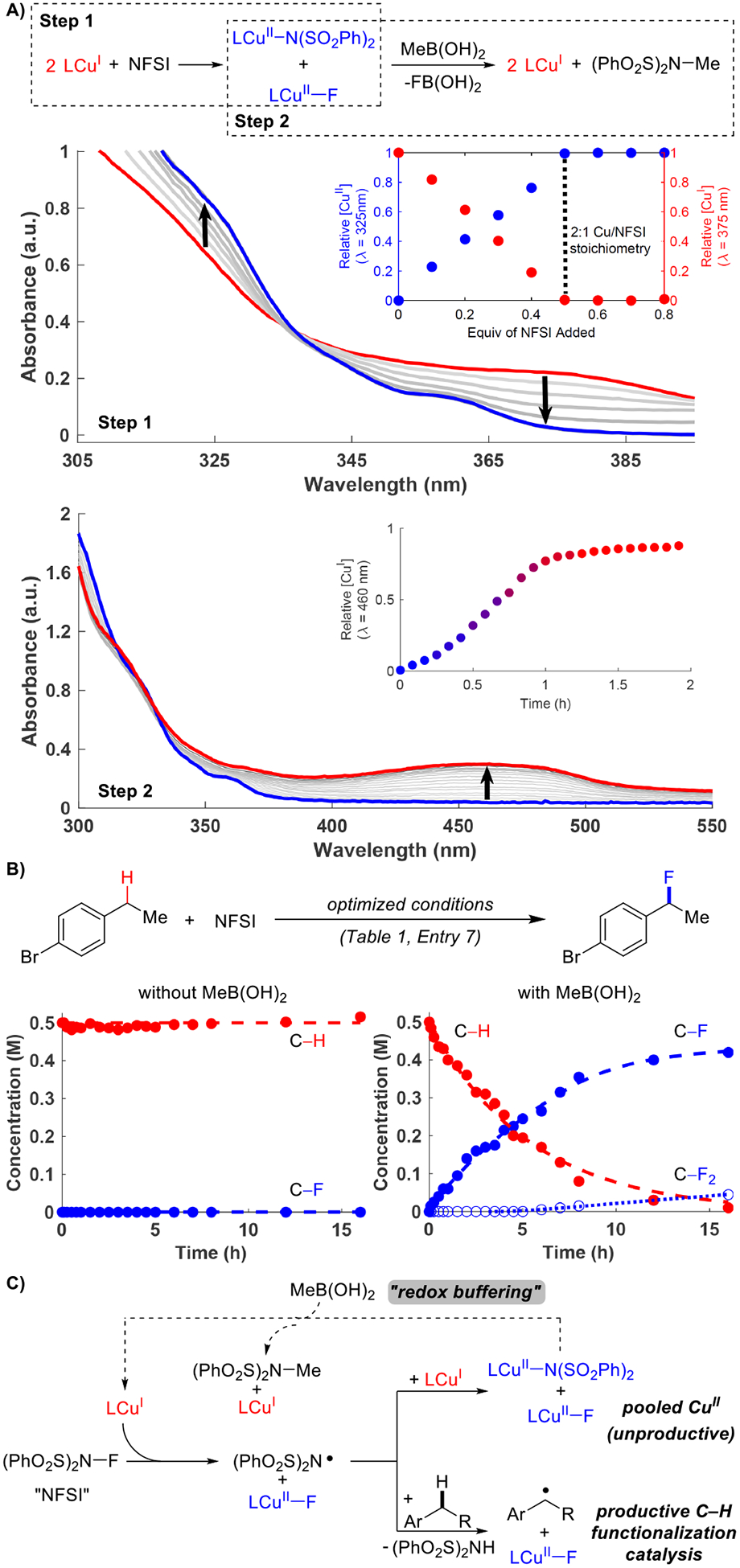
A) Spectroscopic analysis of stoichiometric oxidation of CuI by NFSI and reduction of CuII by MeB(OH)2. B) Reaction time course data demonstrating the effect of MeB(OH)2 redox buffering in the catalytic fluorination reaction. C) Mechanism depicting the redox buffering role of MeB(OH)2 in benzylic fluorination. Conditions: A) Step 1 – 0.33 mM BPhenCuIOAc + 0.1 equiv NFSI (×8) in PhCl. Step 2 – [CuII] (from 0.33 mM BPhenCuI(OAc) + 0.5 equiv NFSI) + 5 equiv MeB(OH)2, 15 equiv Li2CO3 in PhCl. B) See Table 1, entry 7.
The CuII species generated by NFSI is reduced by MeB(OH)2. Addition of 5 equiv of MeB(OH)2 to a solution of CuII generated from a combination of BPhenCuI(OAc) and 0.5 equiv of NFSI in PhCl slowly regenerates CuI over approximately 1 h (Figure 1A, step 2). This process generates Me–N(SO2Ph)2 as a byproduct of the reaction, resembling the previously reported Chan-Lam amidation of alkylboronic acids13 (see Figure S2).
The impact of MeB(OH)2 on the catalytic reaction is clearly evident in Figure 1B. Virtually no reaction is observed in the absence of MeB(OH)2. In contrast, full substrate conversion occurs in the presence of 2 equiv of MeB(OH)2, leading to an 84% yield of the benzyl fluoride. This behavior is rationalized by the ability of MeB(OH)2 to serve as a “redox buffer” for the Cu catalyst, slowly reducing CuII during the reaction. A mechanistic framework for these observations is illustrated in Figure 1C. CuI is oxidized rapidly to CuII by NFSI at the beginning of the reaction, but slow reduction of CuII by MeB(OH)2 (dashed arrow, Figure 1C) generates small amounts of CuI that can react with additional NFSI. Generation of •NSI at this stage can lead to hydrogen atom transfer (HAT) from the benzylic C–H with only limited competitive quenching of •NSI by CuI, due to the low CuI concentration.
The synthetic scope of this reactivity is the focus of a separate study;9 however, testing of representative substrates showed that isolation of benzyl monofluorides can be rather challenging, with poor mass balance and the appearance of new byproducts (see Supporting Information, Section V). Similar challenges are evident from previous C–H fluorination methods,8 and other studies show that benzyl monofluorides undergo facile displacement in the presence of Brønsted and Lewis acids or hydrogen-bond donors.14 The latter insights prompted us to assess the role of Li2CO3 in the reaction, which was also used in a previous benzylic C–H fluorination method.8g A time course for the optimized reaction conditions in Figure 1B (right) may be compared to the time course obtained without added Li2CO3 (Scheme 2A). The rate of substrate conversion is virtually identical in the presence and absence of Li2CO3; however, very little C–F product is observed after the first few hours of the reaction without Li2CO3, and the major products arise from C–O and C–N bond formation. NHSI is a strong acid and will build up as the fluorination reaction proceeds, and it could promote acidolysis of the benzyl fluoride.15 In a control experiment, 1 equiv of NHSI was added to a solution of benzyl fluoride obtained from the catalytic reaction, following filtration through a silica plug to remove the Li2CO3. This reaction results in rapid formation of 1-phenylethanol (Scheme 2B). The hydroxy group presumably originates from adventitious water from the solvent or reagents present in the original reaction mixture. The net C–H fluorination/hydrolysis sequence resembles the recently reported C–H mesylation/hydrolysis strategy to achieve C–H hydroxylation, reported by Ritter and coworkers.16
Scheme 2.
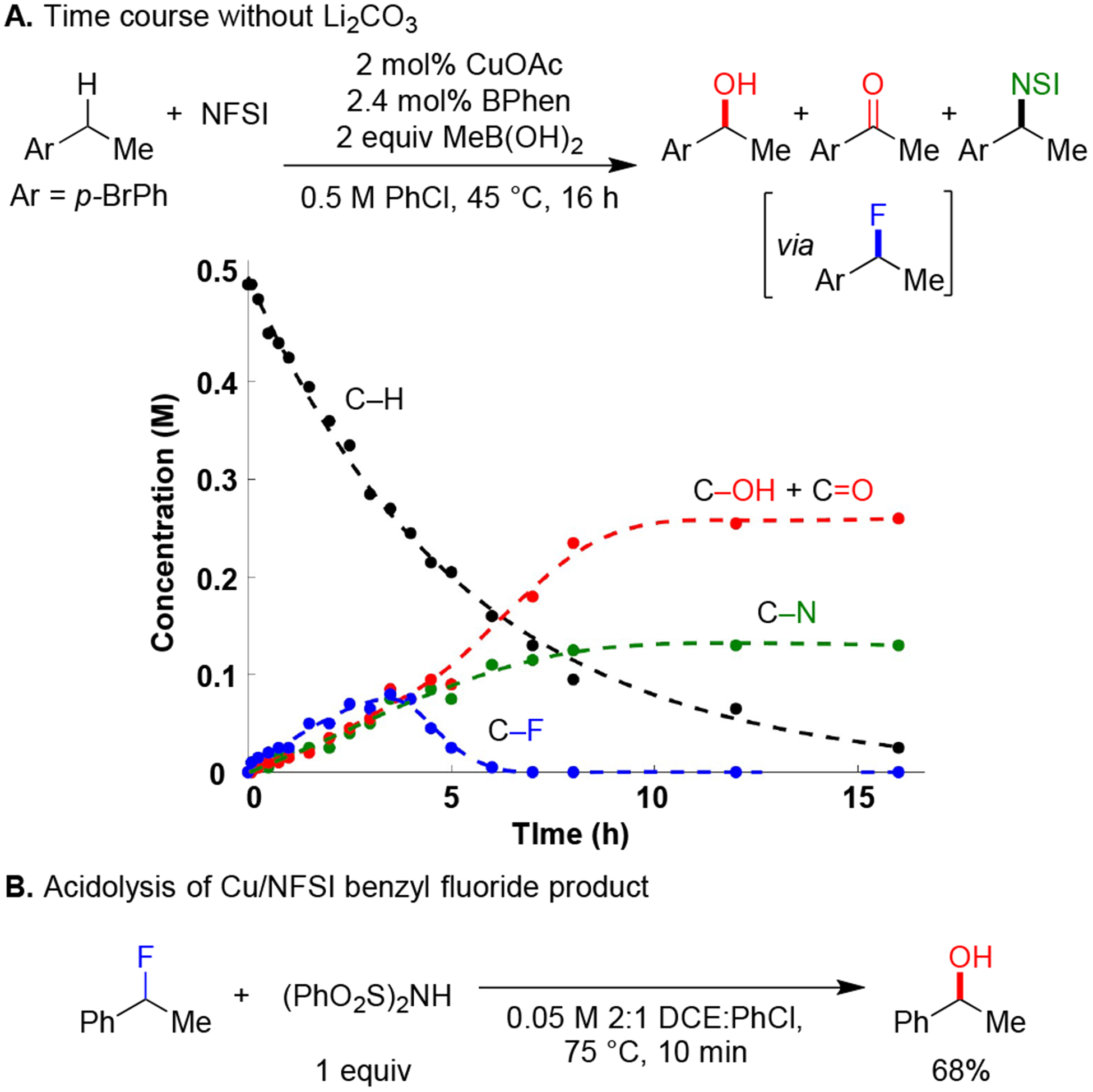
Li2CO3 Effect on Benzyl Fluoride Lability
The results presented herein reveal an unusual switch in selectivity, from C–N to C–F bond formation, in Cu/NFSI-promoted oxidative functionalization of benzylic C–H bonds. Mechanistic studies show how two new reaction additives contribute to formation of the fluorination product. MeB(OH)2 serves as a redox buffer7e that promotes steady-state reduction of CuII to the active CuI species during the catalytic reaction. Li2CO3 serves as a Brønsted base that prevents acid-promoted displacement of the fluoride during the reaction. While the observed substitutional lability of the products undermines the accessibility and practical utility of isolated benzyl monofluorides, this property introduces the possibility of using benzyl fluorides as strategic intermediates in a C–H fluorination/substitution sequence. Development and elaboration of the latter C–H cross-coupling strategy is the focus of a parallel report.9
Supplementary Material
ACKNOWLEDGMENT
The authors thank Scott McCann and Si-Jie Chen (UW-Madison) for valuable discussions leading to the development of redox buffering in Cu/NFSI systems. This work was supported by the NIH (R01 GM126832, R35 GM134929), including a Ruth L. Kirschstein NRSA fellowship (F32 GM129909, to JAB). Spectroscopic instrumentation was partially supported by the NIH (1S10 OD020022-1) and the NSF (CHE-1048642).
Footnotes
Supporting Information
Experimental procedures, characterization data, and NMR spectra.
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- 1.Engle KM; Mei T-S; Wang X; Yu J-Q Bystanding F+ Oxidants Enable Selective Reductive Elimination from High-Valent Metal Centers in Catalysis. Angew. Chem. Int. Ed 2011, 50, 1478–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Roy A; Schneller SW An Unusual Occurrence on Attempted Purine C-8 Electrophilic Fluorination of 5’-Noraristeromycin. Org. Lett 2005, 7, 3889–3891. [DOI] [PubMed] [Google Scholar]; (b) Jing L; Yu X; Guan M; Wu X; Wang Q; Wu X An Efficient Method for Sulfonylation of Amines, Alcohols, and Phenols with N-Fluorobenzenesulfonimide Under Mild Conditions. Chem. Res. Chin. Univ 2018, 34, 191–196. [Google Scholar]; (c) Jie K; Wang Y; Huang L; Guo S; Cai H Convenient Sulfonylation of Imidazoles and Triazoles Using NFSI. J. Sulfur Chem 2018, 39, 465–471. [Google Scholar]
- 3.Gu Q; Vessally E N-Fluorobenzenesulfonimide: A Useful and Versatile Reagent for The Direct Fluorination and Amination of (Hetero)Aromatic C–H Bonds. RSC Adv. 2020, 10, 16756–16768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For recent reviews of fluorination chemistry, see:; (a) Champagne PA; Desroches J; Hamel J-D; Vandamme M; Paquin J-F Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]; (b) Yerien DE; Bonesi S; Postigo A Fluorination Methods in Drug Discovery. Org. Biomol. Chem 2016, 14, 8398–8427. [DOI] [PubMed] [Google Scholar]; (c) Szpera R; Moseley DFJ; Smith LB; Sterling AJ; Gouverneur V The Fluorination of C–H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed 2019, 58, 14824–14848. [DOI] [PubMed] [Google Scholar]
- 5.Li Y; Zhang Q N-Fluorobenzenesulfonimide: An Efficient Nitrogen Source for C–N Bond Formation. Synthesis 2015, 47, 159–174. [Google Scholar]
- 6.Ni Z; Zhang Q; Xiong T; Zheng Y; Li Y; Zhang H; Zhang J; Liu Q Highly Regioselective Copper-Catalyzed Benzylic C–H Amination by N-Fluorobenzenesulfonimide. Angew. Chem. Int. Ed 2012, 51, 1244–1247. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang W; Chen P; Liu G Copper-Catalyzed Arylation of Benzylic C–H Bonds with Alkylarenes as the Limiting Reagents. J. Am. Chem. Soc 2017, 139, 7709–7712. [DOI] [PubMed] [Google Scholar]; (c) Xiao H; Liu Z; Shen H; Zhang B; Zhu L; Li C Copper-Catalyzed Late-Stage Benzylic C(sp3)–H Trifluoromethylation. Chem 2019, 5, 940–949. [DOI] [PubMed] [Google Scholar]; (d) Wang A; DeOliveira CC; Emmert M Non-Directed, Copper Catalyzed Benzylic C–H Amination Avoiding Substrate Excess. ChemRxiv 2019, 10.26434/chemrxiv.8792243. [DOI] [Google Scholar]; (e) Hu H; Chen S-J; Mandal M; Pratik SM; Buss JA; Krska SW; Cramer CJ; Stahl SS Copper-Catalyzed Benzylic C–H Coupling with Alcohols via Radical Relay Enabled by Redox Buffering. Nat. Catal 2020, 3, 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Suh S-E Chen S-J Mandal M; Guzei IA; Cramer CJ; Stahl SS Site-Selective Copper-Catalyzed Azidation of Benzylic C–H Bonds. J. Am. Chem. Soc 2020, 142, 11388–11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For selected primary references describing benzylic fluorination, see:; (a) Bloom S; Pitts CR; Miller DC; Haselton N; Holl MG; Urheim E; Lectka T A Polycomponent Metal-Catalyzed Aliphatic, Allylic, and Benzylic Fluorination. Angew. Chem. Int. Ed 2012, 51, 10580–10583. [DOI] [PubMed] [Google Scholar]; (b) Xia J-B; Zhu C; Chen C Visible Light-Promoted Metal-Free C–H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Difluorination. J. Am. Chem. Soc 2013, 135, 17494–17500. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bloom S; Pitts CR; Woltornist R; Griswold A; Holl MG; Lectka T Iron(II)-Catalyzed Benzylic Fluorination. Org. Lett 2013, 15, 1722–1724. [DOI] [PubMed] [Google Scholar]; (d) Amaoka Y; Nagatomo M; Inoue M Metal-Free Fluorination of C(sp3)−H Bond Using a Catalytic N-Oxyl Radical. Org. Lett 2013, 15, 2160–2163. [DOI] [PubMed] [Google Scholar]; (e) Cantillo D; de Frutos O; Rincón JA; Mateos C; Kappe CO A Continuous-Flow Protocol for Light-Induced Benzylic Fluorinations. J. Org. Chem 2014, 79, 8486–8490. [DOI] [PubMed] [Google Scholar]; (f) Xu P; Guo S; Wang L; Tang P Silver-Catalyzed Oxidative Activation of Benzylic C–H Bonds for the Synthesis of Difluoromethylated Arenes. Angew. Chem. Int. Ed 2014, 53, 5955–5958. [DOI] [PubMed] [Google Scholar]; (g) Nodwell MB; Bagai A; Halperin SD; Martin RE; Knust H; Britton R Direct Photocatalytic Fluorination of Benzylic C–H Bonds with N-Fluorobenzenesulfonimide. Chem. Commun 2015, 51, 11783–11786. [DOI] [PubMed] [Google Scholar]; (h) Groendyke BJ; AbuSalim DI; Cook SP Iron-Catalyzed, Fluoroamide-Directed C–H Fluorination. J. Am. Chem. Soc 2016, 138, 12771–12774. [DOI] [PubMed] [Google Scholar]; (i) Meanwell M; Nodwell MB; Martin RE; Britton R A Convenient Late-Stage Fluorination of Pyridylic C–H Bonds with N-Fluorobenzenesulfonimide. Angew. Chem. Int. Ed 2016, 55, 13244–13248. [DOI] [PubMed] [Google Scholar]; (j) Xiang M; Xin Z-K; Chen B; Tung C-H; Wu L-Z Exploring the Reducing Ability of Organic Dye (Acr+‑Mes) for Fluorination and Oxidation of Benzylic C(sp3)−H Bonds under Visible Light Irradiation. Org. Lett 2017, 19, 3009–3012. [DOI] [PubMed] [Google Scholar]; (k) Hua AM; Mai DN; Martinez R; Baxter RD Radical C–H Fluorination Using Unprotected Amino Acids as Radical Precursors. Org. Lett 2017, 19, 1949–1952. [DOI] [PubMed] [Google Scholar]
- 9.Vasilopoulos A; Golden DL; Buss JA; Stahl SS Copper-Catalyzed C–H Fluorination/Functionalization Sequence Enabling Benzylic C–H Cross Coupling with Diverse Nucleophiles. Org. Lett 2020, Accepted. DOI: 10.1021/acs.orglett.0c02238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bower JK; Cypcar AD; Henriquez B; Stieber SCE; Zhang SC (sp3)–H Fluorination with a Copper(II)/(III) Redox Couple. J. Am. Chem. Soc 2020, 142, 8514–8521. [DOI] [PubMed] [Google Scholar]
- 11.(a) Rueda-Becerril M; Sazepin CC; Leung JCT; Okbinoglu T; Kennepohl P; Paquin J-F; Sammis GM Fluorine Transfer to Alkyl Radicals. J. Am. Chem. Soc 2012, 134, 4026–4029. [DOI] [PubMed] [Google Scholar]; (b) Pitts CR; Bloom S; Woltornist R; Auveshine DJ; Ryzhkov LR; Siegler MA; Lectka T Direct, Catalytic Monofluorination of sp3 C–H Bonds: A Radical-Based Mechanism with Ionic Selectivity. J. Am. Chem. Soc 2014, 136, 9780–9791. [DOI] [PubMed] [Google Scholar]; (c) Li M; Xue X-S; Chen J-P Establishing Cation and Radical Donor Ability Scales of Electrophilic F, CF3, and SCF3 Transfer Reagents. Acc. Chem. Res 2020, 53, 182–197. [DOI] [PubMed] [Google Scholar]
- 12.Haines BE; Kawakami T; Murakami K; Itami K; Musaev DG Cu-Catalyzed Aromatic C–H Imidation with N-Fluorobenzenesulfonimide: Mechanistic Details and Predictive Models. Chem. Sci 2017, 8, 988–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossi SA; Shimkin KW; Xu Q; Mori-Quiroz LM; Watson DA Selective Formation of Secondary Amides via the Copper-Catalyzed Cross-Coupling of Alkylboronic Acids with Primary Amides. Org. Lett 2013, 15, 2314–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Amii H; Uneyama K C–F Bond Activation in Organic Synthesis. Chem. Rev 2009, 109, 2119–2183. [DOI] [PubMed] [Google Scholar]; (b) Hamel J-D; Paquin J-F Activation of C–F Bonds α to C–C Multiple Bonds. Chem. Commun 2018, 54, 10224–10239. [DOI] [PubMed] [Google Scholar]
- 15.(a) Bernstein J; Roth JS; Miller WT Jr. The Preparation and Properties of Some Substituted Benzyl Fluorides. J. Am. Chem. Soc 1948, 70, 2310–2314. [DOI] [PubMed] [Google Scholar]; (b) Swain CG; Spalding RET III. Mechanism of Acid Catalysis of the Hydrolysis of Benzyl Fluoride. J. Am. Chem. Soc 1960, 82, 6104–6107. [Google Scholar]
- 16.Tanwar L; Börgel J; Ritter T Synthesis of Benzylic Alcohols by C– H Oxidation. J. Am. Chem. Soc 2019, 141, 17983–17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.