

Abstract
Endometrial cancer accounts for ~76,000 deaths amongst women worldwide. Disease mortality and the increasing number of new diagnoses make endometrial cancer an important consideration in women’s health, particularly in industrialized countries, where the incidence of this tumor type is highest. Most endometrial cancers are carcinomas, with the remainder being sarcomas. Endometrial carcinomas can be classified into several histological subtypes including endometrioid, serous and clear cell carcinomas. Histological subtyping is currently routinely used to guide prognosis and treatment decisions for endometrial cancer patients, while ongoing studies are evaluating the potential clinical utility of molecular subtyping. In this review we summarize the over-arching molecular features of endometrial cancers and highlight recent studies assessing the potential clinical utility of specific molecular features for early detection, disease risk stratification, and directing the use of targeted therapies.
Introduction
Uterine corpus cancer is the 6th leading cause of cancer death amongst women in the US and the 8th leading cause of cancer-related death amongst European women1,2. Most uterine cancers are endometrial carcinomas (ECs), originating from the uterine epithelium (Fig. 1A). The vast majority of ECs are sporadic, with an estimated 5% occurring in the context of inherited cancer susceptibility syndromes3, most commonly Lynch Syndrome (Box 1)4–6. ECs are classified into various histological subtypes, including endometrioid EC (EEC), serous EC (SEC), clear cell EC (CCEC), mixed EC, and uterine carcinosarcoma [G] (UCS), which differ in their frequency, clinical presentation, prognosis and associated epidemiological risk factors (BOX 1)7–9. Importantly, the incidence of EC is rising in the US and more than 20 other countries10; recent data correcting for hysterectomy [G] rates in the US and Denmark indicates that the incidence of non-EECs, which are generally more clinically aggressive, is increasing while the incidence of EECs has remained stable or decreased over time11,12.
Figure 1. Overview of endometrial carcinoma origin, development, and molecular classification.
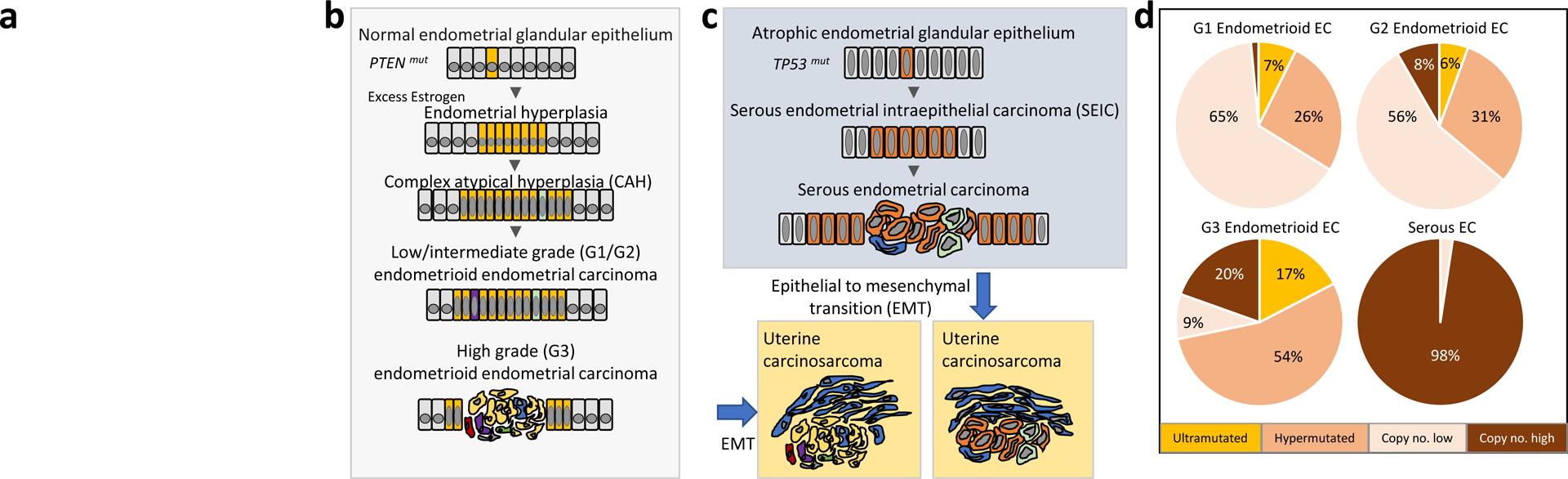
(a) The image in this panel was deleted to comply with Nature Reviews Cancer policy on self-archiving. Schematic depiction of the initiation and progression of endometrioid (B) and serous (C) endometrial carcinomas (ECs) from the normal and atrophic endometrial glandular epithelium, via precursor lesions (CAH and SEIC). Columnar epithelial cells that have acquired somatic mutations are colored; intratumoral heterogeneity is depicted by differentially colored epithelial cells. PTEN mutation and TP53 mutation are, respectively, early events in the etiology of many endometrioid and serous endometrial carcinomas. In some instances, carcinomas, particularly high-grade carcinomas, undergo an epithelial to mesenchymal transition to give rise to uterine carcinosarcomas, which are biphasic tumors consisting of epithelial carcinoma cells and sarcoma cells (blue). (D) Pie charts showing the distribution (% of tumors) of low grade (grade 1 and grade 2) endometrioid EC, high grade (grade 3) endometrioid EC, and serous EC among the four molecular subgroups delineated in The Cancer Genome Atlas15.
Box 1. Epidemiological and genetic risk factors for endometrial cancer.
Epidemiological risk factors:
Increased risk for developing endometrioid endometrial cancer (EC), an estrogen dependent tumor type, is associated with obesity, diabetes, unopposed estrogen use, nulliparity, early menarche, and late menopause8. Increasing age is a risk factor for serous and clear cell ECs8. Tamoxifen use increases risk of developing EC; histological subtypes enriched in users of tamoxifen are serous EC, high-grade endometrioid ECs and carcinosarcomas9.
Genetic risk factors:
Increased genetic risk for developing EC is associated with Lynch Syndrome, Polymerase Proofreading Associated Polyposis, and Cowden Syndrome.
Lynch Syndrome is a highly penetrant, autosomal dominant cancer predisposition syndrome caused by monoallelic germline mutation in a mismatch repair gene, specifically MLH1, MSH2, MSH6 or PMS2, or by germline deletion within EPCAM that leads to epigenetic silencing of the adjacent MSH2 gene4. Mutation carriers are at increased risk of developing colorectal cancer and ECs, the two major component tumors of Lynch Syndrome, as well as cancers of the ovary, stomach, kidney, urinary tract, biliary tract, small intestine and skin4. Approximately 2–6% of ECs are attributed to Lynch Syndrome157.
Polymerase Proofreading Associated Polyposis is an autosomal dominant cancer susceptibility syndrome attributed to germline mutations in the exonuclease domain of POLD1 or POLE. POLD1 mutation carriers are at increased risk of developing attenuated adenomatous polyposis of the colorectum and cancers of the colorectum, endometrium, breast, and brain5. POLE mutation carriers are at increased risk of developing colorectal cancer. Predisposition to EC has not been established in POLE mutation carriers.
Cowden syndrome is a condition in which PTEN mutation carriers have an increased predisposition for developing multiple hamartomas, and cancers of the breast, thyroid, endometrium, colorectum, kidney, and skin6.
EECs constitute more than 80% of newly diagnosed EC cases, are generally estrogen-dependent tumors, and have a mean age at diagnosis of 62 years8. In contrast, SECs and CCECs are relatively uncommon, accounting for ~10% and 3% of newly diagnosed ECs, are generally estrogen-independent, and are diagnosed later in life (mean of 66.5 and 65.6 years, respectively)8. Uterine carcinosarcomas (UCSs), which are biphasic tumors composed of both carcinomatous and sarcomatous cells, represent <2% of ECs13. The prognosis for most newly diagnosed EC patients is good, with a relative 5-year survival rate of 81.1% (2008–2014)14. The generally high survival rate for EC is largely driven by the frequent early detection of EECs, coupled with the effectiveness of surgery for treating many early-stage, low-grade EECs. However, considerably poorer outcomes are associated with high-grade, recurrent, or metastatic EEC, as well as certain non-endometrioid histologies (SEC, CCEC and UCS).
Research conducted over the last two decades, fueled by the need to identify biomarkers to predict disease recurrence and druggable targets, has revealed critical insights into the molecular landscape of ECs (Table 1). We now know that low grade EECs and SECs have distinguishing molecular features, while the mutational profiles of CCECs overlap with that of EECs and SECs15–25. Although UCSs most closely resemble SECs molecularly, these highly aggressive tumors exhibit unique somatic changes including whole genome doubling and variable epithelial-to-mesenchymal transition [G] (EMT) gene signature scores16. Herein, we review the major molecular characteristics of ECs and UCSs, as well as recent efforts to translate this knowledge into clinical actionability.
Table 1.
Somatic aberration [G] frequencies for major driver genes in endometrial carcinomas
| Somatic Aberration | Potential Clinical Actionability | EEC | SEC | UCS | CCEC | Refs |
|---|---|---|---|---|---|---|
| Mutated PTEN |
|
|
2–3% | 11–33% | 0–21% | 15,16,19,21–23,25,38,53,56,63,64,158 |
| Mutated PIK3CA | PI3K-AKT-mTOR inhibition |
|
15–35% | 22–40% | 24–36% | 15–19,21–25,38,43,52,53,56,158 |
| Mutated PIK3R1 | PI3K-AKT-mTOR inhibition |
|
5–8% | 6–20% | 7–18% | 15,16,19,21–23,37,38,56,64 |
| Mutated KRAS | MEK inhibition |
|
2–6% | 10–17% | 2–14% | 15,16,19,21–23,25,38,43,52,53,56,64,158 |
| Mutated FGFR2 | FGFR inhibition |
|
8% | 0–2% | 0% | 15,16,22,38,43,56,64,159 |
| Mutated CTNNB1 | Adverse prognosis in low-grade EEC |
|
0–3% | 0–5% | 0% | 15,16,21,22,25,43,56,158 |
| MSI (MMR-D) |
|
|
0–3% | 3–6% | 11–14% | 15,16,18,19,43,63,144 |
| Mutated ARID1A | Multiple potential synthetic lethal interactions |
|
7–11% | 10–24% | 14–21% | 15,16,19,22,25,56,64,158 |
| Mutated POLE |
|
|
0–2% | 3–4% | 2–7% | 15,18,19,21,56,69,94,95,100,101,151 |
| Mutated TP53 |
|
|
59–93% | 44–91% | 28–46% | 15–19,21,23–25,38,48,52,56,64,112,158,160 |
| Mutated FBXW7 | Undetermined |
|
15–29% | 11–39% | 13–25% | 15–18,21,23,24,52,56 |
| Mutated PPP2R1A | Undetermined |
|
19–43% | 13–28% | 7–21% | 15–19,23–25,52,56,64,158 |
| Amplified ERBB2 | ERBB2 inhibition |
|
26%−44% | 9% | 11% | 15,16,21,52 |
The molecular etiology of endometrial cancers
Endometrioid endometrial cancer
The development of EEC is strongly associated with epidemiological risk factors leading to an excess of estrogen relative to progesterone (Box 1)8,9,26–28. Unopposed estrogen stimulation of the uterine epithelium can result in the outgrowth of endometrial hyperplasia, which may further evolve into complex atypical hyperplasia [G] (CAH), the precursor lesion for EEC (FIG. 1B). Molecular studies of EEC, its precursor lesions, and morphologically normal endometrial glands have inferred the timing of somatic mutations in tumor initiation and progression, which proceeds via branched evolution29–32,33–35. Such studies have shown that PTEN mutation, the most frequent somatic mutation among EECs (Table 1), is an early but insufficient event in the initiation of tumorigenesis. This observation is corroborated and complemented by genetically engineered mouse models of EC, which have demonstrated that biallelic Pten loss leads to development of CAH, whereas biallelic Pten loss together with mutational activation of Pik3ca results in progression of CAH to EC36. These findings add context to the fact that PTEN mutations commonly co-occur with PIK3CA and PIK3R1 mutations in human EECs15,37–40. Genetically-engineered mouse models have also inferred co-operativity between Pten loss and Ctnnb1 (which encodes β-CATENIN) mutation (exon 3 deletion, resulting in β-catenin stabilization) or Mlh1 inactivation. Specifically, Ctnnb1 exon 3 deletion synergizes with biallelic Pten loss and Pik3ca activation to promote EC and myometrial invasion in the setting of ovarian insufficiency [G]41, and endometrial tumorigenesis is accelerated in Pten+/−/Mlh1−/− mice as compared to Pten+/− mice42. In the context of human EC, gain-of-function missense mutations in CTNNB1 exon 3, resulting in β-catenin stabilization, and epigenetic silencing of MLH1, leading to mismatch repair [G] deficiency (MMR-D), are frequent aberrations in EEC that often independently co-occur with PTEN inactivating mutations (Table 1)15,43. The pathogenicity of MMR-D in EC is further underscored by the inherited predisposition to EC in MMR gene mutation carriers in Lynch Syndrome families (Box 1). Another common aberration among EECs is mutational inactivation of the ARID1A tumor suppressor gene [G] (Table 1). Although loss of Arid1a is insufficient to induce endometrial hyperplasia or carcinoma in mouse models44,45, immunohistochemical analyses of human CAHs have shown that endometrial glands with loss of both ARID1A and PTEN expression have higher proliferative indices than adjacent glands with loss of only PTEN46. This finding has led to the proposal that ARID1A acts as a “gatekeeper” to suppress the transition of PTEN-deficient CAH to EEC46.
Serous endometrial cancers
SECs are generally estrogen-independent and arise in the setting of the atrophic endometrium [G] (FIG. 1C). They are high-grade tumors, preceded by serous endometrial intraepithelial carcinoma [G] (SEIC) (FIG. 1C), that are often diagnosed at a late stage with a high risk of recurrence. The occurrence of TP53 mutations and/or p53 stabilization in SEIC, is evidence for this aberration being an early event in SEC pathogenesis47,48. Consistent with this idea, aged transgenic mice with conditional deletion of Trp53 in the genitourinary tract develop SEC, as well as other ECs49. However, this phenotype is in contrast to the lack of endometrial tumors in mice with endometrial-specific deletion of Trp5350. One possible explanation for this difference is the variable ages at which mice were assessed for tumor development in each model (1 year versus 5 months, respectively). The frequent occurrence of FBXW7, PIK3CA, and PPP2R1A somatic mutations (Table 1) as well as CCNE1 amplification in SEC15,17,24,37,51–53, and in adjacent SEIC, indicates that these are also early events in disease pathogenesis24,51. The functional impairment of SEC-associated recurrent mutations in FBXW7, PIK3CA, and PPP2R1A has been established experimentally54,53,55, and the pathogenicity of cyclin E dysregulation in cancer is well-recognized. However, the precise role of these events in the initiation and progression of SEC, both by themselves and in conjunction with TP53 mutations, would be aided by the development of appropriate mouse models.
Uterine carcinosarcomas
Most (but not all) UCSs are believed to be monoclonal, originating from high grade EEC, SEC or other aggressive histotypes that have undergone a metaplastic transition to form the sarcomatous component of these biphasic tumors (FIG. 1B,C). Supporting their derivation from aggressive ECs, the most frequently somatically mutated genes in UCSs are also commonly mutated in other ECs (Table 1)16,18,20,23,56; in fact, UCSs most closely resemble SECs molecularly15,16,18,20,56–58. However, unlike most other subtypes of EC (except grade 3 EEC) where PTEN and TP53 mutations tend to be mutually exclusive15,19, a majority of UCSs with PTEN mutations also have TP53 mutations16,20,56. The co-occurrence of PTEN and TP53 mutations is not likely due to the biphasic nature of UCSs since the carcinoma [G] and sarcoma [G] components of most UCSs share mutations23,56,59,60.
Also distinguishing UCSs from other tumor types, whole-genome-doubling occurs in 90% of UCSs, and UCSs exhibit highly variable EMT transcriptomic gene signature scores16, which may reflect the transient nature of EMT and/or the presence of intermediate EMT states61. Although EMT scores have not been shown to correlate with outcome in UCSs16, EMT has been associated with metastasis and therapy resistance in other tumor types61,62, thus maintaining the possibility that EMT contributes to the poor prognosis for women with UCSs.
Clear cell endometrial cancers
The exact molecular etiology of CCECs remains unclear but multiple studies have shown that clinically diagnosed CCECs share mutational features with both EECs and SECs19,21,63,64 (Table 1). However, a clinical diagnosis of CCEC represents a conundrum because accurate histopathological classification of this tumor is challenging, even for specialty gynecologic pathologists, and some shared molecular features likely reflect a misclassification65. CCECs have been the subject of targeted gene sequencing and exome sequencing studies19,21,63,64,66, but thus far have not been scrutinized using integrated multi-platform analyses akin to methods used by The Cancer Genome Atlas [G] (TCGA).
Molecular classification
The main molecular characteristics of EECs, SECs, and UCSs have been revealed through targeted molecular studies, whole exome sequencing, and TCGA’s integrated genomic analyses (Table 1). While UCSs were characterized by TCGA independently16, TCGA assimilated EECs and SECs into four distinct molecular subgroups with prognostic significance15: The first subgroup is formed by POLE-mutated (ultramutated) tumors, which were characterized by POLE exonuclease domain (ED) mutations, predominantly recurrent POLEP286R or POLEV411L, and an excess of G:C>T:A transversions. Women in this subgroup exhibited the best progression-free survival [G] (PFS). The second subgroup is formed by hypermutated tumors, which were characterized by microsatellite instability [G] (MSI) and hypermethylation of the MLH1 promoter. Forming the third subgroup, copy number low/microsatellite stable (MSS) tumors were characterized by low copy number aberrations, MSS, and exhibited frequent CTNNB1 (β-catenin) mutation. Tumours in the fourth subgroup were copy number high (serous-like) tumors and were characterized by frequent TP53 mutation and high-level somatic copy number alterations. Women in this subgroup exhibited the worst PFS15.
EECs were distributed among all four subgroups, whereas SECs were almost exclusively in the serous-like subgroup (FIG. 1D). Paradoxically, the POLE-mutated subgroup exhibited the best PFS but was enriched for high-grade EECs15. Several subsequent studies confirmed significant associations between POLE-ED mutations and favorable clinical outcomes for high-grade EEC15,67–71. POLE-mutated ECs have high neoantigen [G] loads and markers of enhanced immune response72,73. Whether the favorable prognosis associated with POLE-mutated EC is attributed to these characteristics, aggressive therapeutic regimens administered to high-grade patients, or increased sensitivity to chemotherapy remains unclear72–77. However, recent studies indicate that the favorable prognosis for POLE-mutant ECs is not likely due to differential treatment response or tumor immunogenic phenotype78,79.
Early detection of endometrial cancer
Early detection of EC can increase the likelihood of women achieving disease-free survival [G]. The most common symptom of EC is post-menopausal vaginal bleeding (PMB). However, although 90% of women with EC (irrespective of tumor stage) exhibit PMB, this symptom is not a specific indicator of the disease: only 9% of women with PMB are diagnosed with EC80. Similarly, methods used to screen for EC, most commonly cytology and transvaginal ultrasound, lack specificity22. Thus, accurate screening tests that detect EC in women with early stages of the disease are needed.
In 2013 it was reported that EC-associated mutations could be detected in DNA extracted from specimens collected during routine Papanicolaou (Pap) tests [G]22. This led to the development of a prototype test (the “PapGene” test) that sequenced frequently mutated regions of 12 genes, with the potential to be incorporated into routine medical exams at a cost equivalent to HPV analysis22. In 2018, this test was advanced closer to commercial availability under the name of the “PapSEEK” test, which detects mutations in targeted regions of 18 genes as well as aneuploidy; a test is positive for cancer if a mutation or abnormal chromosome arm number is detected (Fig. 2)81. Of 382 women with EC, 81% tested positive with PapSEEK (78% with stage I/II tumors, 92% with stage III/IV tumors, and 85% with high-grade ECs confined to the endometrium). Furthermore, 93% of 123 EC patients sampled with Tao brushes [G] tested positive with PapSEEK (90% of stage I/II tumors, 98% of stage III/IV tumors, and 89% of high-grade tumors confined to the endometrium). The most commonly mutated genes detected by PapSeek in EC patient Pap and Tao brush samples [G] were PTEN, TP53, PIK3CA, and PIK3R1; the most commonly altered chromosomal arms in Pap samples were 4p, 7q, 8q, and 9q. Importantly, in 125 women without cancer, 0% of Tao brush and 1.4% of Pap samples, respectively, tested positive using PapSEEK, indicating increased specificity over alternate screening methods81. These results confirm earlier reports of sensitivity and specificity ranges for EC detection in Tao brush samples of 95–100% and 66–100%, respectively82–85. The PapSEEK test now needs to be evaluated in prospective studies.
Figure 2. Minimally invasive sampling methods for endometrial cancer (EC) patients. (a.) Tao brush sampling with PapSEEK test:
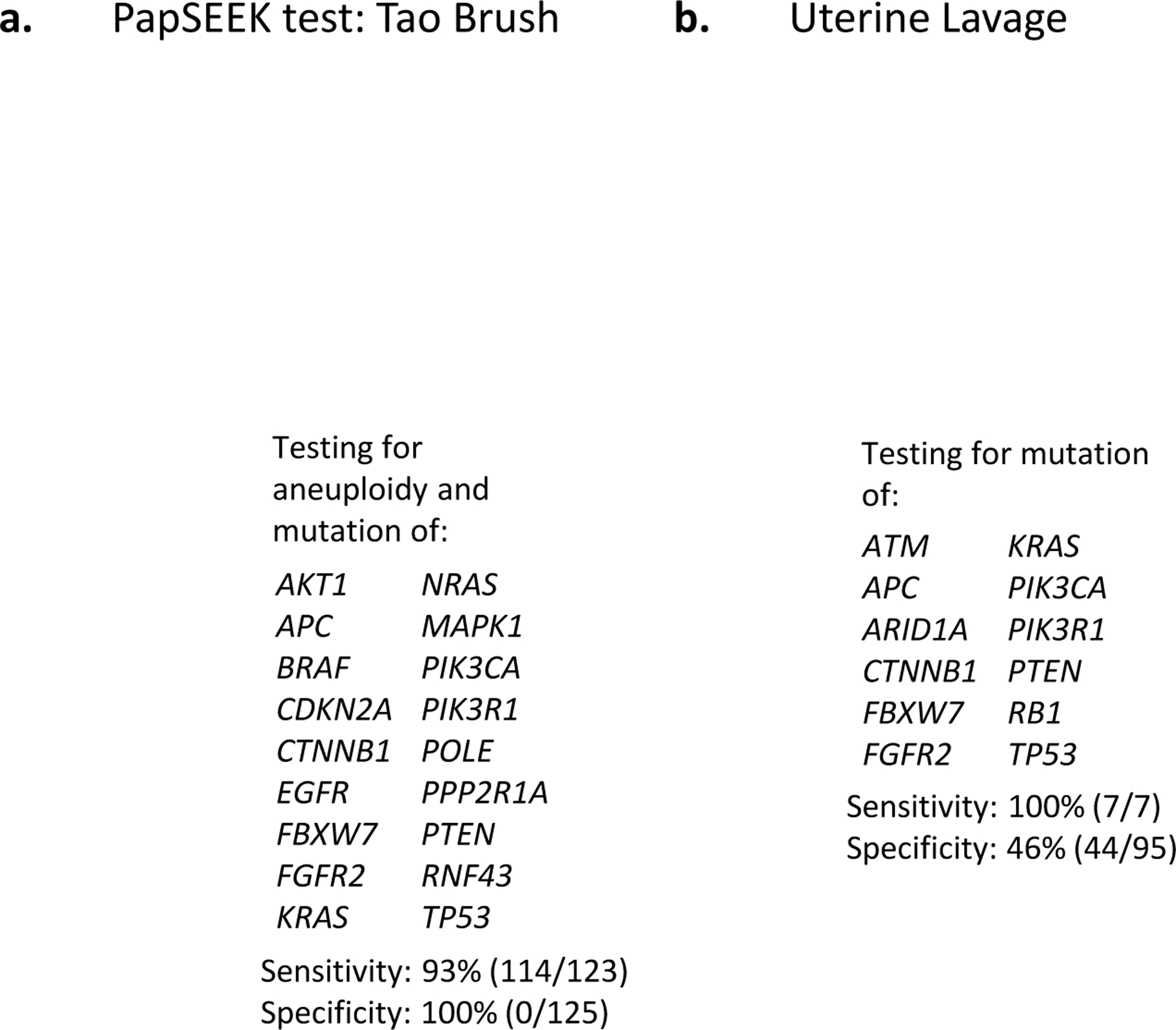
women testing positive for aneuploidy in any of ~38,000 loci of long interspersed nucleotide elements or mutation in any of 18 genes would be sent for confirmatory testing81. PapSEEK testing of Tao brush samples accurately detected EC in 93% of women tested with EC; out of 125 women without EC, none tested positive81. (b.) Uterine lavage samples analyzed on a 12 gene next generation sequencing panel detected cancer in 7 women with EC; mutations in the 12 genes were also detected in 51 of 95 women with a non-cancerous uterus86. Two images in this Figure were deleted to comply with Nature Reviews Cancer policy on self-archiving.
The sensitivity and specificity of PapSEEK testing of Tao brush samples are currently the best reported values, but other promising methods for early detection of EC are also in development. The PapSEEK test has shown increased specificity over the use of next generation sequencing [G] (NGS) on uterine lavage [G] samples86,87 (FIG.2), but it is possible that a “false positive” may reflect the detection of somatic driver aberrations in non-cancerous endometrium, a phenomenon reported by several groups86,88–90, or actually may be accurate early identification of EC. For example, PTEN mutations were detected in the uterine lavage of an asymptomatic woman with no clinical evidence of cancer 10 months prior to the identification of a single microscopic focus of EC91. An endometrial tumor <1mm in diameter contained within a polyp was also identified in the lavage fluid of another woman86, highlighting the sensitivity of genomic analysis of uterine lavage fluid for early detection of EC. Another potentially promising uterine sampling method is vaginal tampon92,93. Increased methylation of 11 genes has been detected in DNA extracted from vaginal tampons and may be particularly useful to identify stage I EECs, although it is unclear what methylation levels would be used to distinguish ECs from non-cancerous endometrium and whether increased methylation would be detectable in women pre-biopsy92,93. Furthermore, it is unclear if other aberrations are detectable from vaginal tampon samples. Despite the needed fine-tuning of this method, the potential use of tampons as screening tools for high risk women is particularly enticing because it could enable samples to be mailed in for testing similar to currently available Cologuard® [G] tests for colorectal cancer screening. Collectively, recent advancements indicate that the addition of genomic analyses to minimally invasive uterine sampling is a move in the right direction toward early detection of women with EC.
Molecular tests for risk stratification
While early detection of EC is ideal, the current reality is that the most clinically aggressive subtypes of EC are commonly diagnosed at advanced stages and identification of women at risk of developing aggressive disease is arguably a more pressing challenge than early detection. These facts have driven the quest to identify actionable genomic aberrations in ECs, which may ultimately be revolutionary for risk stratification and treatment of women with EC. The prognostic significance of TCGA-based molecular subgroups represented a paradigm-shifting development towards the use of molecular information to refine EC risk stratification. Independent groups have confirmed the prognostic significance of the TCGA-based subclasses66,94,95, and have proposed classification systems that are more feasible for routine clinical use than TCGA’s comprehensive molecular analysis (FIG. 3). Although an extensive expanse of literature describes molecular biomarkers for EC risk stratification, here we focus on the two most current pragmatic molecular classification schemes: the Translational Research in Post-Operative Radiation Therapy in EC (TransPORTEC) molecular classification system and the Proactive Molecular Risk Classifier for EC (ProMisE) (FIG. 3).
Figure 3. Molecular-based risk/treatment stratification strategies for endometrial cancer (EC) patients.
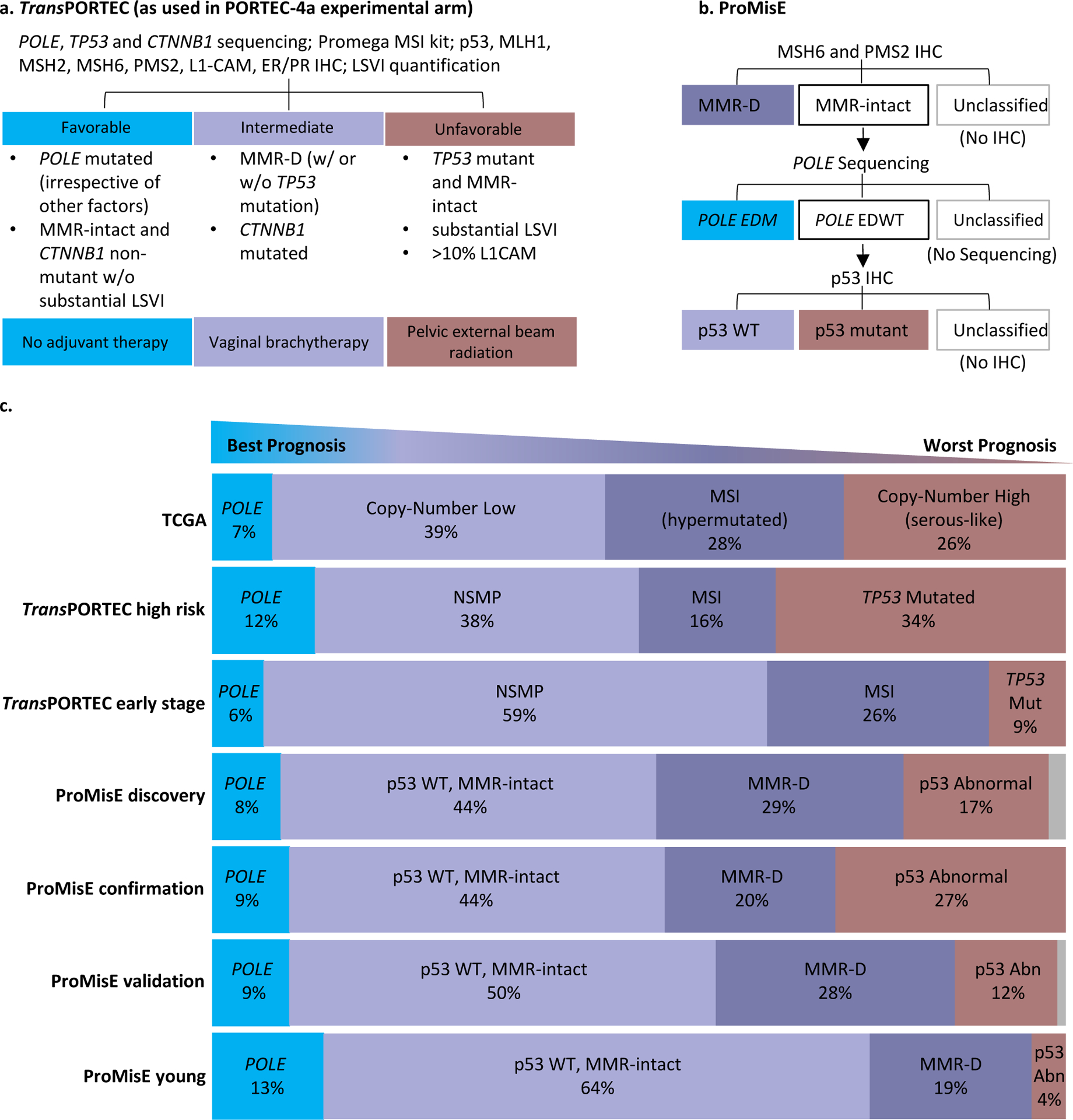
(a.) The TransPORTEC molecular classification system currently being tested in the PORTEC-4a clinical trial97 and (b.) the Proactive Molecular Risk Classifier for EC (ProMisE)100. (c.) Distribution of endometrial cancer patients in molecular subgroups. Each cohort consisted of the following EC patients: TCGA (n=232: 186 EEC, 42 SEC, 4 Mixed ECs)15, TransPORTEC high risk (n=116: 86 high risk EEC, 12 SEC, 18 CCEC)66, TransPORTEC early stage (n=834 early stage EEC)98, ProMisE discovery (n=143: 119 EEC, 15 SEC, 8 mixed, 1 undif; 64 ESMO high risk)94, ProMisE confirmation (n=319; 215 EEC, 5 CCEC, 89 SEC, 10 other; 173 ESMO high risk)99, ProMisE validation (n=452; 397 EEC, 34 SEC, 21 CCEC/mixed; 131 ESMO high risk)100, ProMisE young (n= 257 <50 yo: 225 EEC, 17 NEEC, 15 unknown; 21 ESMO high risk)101.
Abbreviations: Clear Cell Endometrial Cancer (CCEC); Catenin Beta 1 (CTNNB1); Exonuclease Domain Mutation (EDM); Exonuclease Domain Wildtype (EDWT); Endometrioid Endometrial Cancer (EEC); European Society of Medical Oncology (ESMO); Immunohistochemistry (IHC); lymphovascular space invasion [G] (LVSI); MicroSatellite Instable (MSI); Mismatch Repair-Deficient (MMR-D); No Specific Molecular Profile (NSMP); polymerase-ε mutated (POLE); Proactive Molecular Risk Classifier for Endometrial Cancer (ProMisE); Serous Endometrial Cancer (SEC); The Cancer Genome Atlas (TCGA); Translational Research in Post-Operative Radiation Therapy in Endometrial Carcinoma (TransPORTEC); tumor protein 53 (p53); Undifferentiated (undif); wild-type (WT); year-old (yo).
The original TransPORTEC model stratified high-risk EC patients into four subgroups, which are defined as p53 mutant, MSI, POLE-mutant, or No Specific Molecular Profile (NSMP)66. Patients were not classified if molecular testing was only partially performed or if they harbored more than one abnormality. When TransPORTEC was used to classify 116 EC patients deemed high-risk based on clinicopathological features, only patients in the p53 mutant (n=39) and NSMP (n=44) groups were shown to be truly high-risk; they exhibited significantly higher rates of distant metastases and lower 5-year RFS compared to those in the POLE-mutant (n=14) and MSI (n=19) groups (who had favorable prognoses)66 (FIG. 3C). The TransPORTEC molecular classification system was subsequently revised to integrate clinicopathological factors96, and is now being prospectively tested as a means to stratify women with high-intermediate risk EEC for radiotherapy in the phase III PORTEC-4a trial97 (FIG. 3A). While the results of PORTEC-4a are undoubtedly highly anticipated, it is also of great interest to determine whether TransPORTEC could be useful for prospective stratification of EC patients for treatments other than radiotherapy. In the meantime, the TransPORTEC model continues to evolve; the most recent version was refined to incorporate markers of DNA damage repair98, but is not yet being tested clinically.
ProMisE classifies EC patients based on testing of specimens for aberrations in the order of MMR-D, POLE mutation, and p53 mutation99 (FIG. 3B). Patients are not classified if they advance to a step for which they are unable to be tested, and those that harbor more than one aberration are classified based on the first positive test. ProMisE was shown to retrospectively enhance prediction of outcome in first a discovery cohort of 143 EC patients94, then on a confirmation cohort of 319 EC patients when combined with the European Society of Medical Oncology (ESMO) risk-stratification system99. In a validation cohort of 452 ECs, ProMisE was a significant prognostic marker of progression and disease-specific survival, even after adjustment for known risk factors100 (FIG. 3C). Enhanced retrospective prognostic ability of ProMisE was observed when ESMO risk stratification or clinicopathological parameters were added. Most recently, ProMisE was retrospectively significantly correlated with overall and disease-specific survival in a cohort of 257 young (<50 yo) EC patients101. Compared to the other non-age-stratified cohorts tested, this younger cohort was distributed more in p53 WT and POLE mutated subgroups, and less in p53 abnormal and MMR-D (FIG. 3C)101.
The retrospective data for TransPORTEC and ProMisE indicates that either has potential to be implemented as standard practice for risk stratification of EC patients, but neither currently has clinically proven prospective utility. Although both classification schemes utilize similar core molecular features (FIG. 3), differences can be considered. For example, while TransPORTEC completes all molecular testing prior to patient stratification, ProMisE follows sequential molecular testing and stratification. TransPORTEC is already being tested prospectively in the PORTEC-4a trial (described above), but ProMisE was developed, confirmed, and validated following the US Institute of Medicine guidelines and is now ready to be tested in prospective trials100. ProMisE was validated to be performed on diagnostic biopsies; TransPORTEC was also shown to produce concordant results between diagnostic and hysterectomy specimens102, but is being prospectively tested on hysterectomy samples. The TransPORTEC model being tested in PORTEC-4a incorporates molecular testing beyond that used in ProMisE (TP53 and CTNNB1 sequencing, LSVI quantification, and MLH1, MSH2, and L1-CAM immunohistochemistry) (FIG. 3a) although key clinicopathological parameters available at diagnosis are being evaluated for use with ProMisE103. The inclusion of CTNNB1 sequencing reflects findings that CTNNB1 exon 3 mutations have emerged as a prognostic marker for increased risk of disease recurrence among patients with low-grade and early-stage EEC104,105. However, substantive intratumor heterogeneity for CTNNB1 mutations observed in the molecular evolution of low-grade EECs from precursor lesions has been noted, prompting caution on the choice of clinical tissue sampling approaches for this marker34.
Implementation of either TransPORTEC or ProMisE would involve surmounting several challenges that include, but are not limited to, the cost and technical training required to perform and interpret genomic sequencing (with POLE being particularly challenging103), the development of methods for translation of genomic data to patients (most likely with the aid of genetic counselors), as well as risk of patient attrition while awaiting molecular profiling; pilot results of the PORTEC-4a trial indicated an average time of 10.2 days between randomization and molecular profile determination106. Furthermore, neither TransPORTEC or ProMisE currently incorporates histology, stage, or grade. Along these lines, it is important to note that these pragmatic models were originally designed to recapitulate TCGA’s prognostic subgroups of EECs and SECs; therefore, it remains to be determined how prognostic these classifications are for other EC histological subtypes, including mixed and undifferentiated ECs. For now, the current recommendation for reporting of genomic classifiers are to include histology, stage, and grade107.
Matching patients to therapies
The practice of guiding cancer therapy based on molecular aberrations is gaining momentum across the field of oncology and has shown potential to improve patient outcomes, despite logistical hurdles. In addition to those mentioned in the preceding paragraph, common challenges of matching patients to targeted therapies are lack of availability of therapies or clinical trials, impaired geographic accessibility of trials, and lack of insurance coverage108–113. Cost increase, another potential challenge associated with matched targeted therapies, are mainly being attributed to increased treatment duration113. Further complicating the interpretation and translation of genomic results is the fact that therapies targeting identical aberrations in different tumor types have shown differing efficacy; likewise, different aberrations within the same gene have been shown to produce distinctive functional consequences. The contextual importance of mutations in preclinical design and interpretation is highlighted by the variability in synthetic lethal effects observed between EC cell lines with gain-of-function and loss-of-function TP53 mutations114–117. Despite the challenges associated with molecularly guiding therapies, three quarters of 1,281 US physician survey respondents in 2017 reported using NGS tests to guide treatment decisions118, and treatment of solid tumors based on matching actionable mutations to targeted therapies has resulted in improved outcomes of patients with advanced cancers109,110,119–123.
Counter to this optimistic data, clinical trials targeting aberrations in the PI3K pathway or ERBB2 (also known as HER2), which are some of the most common clinically actionable aberrations in advanced ECs112,123, have yielded modest results124. Lack of response could be due in part to initial or acquired drug resistance; in this respect it is noteworthy that a majority (70%; 14/20) of ERBB2 amplified ECs also harbor PI3K pathway aberrations15, raising the potential for combination therapies. Along these lines, whereas preclinical studies have produced variable reports of sensitivity to PARP inhibitors in PTEN-deficient EC125–127, treatment of a mouse model of EC with combined PARP and PI3K inhibitors has resulted in synergistic effects128. Relatedly, in an inducible PTEN knockout mouse model, a CDK4/6 inhibitor exhibited antitumor activity129.
Reliable biomarkers of response to therapies targeting the PI3K pathway or ERBB2 also need to be identified for EC patients. Biomarker identification could potentially be improved if trials are designed to accrue patients and report results by molecular and/or histological subtypes. For example, a recent study that accrued only SEC patients who overexpressed ERBB2, reported decreased risk of progression and increased PFS for patients treated with carboplatin-paclitaxel-trastuzumab [G] compared to those treated with carboplatin-paclitaxel130. Independent of this study, effectiveness of trastuzumab in ERBB2 amplified SEC patients has been reported: one heavily pretreated woman achieved a durable complete response to trastuzumab112, while another woman with recurrent disease achieved a complete response following the addition of trastuzumab to her carboplatin-paclitaxel regimen131. These results are evidence that trials designed to incorporate histological and/or molecular stratification could help identify EC patient populations most likely to respond to targeted therapies.
In this regard, it is noteworthy that in a recent prospective analysis, 47% (16/34) of EC patients who matched to a therapy after NGS panel tumor profiling experienced clinical benefit132, including 40% (2/5) of MSI-H patients treated with immune checkpoint inhibitors and 42% (8/19) of patients matched based on PIK3CA and PTEN mutations112. Of 189 EC patients within this study (75% of which had grade 3 EEC, SEC, CCEC or UCS), 67% had at least one alteration for which a therapy was either FDA-approved or under clinical investigation. The most common clinically actionable aberrations among the entire cohort were PIK3CA or PTEN mutation, MSI, and ERBB2 amplification112. Importantly, of 4 patients with matched primary and metastatic samples within this study, the mutational profiles differed between primary and metastatic sites; in 2 cases, metastases acquired potentially actionable mutations in MTOR and PIK3R1114. Indeed, discordance in mutations, and changes in the dominant mutational signature, between matched primary and metastatic ECs have been reported133,134,135. EC metastases gain aberrations in genes in multiple functional groupings including the PI3K pathway, WNT signaling, RAS-RAF pathways, transcriptional regulation, DNA damage response, and FBXW7-related genes (FIG. 4). Additionally, recurrent or metastatic EECs exhibit ~7% higher frequencies of MSI and/or MMR-D compared to matched primary tumors136,137. Discordance between aberrations found between primary tumors and metastases may reflect lack of representation of tumor heterogeneity in primary tumor biopsies, or the acquisition of novel aberrations, potentially due to a change in microenvironment encountered by metastatic lesions. Regardless of the biological explanation for this descrepancy, comprehensive molecular profiling of metastatic lesions may be key for treatment stratification of EC patients. Encouragingly, a recent mutational analysis of metastases from 20 untreated cancer patients (including 4 ECs lacking POLE mutation) indicated that all metastases within a patient share functional driver mutations138. If this holds true for other aberrations and for patients in relapse, it would decrease the need to evaluate multiple metastases from a single patient.
Figure 4. Functional grouping of genes in which aberrations are acquired in metastases of endometrial cancer (EC).
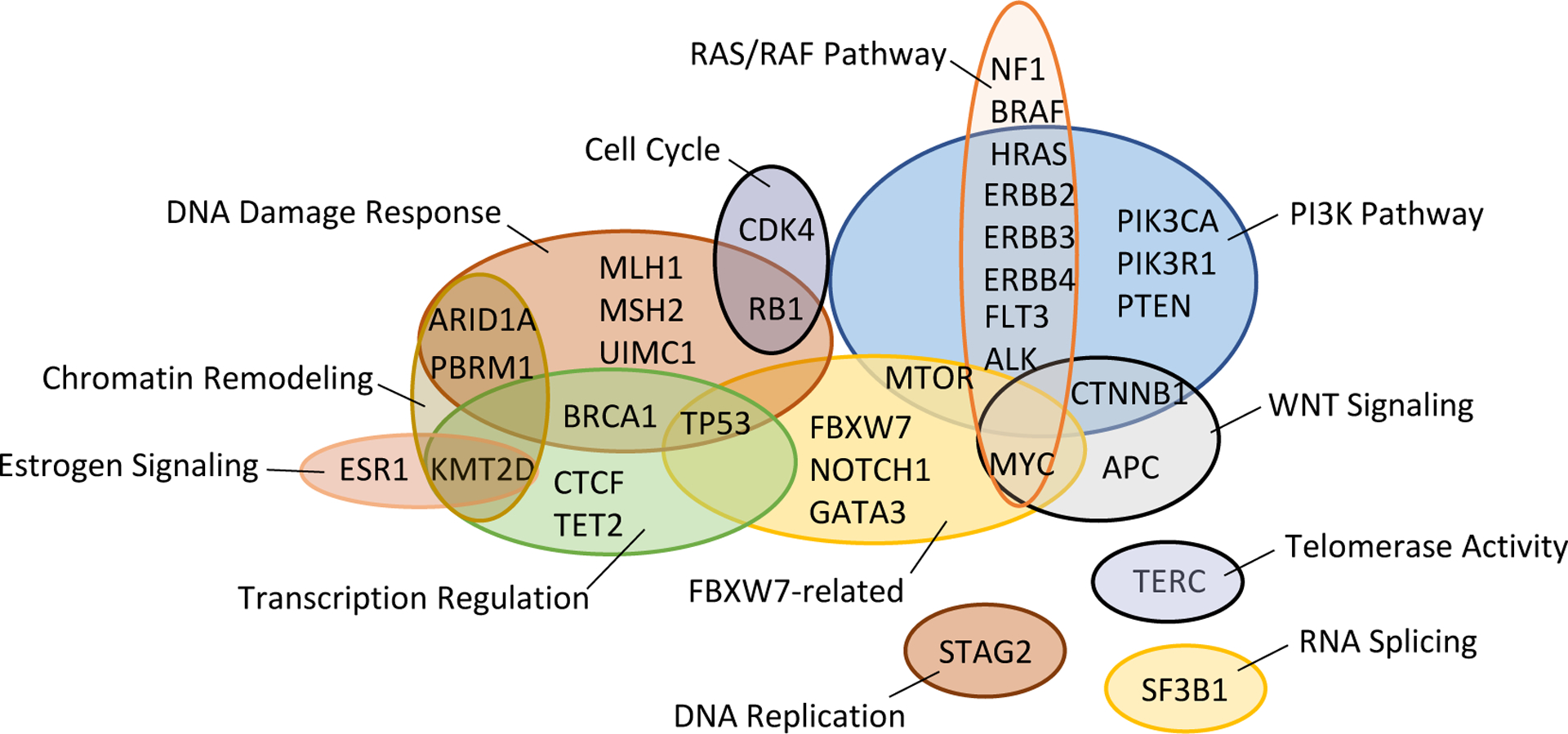
Targeted treatment of additional EC cohorts is needed to help assess the utility of matched therapies in this patient population. Treatment arms of the National Cancer Institute’s Molecular Analysis for Therapy of Choice (MATCH) trial comprise one such cohort139. Another is patients enrolled in the American Society of Clinical Oncology’s (ASCO’s) Targeted Agent and Profiling Utilization Registry (TAPUR) trial140. Importantly, and relevant to the next section of this review, EC patients that are MMR-D that enroll in the MATCH trial and those in the TAPUR trial that harbor POLE/POLD1 mutations, have a high mutational load, or are MSI-H have the potential to match to immunotherapies.
Immunotherapy for endometrial cancer
As of November 2018, clinicaltrials.gov141, which encompasses the US and 20 other countries worldwide, listed over 50 clinical trials testing various forms of immunotherapy for which advanced EC patients were potentially eligible (Supplementary Table 1). Immune therapies have shown particular efficacy in solid tumors that are MSI-H, MMR-D and/or those with high concentrations of Tumor Infiltrating Lymphocytes [G] (TILs). In fact, the programmed cell death 1 (PD1) antibody pembrolizumab [G], received US food and drug administration (FDA) approval in 2017 for treatment of MSI-H or MMR-D cancers, regardless of tumor type. This landmark approval could be particularly beneficial for EC patients, given that 16–17% are MMR-D as detected by NGS112,142, ~34% of EECs have MSI15,143–145, 48–100% express PD1 ligand 1 (PDL1) or PDL2146,147, and equivalently high numbers of TILs are found in subsets of all molecular subgroups of EC79.
Pembrolizumab has been remarkably effective in small numbers of EC patients (Table 2), but definitive biomarkers of response remain elusive, a need several clinical trials are currently attempting to address (Supplementary Table 1). Treatment with pembrolizumab achieved a noteworthy 53% (8/15) overall response rate (ORR) in MMR-D EC patients142, and 43% (3/7) ORR when combined with the indoleamine 2,3-dioxygenase (IDO1) inhibitor epacadostat in ECs with unreported biomarker status148. Two separate cohorts of advanced EC patients each responded with ORRs of 13% to single-agent pembrolizumab and the PD-L1 antibody atezolizumab [G]. The combined results of these cohorts may indicate that hypermutation or high TILs combined with PD-L1 positivity may predict response to PD-1 blockade: of patients responding to atezolizumab, one exhibited 70% TIL, while the other was hypermutated and a patient that exhibited a prolonged (>14 month) partial response to pembrolizumab harbored POLE mutations; all three patients were PD-L1 positive (Table 2)149,150.
Table 2.
Immunotherapy clinical trial results for endometrial cancer (EC) patients
| Immunotherapy | Patient Population | ORR | Responding Biomarker Status (response) | Refs |
|---|---|---|---|---|
| Anti-PD1 (Pembrolizumab) | MMR status: 100% (15/15) MMR-D |
53% (8/15) | All MMR-D; other biomarker status not reported 3 (CR) 5 (PR) 3 (SD) |
142,161 |
| Anti-PD1 (Pembrolizumab) plus IDO1 inhibitor (epacadostat) | No information for EC patients | 43% (3/7) | Biomarker status not reported 1 (CR) 2 (PR) |
148,162 |
| Anti-PD1 (Pembrolizumab) | MSI status: 5% (1/19) MSI-H 95% (18/19) MSS Histology: 74% (17/23) EEC 9% (2/23) SEC 4% (1/23) UCS 13% (3/23) other PDL1 status: 100% (23/23) + |
13% (3/23) | 1 PD-L1+, POLE muts (PR >14months) 1 PD-L1+, MSS (PR) 1 PD-L1+ (PR) |
146,149,163 |
| Anti-PDL1 (Atezolizumab) | MSI status: 6% (1/15) MSI-H 47% (7/15) MSS 47% (7/15) unknown histology: 33% (5/15) EEC 33% (5/15) SEC 7% (1/15) Leiomyosarcoma 27% (4/15) Unknown PDL1 status: 33% (5/15) + 67% (10/15) - |
13% (2/15) | 1 PD-L1+, MSS, 70% TIL (PR) 1 PD-L1+, MSI, 10% TIL, hypermutated (PR) |
150,164 |
IDO1, Indoleamine 2, 3-dioxygenase 1; MMR= Mismatch Repair; MSI= Microsatellite Instability; EEC= endometrioid EC; SEC= serous EC; UCS= uterine carcinosarcoma; PD1, programmed cell death 1; PDL1= PD1 Ligand 1; ORR=overall response rate; CR= complete response; PR= partial response; SD= stable disease.
Information revealed in recent studies considering ECs along with other cancer types, so-called “pan-cancer studies”, has supported a potential importance of immune response in ECs and may also aid in identification of novel treatment strategies151. For example, unsupervised clustering of TCGA’s Pan-Gyn cohort [G] based on 16 molecular features revealed that 16.5% of SEC-like EECs, SECs, and UCSs group within a cluster characterized by high leucocyte infiltration, which supports immunotherapy as a potential treatment option. It was further speculated that tumors within two other clusters encompassing 32.5% and 36.9% of SEC-like EECs, SECs, and UCSs might respond to HER2 targeted therapy (discussed above as a potential promising treatment strategy for ERBB2 amplified SECs) or therapies targeting the DNA damage response151. A second pan-cancer clustering based on 5 immune suppression gene signatures revealed that the vast majority of CN-high ECs, SEC-like ECs, and UCSs populated “wound healing” and “IFNγ dominant” clusters, raising the possibility that molecular targets involved in the physiological response to wounds or IFNγ signaling could be therapeutically relevant for clinically aggressive ECs152. Finally, a large-scale functional genomics screen of pan-cancer cell lines revealed that WRN (Werner syndrome RecQ like helicase) is a synthetic lethal target in MSI (but not POLE mutated153) ECs153–156. These discoveries open up promising new avenues for future preclinical exploration of rational drug development for ECs.
Concluding remarks
The current molecular portraits of the most common ECs, and of rare but clinically aggressive forms of the disease, have revealed shared and distinguishing features, as well as prognostically distinct subgroups. This knowledge has inspired ongoing efforts to develop diagnostic tests to facilitate the early detection of EC and clinically feasible molecular classifiers that may be used for disease risk stratification, to prevent both under- and over-treatment of women with EC. Future challenges in the field include: overcoming difficulties associated with incorporation of molecular subtyping in the clinic; more extensive genomic characterization of CCECs and of EC metastases; functional characterization of mutations in novel driver genes; proteomic studies to provide a global view of the net impact of genomic, transcriptomic and translational perturbations in ECs; and high throughput screens for druggable targets and synthetic lethal interactions in this disease. It is hoped that these efforts, together with strategies to reduce obesity, will ultimately reduce the impact of EC on women’s health.
Supplementary Material
Acknowledgements
We apologize to those authors whose work we could not cite due to space limitations. This work was supported by the Intramural Research Program of the National Human Genome Research Institute at NIH (HG200338 and HG200379) to DWB.
Glossary
- atezolizumab
Humanized, monoclonal antibody of programmed cell-death ligand 1.
- atrophic endometrium
Thin layer of nonpoliferative epithelial cells lining the uterus; characteristic of postmenopausal women
- carboplatin
Chemotherapy drug that inhibits cell growth and/or causes apoptosis by inducing DNA-DNA and DNA-protein crosslinks.
- Carcinoma
Cancer caused by uncontrolled proliferation of epithelial cells.
- Carcinosarcoma
Tumor comprised of both carcinoma and sarcoma.
- clear cell endometrial cancer
(CCEC) rare histopathological subtype of endometrial cancer that typically arises from atrophic endometrium and includes large clear eosinophilic cells (basic cells that stain with the acidic dye eosin).
- Cologuard® test
Colorectal cancer screening test which enables patients to collect stool samples in-home; samples are mailed to a lab where they are analyzed for presence of blood and DNA abnormalities.
- complex atypical hyperplasia of the endometrium
(CAH) precancerous changes in the epithelial cells lining the uterus characterized by abnormal growth and acquisition of somatic genomic aberrations.
- driver genes
Pathogenic aberrations of these genes contribute to the initiation and/or progression of cancer.
- disease-free survival
Length of time a patient lives without signs of disease.
- endometrioid endometrial cancer
(EEC) most common histopathologic subtype of endometrial cancer.
- epithelial-to-mesenchymal transition
(EMT) process by which epithelial cells acquire characteristics of mesenchymal cells, including but not limited to, decreased cell-to-cell adhesion, decreased polarity and increased motility.
- Hysterectomy
Surgical removal of the uterus.
- lymphovascular space invasion
(LSVI) spreading of cancer to the lymphatic system or blood vessels.
- microsatellite instability
(MSI) alteration of the number of short, repeated sequences of DNA because of a defect in DNA mismatch repair.
- mismatch repair
(MMR) type of DNA repair that corrects base-base mismatches and insertions/deletions.
- Neoantigen
Antigens not previously recognized by the immune system.
- next generation sequencing
(NGS) high-throughput technologies (also known as massively parallel or deep sequencing) that enable faster determination of DNA or RNA base pair codes than previously-used technologies (e.g. Sanger Sequencing).
- ovarian insufficiency
Loss of normal ovarian function.
- Paclitaxel
Chemotherapy drug that binds tubulin and inhibits cell division; also induces apoptosis through binding and inhibition of Bcl-2 (B-cell leukemia 2).
- Papanicolaou (Pap) test
Routine screening tool in which cervical cells are collected using a small brush and analyzed microscopically for signs of disease (e.g. irregular cell morphology).
- pap brush
Flexible brush used to sample the inside of the cervix.
- Pembrolizumab
Humanized, monoclonal antibody of programmed cell-death ligand 1.
- progression-free survival
(PFS) length of time a patient lives without objective worsening of disease.
- Sarcoma
Cancer caused by uncontrolled proliferation of connective tissue.
- serous endometrial cancer
(SEC) rare histopathologic subtype of endometrial cancer that typically arises in atrophic endometrium, usually with well-formed papillae.
- serous endometrial intraepithelial carcinoma
(SEIC) noninvasive malignant precursor to SEC (serous endometrial cancer).
- somatic aberration
Genomic change that occurs spontaneously; is not present in germline.
- Synthetic lethality
Occurs when multiple genomic aberrations combine to cause cell death, while the independent aberrations do not.
- Tao brush
Flexible brush used to sample the inside of the uterus.
- The Cancer Genome Atlas
(TCGA) NIH (National Institutes of Health)-funded initative that molecularly characterized over 20,000 primary cancer and matched normal samples covering 33 cancer types.
- The Cancer Genome Atlas’ Pan-Gyn cohort
1,087 invasive breast carcinomas, 308 endocervical adenocarcinomas, 579 high-grade serous ovarian cystadenocarcinomas, 548 uterine corpus endometrial carcinomas, and 57 uterine carcinosarcomas molecularly characterized by TCGA.
- Trastuzumab
Recombinant human monoclonal HER2 (human epidermal growth factor receptor 2) antibody.
- tumor infiltrating lymphocytes
White blood cells (immune cells) found within tumor tissue.
- tumor suppressor gene
Gene which normally functions to prevent uncontrolled growth of cells.
- uterine lavage
Process by which the uterus is flushed with a sterile solution.
Footnotes
Competing interests
Dr. Bell receives royalty income from US patent No.7,294,468 “Method to determine responsiveness of cancer to epidermal growth factor receptor targeting treatments”, which is licensed to Esoterix Genetic Labs LLC. Dr. Urick has no competing interests.
References
- 1.American Cancer Society. Cancer facts & figures 2019. Atlanta:American Cancer Society; (2019). [Google Scholar]
- 2.Amant F, Mirza MR, Koskas M & Creutzberg CL Cancer of the corpus uteri. Int. J. Gynaecol. Obstets 143 Suppl 2, 37–50 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Gruber SB & Thompson WD A population-based study of endometrial cancer and familial risk in younger women. Cancer and steroid hormone study group. Cancer Epidemiol. Biomarkers Prev 5, 411–417 (1996). [PubMed] [Google Scholar]
- 4.Lynch HT, Snyder CL, Shaw TG, Heinen CD & Hitchins MP Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 15, 181–194 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Palles C, Latchford A & Valle L Adenomatous Polyposis Syndromes: Polymerase Proofreading-Associated Polyposis In: Valle L, Gruber S, Capellá G (eds) Hereditary Colorectal Cancer. Springer, Cham; (2018). [Google Scholar]
- 6.Ngeow J, Stanuch K, Mester JL, Barnholtz-Sloan JS & Eng C Second malignant neoplasms in patients with Cowden syndrome with underlying germline PTEN mutations. J. Clin. Oncol 32, 1818–1824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaber C, Meza R, Ruterbusch JJ & Cote ML Endometrial cancer trends by race and histology in the USA: Projecting the number of new cases from 2015 to 2040. J Racial Ethn. Health Disparitie (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Setiawan VW et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol 31, 2607–2618 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinton LA et al. Etiologic heterogeneity in endometrial cancer: evidence from a Gynecologic Oncology Group trial. Gynecol. Oncol 129, 277–284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lortet-Tieulent J, Ferlay J, Bray F & Jemal A International patterns and trends in endometrial cancer incidence, 1978–2013. J. Natl. Cancer Inst 110, 354–361 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Clarke MA, Devesa SS, Harvey SV & Wentzensen N Hysterectomy-corrected uterine corpus cancer incidence trends and differences in relative survival reveal racial disparities and rising rates of nonendometrioid cancers. J. Clin. Oncol, JCO1900151, doi: 10.1200/JCO.19.00151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faber MT, Frederiksen K, Jensen A, Aarslev PB & Kjaer SK Time trends in the incidence of hysterectomy-corrected overall, type 1 and type 2 endometrial cancer in Denmark 1978–2014. Gynecol. Oncol 146, 359–367 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Dedes KJ, Wetterskog D, Ashworth A, Kaye SB & Reis-Filho JS Emerging therapeutic targets in endometrial cancer. Nat. Rev. Clin. Oncol 8, 261–271 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Noone AM et al. SEER cancer statistics review, 1975–2015, National Cancer Institute. Bethesda, MD,https://seer.cancer.gov/csr/1975_2015/, based on November 2017 SEER data submission, posted to the SEER web site, April 2018. [Google Scholar]
- 15.Kandoth C et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Landmark TCGA study that reported the molecular genomic landscape of endometrioid and serous endometrial carcinomas, defining four distinct molecular subgroups.
- 16.Cherniack AD et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 31, 411–423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Landmark TCGA study that reported the molecular genomic landscape of 57 uterine carcinosarcomas.
- 17.Le Gallo M et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genetics 44, 1310–1315 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Gallo M et al. The FOXA2 transcription factor is frequently somatically mutated in uterine carcinosarcomas and carcinomas. Cancer 124, 65–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Gallo M et al. Somatic mutation profiles of clear cell endometrial tumors revealed by whole exome and targeted gene sequencing. Cancer 123, 3261–3268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones S et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat. Comm 5, 5006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLair DF et al. The genetic landscape of endometrial clear cell carcinomas. J. Pathol 243, 230–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinde I et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci. Transl. Med 5, 167ra164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; First study to show that endometrial cancer- associated mutations could be detected during routine Pap tests; led to the development of the prototype “PapGene” test (2013).
- 23.Zhao S & Santin AD Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial–mesenchymal transition. Proc. Natl. Acad. Sci. USA 113, 12238–12243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn E et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J. Nat. Cancer Inst 104, 1503–1513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McConechy MK et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol 228, 20–30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felix AS et al. Factors associated with Type I and Type II endometrial cancer. Cancer Causes & Control 21, 1851–1856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherman ME et al. Risk factors and hormone levels in patients with serous and endometrioid uterine carcinomas. Mod. Pathol 10, 963–968 (1997). [PubMed] [Google Scholar]
- 28.Yang HP et al. Endometrial cancer risk factors by 2 main histologic subtypes: the NIH-AARP Diet and Health Study. Am. J. Epidemiol 177, 142–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mutter GL, Monte NM, Neuberg D, Ferenczy A & Eng C Emergence, involution, and progression to carcinoma of mutant clones in normal endometrial tissues. Cancer Res 74, 2796–2802 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levine RL et al. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 58, 3254–3258 (1998). [PubMed] [Google Scholar]
- 31.Mutter GL et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J. Natl. Cancer Inst 92, 924–930 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Lin MC, Burkholder KA, Viswanathan AN, Neuberg D & Mutter GL Involution of latent endometrial precancers by hormonal and nonhormonal mechanisms. Cancer 115, 2111–2118 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russo M et al. Clonal evolution in paired endometrial intraepithelial neoplasia/atypical hyperplasia and endometrioid adenocarcinoma. Hum. Pathol 67, 69–77 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Lazo de la Vega L et al. Multiclonality and marked branched evolution of low-grade endometrioid endometrial carcinoma. Mol. Cancer Res 17, 731–740 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Mota A et al. Genetic analysis of uterine aspirates improves the diagnostic value and captures the intra-tumor heterogeneity of endometrial cancers. Mod. Pathol 30, 134–145 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Joshi A, Miller C Jr., Baker SJ & Ellenson LH Activated mutant p110alpha causes endometrial carcinoma in the setting of biallelic Pten deletion. Am. J. Pathol 185, 1104–1113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urick ME et al. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res.71, 4061–4067 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung LW et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 1, 170–185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oda K et al. PIK3CA cooperates with other phosphatidylinositol 3’-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 68, 8127–8136 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Oda K, Stokoe D, Taketani Y & McCormick F High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 65, 10669–10673 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Terakawa J et al. Ovarian insufficiency and CTNNB1 mutations drive malignant transformation of endometrial hyperplasia with altered PTEN/PI3K activities. Proc. Natl. Acad. Sci. U S A, doi: 10.1073/pnas.1814506116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H et al. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am. J. Pathol 160, 1481–1486 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byron SA et al. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS One 7, e30801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Khatri S, Broaddus R, Wang Z & Hawkins SM Deletion of Arid1a in reproductive tract mesenchymal cells reduces fertility in female mice. Biol. Reprod 94, 93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim TH et al. ARID1A Is essential for endometrial function during early pregnancy. PLoS Genet 11, e1005537 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ayhan A et al. Increased proliferation in atypical hyperplasia/endometrioid intraepithelial neoplasia of the endometrium with concurrent inactivation of ARID1A and PTEN tumour suppressors. J. Pathol. Clin. Res 1, 186–193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherman ME, Bur ME & Kurman RJ p53 in endometrial cancer and its putative precursors: evidence for diverse pathways of tumorigenesis. Hum. Pathol 26, 1268–1274 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Lax SF, Kendall B, Tashiro H, Slebos RJ & Hedrick L The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer 88, 814–824 (2000). [PubMed] [Google Scholar]
- 49.Wild PJ et al. p53 suppresses type II endometrial carcinomas in mice and governs endometrial tumour aggressiveness in humans. EMBO Mol. Med 4, 808–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daikoku T et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 68, 5619–5627 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhn E, Bahadirli-Talbott A & Shih Ie M Frequent CCNE1 amplification in endometrial intraepithelial carcinoma and uterine serous carcinoma. Mod. Pathol 27, 1014–1019 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Zhao S et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Nat. Acad. Sci. USA 110, 2916–2912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rudd ML et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin. Cancer Res 17, 1331–1340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haesen D et al. Recurrent PPP2R1A mutations in uterine cancer act through a dominant-negative mechanism to promote malignant cell growth. Cancer Res. 76, 5719–5731 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Urick ME & Bell DW In vitro effects of FBXW7 mutation in serous endometrial cancer: Increased levels of potentially druggable proteins and sensitivity to SI-2 and dinaciclib. Mol. Carcinog 57, 1445–1457 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McConechy MK et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J. Pathol. Clin. Res 1, 173–185 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiyoda T et al. Expression profiles of carcinosarcoma of the uterine corpus-are these similar to carcinoma or sarcoma? Genes Chromosom. Cancer 51, 229–239 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Jin Z et al. Carcinosarcomas (malignant mullerian mixed tumors) of the uterus and ovary: a genetic study with special reference to histogenesis. Int. J. Gynecol. Pathol 22, 368–373 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Abeln EC et al. Molecular genetic evidence for the conversion hypothesis of the origin of malignant mixed mullerian tumours. J. Pathol 183, 424–431 (1997). [DOI] [PubMed] [Google Scholar]
- 60.Wada H et al. Molecular evidence that most but not all carcinosarcomas of the uterus are combination tumors. Cancer Res. 57, 5379–5385 (1997). [PubMed] [Google Scholar]
- 61.Nieto MA, Huang RY, Jackson RA & Thiery JP EMT: 2016. Cell 166, 21–45 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Thiery JP, Acloque H, Huang RY & Nieto MA Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009). [DOI] [PubMed] [Google Scholar]
- 63.An HJ, Logani S, Isacson C & Ellenson LH Molecular characterization of uterine clear cell carcinoma. Mod. Pathol 17, 530–537 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Hoang LN et al. Targeted mutation analysis of endometrial clear cell carcinoma. Histopathology 66, 664–674 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Han G et al. Endometrial carcinomas with clear cells: A study of a heterogeneous group of tumors including interobserver variability, mutation analysis, and immunohistochemistry with HNF-1beta. Int. J. Gynecol. Pathol 34, 323–333 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Stelloo E et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol 28, 836–844 (2015). [DOI] [PubMed] [Google Scholar]
- 67.McConechy MK et al. Endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Clin. Cancer Res 22, 2865–2873 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Church DN et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J. Natl. Cancer Inst 107, 402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meng B et al. POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol. Oncol 134, 15–19 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Hussein YR et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Mod. Pathol 28, 505–514 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Billingsley CC et al. Polymerase varepsilon (POLE) mutations in endometrial cancer: clinical outcomes and implications for Lynch syndrome testing. Cancer 121, 386–394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Gool IC et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin. Cancer Res 21, 3347–33557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bellone S et al. Polymerase epsilon (POLE) ultra-mutated tumors induce robust tumor-specific CD4+ T cell responses in endometrial cancer patients. Gynecol. Oncol 138, 11–17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shukla SA, Howitt BE, Wu CJ & Konstantinopoulos PA Predicted neoantigen load in non-hypermutated endometrial cancers: Correlation with outcome and tumor-specific genomic alterations. Gynecol. Oncol. Rep 19, 42–45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Howitt BE et al. Association of Polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 1, 1319–1323 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Eggink FA et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology 6, e1264565 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bellone S et al. Polymerase epsilon (POLE) ultra-mutation in uterine tumors correlates with T lymphocyte infiltration and increased resistance to platinum-based chemotherapy in vitro. Gynecol. Oncol 144, 146–152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Gool IC et al. Adjuvant treatment for POLE proofreading domain-mutant cancers: sensitivity to radiotherapy, chemotherapy, and nucleoside analogues. Clin. Cancer Res 24, 3197–3203 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Talhouk A et al. Molecular subtype not immune response drives outcomes in endometrial carcinoma. Clin. Cancer Res 25, 2537–2548 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Clarke MA et al. Association of endometrial cancer risk with postmenopausal bleeding in women: a systematic review and meta-analysis. JAMA Intern. Med 178, 1210–1222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y et al. Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Sci. Transl. Med 10, eaap8793 (2018). Publication demonstrating detection of early stage endometrial cancers from samples collected during routine Pap tests using the “PapSEEK” test (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Del Priore G et al. Endometrial brush biopsy for the diagnosis of endometrial cancer. J Reprod. Med 46, 439–443 (2001). [PubMed] [Google Scholar]
- 83.Kipp BR et al. Direct uterine sampling with the Tao brush sampler using a liquid-based preparation method for the detection of endometrial cancer and atypical hyperplasia: a feasibility study. Cancer 114, 228–235 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Maksem J, Sager F & Bender R Endometrial collection and interpretation using the Tao brush and the CytoRich fixative system: a feasibility study. Diagn. Cytopathol 17, 339–346 (1997). [DOI] [PubMed] [Google Scholar]
- 85.Wu HH, Casto BD & Elsheikh TM Endometrial brush biopsy. An accurate outpatient method of detecting endometrial malignancy. J. Reprod. Med 48, 41–45 (2003). [PubMed] [Google Scholar]
- 86.Nair N et al. Genomic Analysis of uterine lavage fluid detects early endometrial cancers and reveals a prevalent landscape of driver mutations in women without histopathologic evidence of cancer: a prospective cross-sectional study. PLoS Med. 13, e1002206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maritschnegg E et al. Lavage of the uterine cavity for molecular detection of mullerian duct carcinomas: a proof-of-concept study. J. Clin. Oncol 33, 4293–4300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suda K et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Reports 24, 1777–1789 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Anglesio MS et al. Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med 376, 1835–1848 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo SW Cancer driver mutations in endometriosis: Variations on the major theme of fibrogenesis. Reprod. Med. Bio l 17, 369–397 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martignetti JA et al. Detection of endometrial precancer by a targeted gynecologic cancer liquid biopsy. Cold Spring Harb. Mol. Case Stud 4, pii: a003269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bakkum-Gamez JN et al. Detection of endometrial cancer via molecular analysis of DNA collected with vaginal tampons. Gynecol. Oncol 137, 14–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fiegl H et al. Methylated DNA collected by tampons--a new tool to detect endometrial cancer. Cancer Epidemiol. Biomarkers Prev 13, 882–888 (2004). [PubMed] [Google Scholar]
- 94.Talhouk A et al. A clinically applicable molecular-based classification for endometrial cancers. Br. J. Cancer 113, 299–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wortman BG et al. Ten-year results of the PORTEC-2 trial for high-intermediate risk endometrial carcinoma: improving patient selection for adjuvant therapy. Br. J. Cancer 119, 1067–1074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stelloo E et al. Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer-combined analysis of the PORTEC cohorts. Clin. Cancer Res 22, 4215–4224 (2016). [DOI] [PubMed] [Google Scholar]; Description of the TransPORTEC molecular classification system that is currently being tested in clinical trials of endometrial cancer patients.
- 97.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03469674. (2018).
- 98.Auguste A et al. Refinement of high-risk endometrial cancer classification using DNA damage response biomarkers: a TransPORTEC initiative. Mod. Pathol 31, 1851–1861 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Talhouk A et al. Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer. Cancer 123, 802–813 (2017). [DOI] [PubMed] [Google Scholar]
- 100.Kommoss S et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol 29, 1180–1188 (2018). Description of the “locked down” ProMisE molecular classifier that is now ready for clinical evaluation (2018). [DOI] [PubMed] [Google Scholar]
- 101.Britton H et al. Molecular classification defines outcomes and opportunities in young women with endometrial carcinoma. Gynecol. Oncol 153, 487–495 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Stelloo E et al. High concordance of molecular tumor alterations between pre-operative curettage and hysterectomy specimens in patients with endometrial carcinoma. Gynecol. Oncol 133, 197–204 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Talhouk A & McAlpine JN New classification of endometrial cancers: the development and potential applications of genomic-based classification in research and clinical care. Gynecol. Oncol. Res. Pract 3, 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurnit KC et al. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod. Pathol 30, 1032–1041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Myers A, Barry WT, Hirsch MS, Matulonis U & Lee L Beta-catenin mutations in recurrent FIGO IA grade I endometrioid endometrial cancers. Gynecol. Oncol 134, 426–427 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Wortman BG et al. Molecular-integrated risk profile to determine adjuvant radiotherapy in endometrial cancer: Evaluation of the pilot phase of the PORTEC-4a trial. Gynecol. Oncol 151, 69–75 (2018). [DOI] [PubMed] [Google Scholar]
- 107.Soslow RA et al. Endometrial carcinoma diagnosis: Use of FIGO grading and genomic subcategories in clinical practice: recommendations of the international society of gynecological pathologists. Int. J. Gynecol. Pathol 38 Suppl 1, S64–S74 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meric-Bernstam F et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J. Clin. Oncol 33, 2753–2762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stockley TL et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 8, 109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wheler JJ et al. cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 76, 3690–3701 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soumerai TE et al. Clinical utility of prospective molecular characterization in advanced endometrial cancer. Clin. Cancer Res 24, 5939–5947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chawla A et al. Estimated cost of anticancer therapy directed by comprehensive genomic profiling in a single-center study. JCO Precis. Oncol 2, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Meng X et al. AZD1775 Increases sensitivity to olaparib and gemcitabine in cancer cells with p53 mutations. Cancers (Basel) 10, pii: E149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meng X et al. Strategies for molecularly enhanced chemotherapy to achieve synthetic lethality in endometrial tumors with mutant p53. Obstet. Gynecol. Int 2013, 828165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ebeid K et al. Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat. Nanotechnol 13, 72–81 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meng X et al. Induction of mitotic cell death by overriding G2/M checkpoint in endometrial cancer cells with non-functional p53. Gynecol. Oncol 128, 461–469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Freedman AN et al. Use of next-generation sequencing tests to guide cancer treatment: results from a nationally representative survey of oncologists in the United States. JCO Precis. Oncol 2, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rodon J et al. WINTHER: An international WIN Consortium precision medicine trial using genomic and transcriptomic analysis in patients with advanced malignancies. J. Clin. Oncol 36 12011 (2018). [Google Scholar]
- 120.Massard C et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 7, 586–595 (2017). [DOI] [PubMed] [Google Scholar]
- 121.Schwaederle M et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: a meta-analysis. JAMA Oncol. 2, 1452–1459 (2016). [DOI] [PubMed] [Google Scholar]
- 122.Jardim DL et al. Impact of a biomarker-based strategy on oncology drug development: A meta-analysis of clinical trials leading to FDA approval. J. Natl. Cancer Inst 107 pii: djv253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tsimberidou AM et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin. Cancer Res 18, 6373–6383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lheureux S & Oza AM Endometrial cancer-targeted therapies myth or reality? Review of current targeted treatments. Eur. J. Cancer 59, 99–108 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Dedes KJ et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med 2, 53ra75 (2010). [DOI] [PubMed] [Google Scholar]
- 126.Mendes-Pereira AM et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med 1, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miyasaka A et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer 14, 179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bian X et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 37, 341–351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dosil MA et al. Palbociclib has antitumour effects on Pten-deficient endometrial neoplasias. J. Pathol 242, 152–164 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Fader AN et al. Randomized phase ii trial of carboplatin-paclitaxel versus carboplatin-paclitaxel-trastuzumab in uterine serous carcinomas that overexpress human Epidermal Growth Factor Receptor 2/neu. J Clin. Oncol 36, 2044–20516 (2018). [DOI] [PubMed] [Google Scholar]
- 131.Musselman K et al. Identification of a therapeutic target using molecular sequencing for treatment of recurrent uterine serous adenocarcinoma. Gynecol. Oncol. Rep 28, 54–57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01775072. (2019). [DOI] [PubMed]
- 133.Bergstrom CP, Geest K, O’Gara R, Corless CL & Morgan TK Discordant mutations in paired primary and metastatic endometrial adenocarcinomas identified by semiconductor-based sequencing for rapid cancer genotyping. Reprod. Sci 23, 1575–1579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gibson WJ et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet 48, 848–55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ashley CW et al. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol. Oncol 152, 11–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Soslow RA et al. Clinicopathologic analysis of matched primary and recurrent endometrial carcinoma. Am. J. Surg. Pathol 36, 1771–1781 (2012). [DOI] [PubMed] [Google Scholar]
- 137.Ta RM, Hecht JL & Lin DI Discordant loss of mismatch repair proteins in advanced endometrial endometrioid carcinoma compared to paired primary uterine tumors. Gynecol. Oncol 151, 401–406 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Reiter JG et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 361, 1033–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Barroilhet L & Matulonis U The NCI-MATCH trial and precision medicine in gynecologic cancers. Gynecol. Oncol 148, 585–590 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Mangat PK et al. Rationale and design of the targeted agent and profiling utilization registry study. JCO Precis. Oncol doi:DOI: 10.1200/PO.18.00122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/. (2019). [DOI] [PubMed]
- 142.Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Study describing the efficacy of single-agent pembrolizumab in mismatch repair-deficient endometrial cancer patients.
- 143.Cosgrove CM et al. Epigenetic silencing of MLH1 in endometrial cancers is associated with larger tumor volume, increased rate of lymph node positivity and reduced recurrence-free survival. Gynecol. Oncol 146, 588–595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McMeekin DS et al. clinicopathologic significance of mismatch repair defects in endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. J Clin. Oncol 34, 3062–3068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zighelboim I et al. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J Clin. Oncol 25, 2042–2048 (2007). [DOI] [PubMed] [Google Scholar]
- 146.Ott PA et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1-positive endometrial cancer: results from the KEYNOTE-028 Study. J Clin. Oncol 35, 2535–2541 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Vanderstraeten A, Luyten C, Verbist G, Tuyaerts S & Amant F Mapping the immunosuppressive environment in uterine tumors: implications for immunotherapy. Cancer Immunol. Immunother 63, 545–557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mitchell TC et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase i results from a multicenter, open-label phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol doi: 10.1200/JCO.2018.78.9602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mehnert JM et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Invest 126, 2334–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fleming GF et al. Clinical activity, safety and biomarker results from a phase Ia study of atezolizumab (atezo) in advanced/recurrent endometrial cancer (rEC). J. Clin. Oncol 35, 15_suppl, 5585–5585 (2017). [Google Scholar]
- 151.Berger AC et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 33, 690–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830 e814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chan EM et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Behan FM et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Kategaya L, Perumal SK, Hager JH & Belmont LD Werner syndrome helicase is required for the survival of cancer cells with microsatellite instability. iScience 13, 488–497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lieb S et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. Elife 8, doi: 10.7554/eLife.4333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wong A & Ngeow J Hereditary syndromes manifesting as endometrial carcinoma: how can pathological features aid risk assessment? Biomed. Res. Int 2015, 219012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McConechy MK et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod. Pathol 27, 128–1347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jeske YW et al. FGFR2 mutations are associated with poor outcomes in endometrioid endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol 145, 366–373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tashiro H et al. p53 gene mutations are common in uterine serous carcinoma and occur early in their pathogenesis. Am. J. Pathol 150, 177–185 (1997). [PMC free article] [PubMed] [Google Scholar]
- 161.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01876511. (2019). [DOI] [PubMed]
- 162.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02178722. (2019). [DOI] [PubMed]
- 163.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02054806. (2019). [DOI] [PubMed]
- 164.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01375842. (2018). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.