

Abstract
Applications in early-phase cancer trials have motivated the development of many statistical designs since the late 1980s, including dose-finding methods, futility screening, treatment selection, and early stopping rules. These methods are often proposed to address the conventional cytotoxic therapeutics for neoplastic diseases and cancer. Recent advances in precision medicine have motivated novel trial designs, most notably the idea of master protocol (eg, platform trial, basket trial, umbrella trial, N-of-1 trial), for the evaluation of molecularly targeted cancer therapies. In this article, we review the concepts and methodology of early-phase cancer trial designs with a focus on dose finding and treatment screening and put these methods in the context of platform trials of molecularly targeted cancer therapies. Because most cancer trial designs have been developed for cytotoxic agents, we will discuss how these time-tested design principles hold relevance for targeted cancer therapies, and we will delineate how a master protocol may serve as an efficient platform for safety and efficacy evaluations of novel targeted therapies.
EARLY-PHASE PLATFORM TRIALS
Evaluation of cancer therapeutics has traditionally been conducted in distinct phases of clinical trials that examine one treatment (a single agent or a combination of agents) in a single cancer population at a time. As a result of the advances in molecular biology and genomic technology, cancer drug development has undergone an evolutional shift over the past decade. The widespread availability of genomic profiling and next-generation genomic sequencing has enabled comprehensive characterization of tumors on the basis of cancer biology. Tumors once thought to be of the same histologic type may now be considered heterogeneous and differentiated on the basis of genomic biomarkers and may be treated differently according to the biomarker expression profile. Because the subject subgroups defined by specific genetic abnormalities of the tumors may only represent a small fraction of the disease population, the concept of precision oncology trials for targeted therapies has created enormous challenges in recruiting patients with rare genomic subtypes. This recruitment challenge, coupled with the need to test multiple targeted therapies in relatively small molecularly defined patient subgroups in an efficient manner, has led to the rethinking of the cancer drug development paradigm. To meet these challenges, the concept of master protocols has been introduced to increase the speed of oncologic therapy development and evaluation. A master protocol generally refers to an overall framework under which multiple treatments are evaluated concurrently in substudies within the same trial structure. Patient specimens would typically undergo a centralized biomarker profiling, and on the basis of this evaluation, patients are assigned to a substudy within the master protocol. Figure 1 depicts the general structure of a master protocol. The main goal of constructing a master protocol is to enable sharing of operational infrastructure and key design elements across substudies so as to achieve better coordination and efficiency than can be achieved in single trials designed and conducted independently in biomarker-defined subpopulations.1-5
FIG 1.

Master protocol design schema.
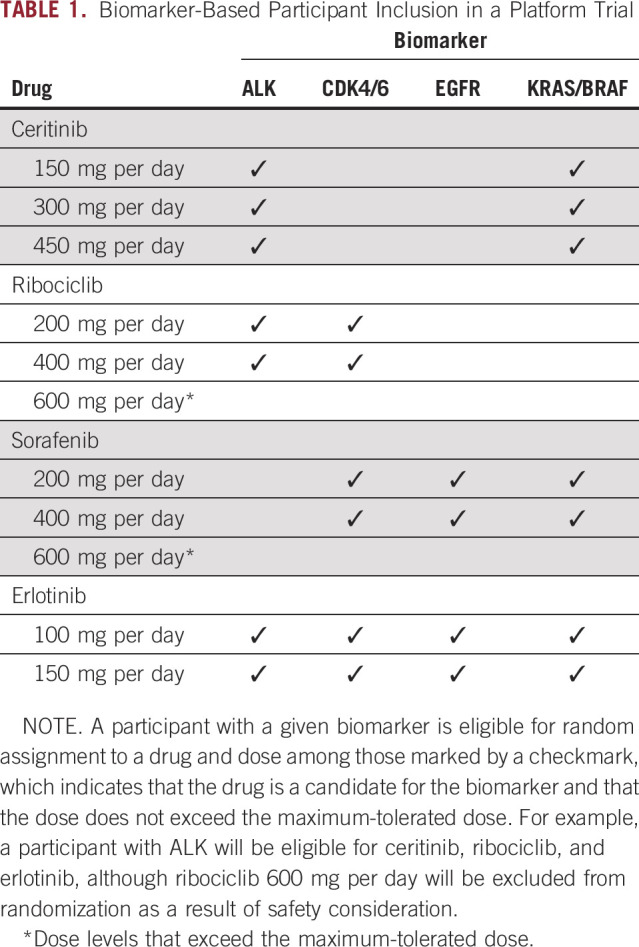
A platform trial is a randomized trial in a single histology that involves multiple treatments and multiple biomarkers under a master protocol. A platform trial does not test a specific hypothesis by matching the targeted therapies to their putative biomarkers. Instead, rather than presuming we know which therapy is appropriate for which biomarker stratum, randomization is used for treatment assignment within each biomarker stratum. Table 1 gives a schema of biomarker-based inclusion criteria in a hypothetical platform trial. Two well-known examples of platform trials are the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial in advanced non–small-cell lung cancer6,7 and the Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging and Molecular Analysis 2 (I-SPY2) trial for neoadjuvant treatment of women with locally advanced breast cancer.8,9 Methodologic innovations in a platform trial may include midtrial outcome-adaptive randomization to favor treatments with higher response rates within the respective biomarker-defined stratum.10,11 The treatment effects of various experimental therapies are often estimated according to a Bayesian hierarchical model by borrowing outcome information across molecularly defined biomarker subgroups.10,11 At the same time, Bayesian decision rules may be incorporated to determine when and whether therapies with low probabilities of success should be discontinued and therapies with high probabilities of future success should advance to subsequent confirmatory studies; in this way, novel therapies may be added or dropped in a perpetual manner.10,12,13 Because of the need for midtrial adaptations, platform trials often use short-term end points, such as absence of disease progression at 8 weeks (as in BATTLE) or pathologically confirmed complete response (as in I-SPY2). Although the Bayesian inferential structure provides a convenient and efficient framework for analysis, it does not protect the type I error rate against multiple comparisons in the conventional sense. As such, phase II platform trials are generally regarded as exploratory, and positive findings from these trials will require independent confirmation for the promising drug-biomarker stratum in subsequent phase III trials.
TABLE 1.
Biomarker-Based Participant Inclusion in a Platform Trial
CONTEXT
Key Objective
This article provides a critical review of early-phase cancer trial statistical methods and discusses their practicality under the novel platform trial paradigm for evaluating molecularly targeted therapies.
Knowledge Generated
Although many trial design principles for cytotoxic agents still hold relevance for molecularly targeted therapies, new design and analysis issues, including delayed toxicities and efficacy-tolerability trade-offs, should be considered when designing studies involving these agents. Early-phase platform trials facilitate efficiency gains by allowing within-trial information sharing of safety and efficacy data.
Relevance
This article provides a practical guide for early-phase platform trial designs in the era of precision medicine. Novel statistical methods continue to be needed to harness the information garnered in these new-generation trial designs to help expedite oncologic drug development and approval.
Cancer trial designs have been well established for cytotoxic agents. Specifically, phase I trials are dose-finding studies that examine safety of the drug and estimate the maximum-tolerated dose (MTD),14 and phase II trials are designed to screen new drugs on the basis of pilot efficacy such as clinical response.15,16 The general objective of early-phase cancer trials is to identify and recommend a promising dose regimen of a new agent for confirmatory investigation in multicenter phase III randomized clinical trials. With the new pathway for accelerated approval,17 it is arguably even more critical to be able to generate rigorous evidence in these early-phase trials. In this article, we review these time-tested design concepts for cytotoxic agents, describe their relevance to the evaluation of molecularly targeted cancer therapies, and outline new design and analysis issues under the novel master protocol framework.
TRADITIONAL PARADIGM OF EARLY-PHASE DOSE AND TREATMENT SCREENING
The MTD of a new agent is defined as a dose that produces a certain percentage of dose-limiting toxicities (DLTs), and it is often used as a recommended phase II dose. The rationale for using toxicity as a surrogate for efficacy can be traced back to the suggestion of using nitrogen mustards in the treatment of neoplastic diseases in the 1960s.18 The most commonly used statistical design for a phase I oncology dose-finding trial is the 3+3 design. This is a rule-based design in which the number of dose levels is fixed in advance and the dose escalation or de-escalation decision is based on the number of DLTs that patients experience at a given dose. The 3+3 design has received extensive examinations and has been demonstrated to produce poor statistical properties, including treating many patients at doses that are below the biologically active level and imprecise estimation of DLT probability because of the relatively small number (three to six) of patients treated at each dose cohort.19-21
To address the deficiencies of the 3+3 design, many dose-finding designs have been proposed since the 1990s that purport to improve statistical accuracy and trial efficiency in the context of chemotherapy trials.22 The most prominent work among these is an adaptive design known as the continual reassessment method (CRM).23,24 The CRM uses a mathematical model to relate the dose levels to the probability of DLT. The process starts with a target DLT rate (eg 25%) and an initial dose-toxicity curve posited by the investigators on the basis of prior knowledge of the toxicity profiles of the drug. After the first patient has been treated at the dose determined by the initial dose-toxicity curve, the toxicity information is combined statistically with the initial curve, and an updated dose-toxicity curve is estimated using a Bayesian methodology. The next patient is then assigned adaptively to the dose level for which the estimated toxicity rate is closest to the target toxicity rate. This process is repeated every time a new patient is enrolled, and the final estimated dose-toxicity curve is used to identify the MTD. Figure 2 gives a graphical illustration of the updates of dose-toxicity curves, with a 25% target DLT rate. The CRM has been shown to improve the precision in estimating the MTD and to increase the number of patients assigned to the selected MTD when compared with the standard 3+3 method.25-27 The latter property has important ethical implications because traditional methods will likely treat many patients at low and, hence, inefficacious doses. In this regard, the CRM fulfills the therapeutic purpose of phase I cancer trials, which are often the last resort of the patients who have exhausted other alternative treatment options. During its early inception, the CRM drew some criticisms particularly with regard to the possibility of exposing patients to doses that are likely to cause DLT.28,29 To address these concerns, several trials used a hybrid approach that combined the model-based CRM with a rule-based initial dose-escalation plan (ClinicalTrial.gov identifiers: NCT03141203, NCT03733990, and NCT03028766).28-31
FIG 2.

Example of dose-toxicity curves in the continual reassessment method (CRM) paradigm. The dashed (gray) curves represent the initial curve posited by the investigators. The colored solid curves represent updated curves after the toxicity data from a certain number of patients have been observed. The target dose-limiting toxicity (DLT) rate is assumed to be 25% (horizontal line). The vertical dotted lines point to the optimal dose level to which patients will be assigned.
Once an MTD is identified, a new chemotherapy will be studied in phase II trials, which are proof-of-concept studies looking for early indication of antitumor activity. This objective is often achieved with a single-arm trial design using clinical response as an end point, with the possibility of stopping a trial early as a result of futility.16,23,32,33 For instance, to have 80% statistical power to reject a 25% response rate in favor of a 45% response rate of a new drug at 5% significance, a single-arm, fixed-sample size trial will need to enroll 36 patients and seek to observe 14 responses in the enrolled patients. Alternatively, a Simon two-stage design16 will first enroll 17 patients and then enroll an additional 24 patients (ie, a total of 41 patients) only if there are at least six responses in the first 17 patients. In other words, if there are five or fewer responses in the first 17 patients, the new drug will be declared futile without additional investigation. Because of the provision for futility stopping, the Simon design will, on average, treat fewer patients with inefficacious drugs, thus allocating resources to the more promising ones. However, to demonstrate promise of a new drug, an adaptive design with futility stopping will always require a larger maximum sample size than a fixed trial (as in the example just provided). Thus, whether using such an adaptive trial design in a single-arm study is advantageous depends on the context and perspectives of the investigators.34
DESIGN AND ANALYSIS ISSUES IN PLATFORM TRIALS
Late-Onset Toxicities and Cumulative Toxicities
The MTD is inherently associated with a fixed observation window within which DLTs are expected to occur. As such, dose assignments are often made based on DLTs observed during this window in a phase I trial. In the cytotoxic chemotherapy paradigm, toxic effects are acute, and hence, it is adequate to assess DLTs occurring in the first cycle of treatment. For molecularly targeted agents and immunotherapies, the mechanism of action typically involves targeting the cellular survival or inhibiting the growth signaling pathway. These noncytotoxic agents tend to induce long-lasting mild toxicities, and hence, the assumption of acute toxicities may not apply.35 For some agents, prolonged administration may be required to achieve desirable therapeutic benefit, thus increasing the likelihood of late-onset or cumulative toxicities that may manifest many months after the initiation of treatment. Therefore, for phase I trials of targeted therapies, it is critical to identify the MTD with respect to a longer observation window, so as to ensure future investigations are limited to a tolerated dose range.
The 3+3 design and CRM-type designs dictate that accrual of new patients be suspended until all enrolled patients have been fully evaluated for DLT. When delayed toxicities are anticipated, the trial length may be prohibitively long because of the need for frequent accrual suspension and the lengthy DLT observation window.36,37 A dose-finding design that does not require suspension of patient accrual while waiting for DLT information on previously treated patients to mature is the time-to-event continual reassessment method (TITE-CRM).38 The dose allocation scheme of the TITE-CRM follows closely the original CRM paradigm but additionally leverages the time-to-toxicity and partial follow-up information in all enrolled patients when estimating the MTD. The TITE-CRM has the potential to substantially reduce the trial length compared with the standard methods. Since its introduction, the TITE-CRM design has been successfully implemented in trials at major academic cancer centers,39-48 as well as US National Cancer Institute–sponsored cooperative groups trials such as Radiation Therapy Oncology Group 0813 trial (ClinicalTrials.gov identifier: NCT00750269). In situations where patients become available to a trial at a rapid rate, the TITE-CRM could accrue many patients at a given dose level before anyone has been observed for a meaningfully long period. This could cause problems in two possible ways. First, if the current dose is truly safe, the TITE-CRM will treat more patients at the current dose than necessary before higher doses are explored, thus resulting in loss in efficiency. Second, if the current dose exhibits late-onset toxicities, the trial could expose many patients to a highly toxic dose before any safety issues appear. Therefore, it may be useful to impose a waiting window between cohorts of patients, so as to allow adequate follow-up before adaptation is made.49-51
Tolerability and Efficacy Trade-Off
For molecularly targeted agents and immunotherapies, it may not be adequate to only consider toxicity and tolerability in a dose-finding trial. First, acute toxicity is no longer a surrogate for the agent’s therapeutic activity, as in cytotoxic agents. Second, a higher dose of a new compound may be less efficacious than a lower dose52 or may see limited benefits on the basis of considerations of pharmacokinetic properties.53 Hence, the MTD may not be an ideal recommended phase II dose, although it is still useful to define the upper safety limit by the MTD. Third, DLTs may not be observed with these agents, and MTD may not be determined at the conclusion of dose escalation.54,55 These are the scenarios where both toxicity and activity end points should be used to guide dose recommendations.54,55 Yan et al56 proposed an adaptive Bayesian phase I/II EffTox design that guides dose selection on the basis of the trade-offs between the probabilities of treatment toxicity and efficacy. Paoletti and Postel-Vinay55 discussed the ideal setting for the EffTox design and contended that such design is best suited when a homogeneous disease population is well-defined for drug sensitivity and an early biomarker for efficacy is available. Houede et al57 model clinical utilities on the basis of both toxicity and clinical response in a combination therapy trial, which aims to choose the optimal dose pair of a chemotherapy and a biologic agent. Finally, Li et al58 proposed an approach to identify the best drug at an efficacious and safe dose for a given biomarker subtype in an early-phase platform trial. They used the CRM to estimate the MTD of a drug and hierarchical Bayesian modeling to estimate the efficacy of a drug administered at multiple doses. As cancer trials increasingly focus on combination therapies, it is critical to leverage efficacy information as well as toxicity in dosing decisions.
Treatment Screening in a Randomized Platform Trial
In a single-arm, proof-of-concept trial, the interpretation of the results and conclusion of the trial depend on the specification of what would constitute a poor (null) response rate on the basis of historical data or a priori clinical experience. The choice of a single null response, however, can be elusive in the early-phase screening of targeted therapy. Although response defined by tumor shrinkage is an indicator of activity for cytotoxic drugs, progression-free survival (PFS) may be more appropriate for early indication of efficacy of a targeted therapy whose mechanism does not necessarily shrink tumors. PFS is a less specific end point than tumor shrinkage because untreated patients may survive without progression for a period of time, whereas patients not treated with chemotherapy are unlikely to experience response. As a result, heterogeneity in PFS experience may render historical data insufficient to define a null PFS rate for screening purposes. Furthermore, as precision medicine challenges the fundamental assumption of tumor homogeneity, the appropriate null response rate may vary with biomarker expression.
In a platform trial where several drugs are considered for a biomarker subtype (Table 1), drug screening can be based on randomized comparison and ranking, thereby eliminating some subjectivity in choosing a null response rate. Simon et al59 propose ranking several randomized experimental drugs concurrently when a standard therapy is not available. Others have proposed adaptive screening strategies such as a two-stage design,60,61 whereby the first screening stage aims to select a promising drug among several for additional evaluation, and continuous monitoring using the sequential probability ratio test.62 These innovative adaptive designs have been shown to increase the final dose selection accuracy while treating more patients with better drugs during the trial and can accommodate situations where there is a concurrent standard therapy. Indeed, the infrastructure developed under a master protocol, in conjunction with the use of these innovative adaptive designs, facilitates unbiased randomized comparisons of potential drug candidates in a platform trial in a timely and efficient manner.
Pooling Information Across Subtypes in a Platform Trial
In an early-phase platform trial, where a drug may be given to patients across biomarker subtypes, the safety profile of the drug can be evaluated using pooled information across subgroups. Specifically, it is often reasonable to assume there is a common MTD for all biomarker subtypes when the biomarker is agnostic to adverse reactions to the drug.58 The concept of combining toxicity information from different diseases is not new, because phase I cancer trials of chemotherapy are often conducted in patients with heterogeneous malignancies (eg, solid tumors). However, implementing this concept in the context of a platform trial provides much needed relevant safety information of the drug in patients with rare biomarker subtypes.
When assessing a drug’s efficacy in a given biomarker subtype, a statistically unbiased approach is to consider the observed response or PFS rate using only data of patients with that subtype. A Bayesian analysis, for instance, could be adopted by assuming a conjugate β prior on each response rate. This analytical approach may make it difficult to understand the drug’s effect in rare biomarker subtypes. A potential advantage of running a platform trial is that a targeted therapy’s efficacy for a given biomarker can potentially be analyzed based on its efficacy in other subtypes of the same histology, when the drug is hypothesized to work through a common molecular mechanism in all subtypes. Such borrowing strength can be achieved in a Bayesian analysis by putting a hyperprior distribution on the hyperparameters of the β prior. Because this analysis adds another level in the hierarchy of Bayesian analysis, it is called hierarchical Bayesian modeling (HBM). Several authors have examined and proposed the use of HBM in platform trials.58,63,64 It has been noted that borrowing information using HBM can be counterproductive when the drug’s effects vary across biomarker subtypes.63 This is conceivable when a drug has nonspecific anticancer activity that is not specific to the molecule. Although ongoing work has been suggested to model multiple sources of exchangeable drug effects in conjunction with HBM,64 it is critical to evaluate the clinical situations as to whether there is a strong reason for expecting a common drug effect across subgroups.
DISCUSSION
There are many potential efficiency gains associated with a platform trial.2,5 First, the use of a common genomic screening platform to identify patients eligible for the trial often results in shorter recruitment time and lower screen failure rate. This means that more patients will have the opportunity to participate in investigational research and gain access to potentially beneficial drugs. The common assay standard operating procedure also ensures consistent and high-quality tissue material collection and submission, thus enhancing preanalytical and analytical validity of the biomarker measurement. Second, the use of centralized governance bodies under a master protocol (eg, the scientific review committee, institutional review board, or data and safety monitoring committee) can ensure streamlined and efficient oversight for all substudies. In particular, the trial efficiency is enhanced by up-front standardization of the development, approval, and monitoring process across studies. For example, the Trial Innovation Network represents a collaborative national network that aims to enhance operational efficiency by leveraging the resources and expertise of the Clinical and Translational Science Award.65 Third, the speed of drug development and evaluation is improved by the common infrastructure that allows opening or closure of substudies of new agents and biomarkers more expeditiously.
Other related trial designs for biomarker-based cancer drug development under the broad definition of a master protocol include basket trials and umbrella trials.1,3,4 A basket trial evaluates a targeted therapy on multiple disease subtypes; specifically, each basket evaluates the therapeutic effect of a targeted therapy for several cancer types that have a common molecular biomarker or genetic alteration. Basket trials are typically nonrandomized; on the basis of a prespecified genetic alteration, patients are assigned to a regimen that is expected to be active for their tumor. In contrast, an umbrella trial generally restricts patient enrollment to a single tumor type. A patient’s tumor undergoes central genetic testing, and the patient is triaged to one of several molecularly defined substudies where the patient receives a matched targeted therapy. These biomarker-defining strata may be single-arm phase II or phase II/III trials that randomly assign patients to either the matched targeted therapy or some standard therapy (or placebo). An example of an umbrella trial is the Adjuvant Lung Cancer Enrichment Marker Identification and Sequencing Trial (ALCHEMIST) for early-stage non–small-cell lung cancer.66
Although these novel trials provide a platform to answer clinical questions that traditional clinical trial design may not afford, it is important to appreciate that these gains are accompanied by increased logistical challenges and coordination efforts.67 For example, a platform trial requires tremendous resources and time commitment to establish the common trial infrastructure. Often, a platform trial would involve multiple agents developed by different industry partners; the contract negotiation with multiple stakeholders in terms of data sharing or coordination across substudies will likely pose additional communication challenges. Finally, the assay platform used to screen patients for eligibility and assignment to substudies will also need to be cleared with an investigational device exemption by the US Food and Drug Administration, further adding regulatory complexities.
Despite these challenges, the opportunities afforded by the new-generation platform trials are enormous. From a study design perspective, as we have illustrated, a single platform trial under a master protocol can facilitate information sharing of safety and efficacy data across disease subtypes in the statistical analysis.58 To date, most existing statistical methods have been proposed that deal with safety and efficacy questions independently (eg, CRM for safety dose finding, HBM for efficacy evaluation). New statistical paradigms may involve assembling these method modules into an omnibus design that ensures statistical rigor and harnessing the information necessary for an informed decision or drug approval.
Footnotes
Supported, in part, by National Institutes of Health/National Center for Advancing Translational Sciences Grant No. UL1 TR000040 (Y.K.C.).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Ying Kuen Cheung
Consulting or Advisory Role: Remedy Pharmaceuticals, Shire
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hirakawa A, Asano J, Sato H, et al. Master protocol trials in oncology: Review and new trial designs. Contemp Clin Trials Commun. 2018;12:1–8. doi: 10.1016/j.conctc.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Redman MW, Allegra CJ. The master protocol concept. Semin Oncol. 2015;42:724–730. doi: 10.1053/j.seminoncol.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renfro LA, Sargent DJ. Statistical controversies in clinical research: Basket trials, umbrella trials, and other master protocols—A review and examples. Ann Oncol. 2017;28:34–43. doi: 10.1093/annonc/mdw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simon R. Critical review of umbrella, basket, and platform designs for oncology clinical trials. Clin Pharmacol Ther. 2017;102:934–941. doi: 10.1002/cpt.814. [DOI] [PubMed] [Google Scholar]
- 5.Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377:62–70. doi: 10.1056/NEJMra1510062. [DOI] [PubMed] [Google Scholar]
- 6.Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: Personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Lee JJ. An overview of the design and conduct of the BATTLE trials. Linchuang Zhongliuxue Zazhi. 2015;4:33. doi: 10.3978/j.issn.2304-3865.2015.06.07. [DOI] [PubMed] [Google Scholar]
- 8.Barker AD, Sigman CC, Kelloff GJ, et al. I-SPY 2: An adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009;86:97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 9.Park JW, Liu MC, Yee D, et al. Adaptive randomization of neratinib in early breast cancer. N Engl J Med. 2016;375:11–22. doi: 10.1056/NEJMoa1513750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berry DA. The Brave New World of clinical cancer research: Adaptive biomarker-driven trials integrating clinical practice with clinical research. Mol Oncol. 2015;9:951–959. doi: 10.1016/j.molonc.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. doi: 10.1177/1740774508091815. Zhou X, Liu S, Kim ES, et al: Bayesian adaptive design for targeted therapy development in lung cancer: A step toward personalized medicine. Clin Trials 5:181-193, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobbs BP, Chen N, Lee JJ. Controlled multi-arm platform design using predictive probability. Stat Methods Med Res. 2018;27:65–78. doi: 10.1177/0962280215620696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. doi: 10.1177/1740774515626362. Saville BR, Berry SM: Efficiencies of platform clinical trials: A vision of the future. Clin Trials 13:358-366, 2016. [DOI] [PubMed] [Google Scholar]
- 14.American Society of Clinical Oncology Critical role of phase I clinical trials in cancer treatment. J Clin Oncol. 1997;15:853–859. doi: 10.1200/JCO.1997.15.2.853. [DOI] [PubMed] [Google Scholar]
- 15.Ratain MJ, Mick R, Schilsky RL, et al. Statistical and ethical issues in the design and conduct of phase I and II clinical trials of new anticancer agents. J Natl Cancer Inst. 1993;85:1637–1643. doi: 10.1093/jnci/85.20.1637. [DOI] [PubMed] [Google Scholar]
- 16.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 17. doi: 10.1158/1078-0432.CCR-16-0663. Chuk MK, Chang JT, Theoret MR, et al: FDA approval summary: Accelerated approval of pembrolizumab for second-line treatment of metastatic melanoma. Clin Cancer Res 23:5666-5670, 2017. [DOI] [PubMed] [Google Scholar]
- 18.Gilman A. The initial clinical trial of nitrogen mustard. Am J Surg. 1963;105:574–578. doi: 10.1016/0002-9610(63)90232-0. [DOI] [PubMed] [Google Scholar]
- 19.Korn EL, Midthune D, Chen TT, et al. A comparison of two phase I trial designs. Stat Med. 1994;13:1799–1806. doi: 10.1002/sim.4780131802. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y, Shih WJ. Statistical properties of the traditional algorithm-based designs for phase I cancer clinical trials. Biostatistics. 2001;2:203–215. doi: 10.1093/biostatistics/2.2.203. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberger WF, Haines LM. Competing designs for phase I clinical trials: A review. Stat Med. 2002;21:2757–2770. doi: 10.1002/sim.1229. [DOI] [PubMed] [Google Scholar]
- 22. doi: 10.1214/10-STS334. Cheung YK: Stochastic approximation and modern model-based designs for dose-finding clinical trials. Stat Sci 25:191-201, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheung Y: The time-to-event CRM, in Chow S (ed): Dose Finding by the Continual Reassessment Method. Boca Raton, FL, Chapman & Hall/CRC, 2011, pp 119-136. [Google Scholar]
- 24.O’Quigley J, Pepe M, Fisher L. Continual reassessment method: A practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. [PubMed] [Google Scholar]
- 25.O’Quigley J, Chevret S. Methods for dose finding studies in cancer clinical trials: A review and results of a Monte Carlo study. Stat Med. 1991;10:1647–1664. doi: 10.1002/sim.4780101104. [DOI] [PubMed] [Google Scholar]
- 26. doi: 10.1191/1740774506cn134oa. Garrett-Mayer E: The continual reassessment method for dose-finding studies: A tutorial. Clin Trials 3:57-71, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst. 2009;101:708–720. doi: 10.1093/jnci/djp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heyd JM, Carlin BP. Adaptive design improvements in the continual reassessment method for phase I studies. Stat Med. 1999;18:1307–1321. doi: 10.1002/(sici)1097-0258(19990615)18:11<1307::aid-sim128>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 29.Piantadosi S, Fisher JD, Grossman S. Practical implementation of a modified continual reassessment method for dose-finding trials. Cancer Chemother Pharmacol. 1998;41:429–436. doi: 10.1007/s002800050763. [DOI] [PubMed] [Google Scholar]
- 30.Elkind MS, Sacco RL, Macarthur RB, et al. High-dose lovastatin for acute ischemic stroke: Results of the phase I dose escalation neuroprotection with statin therapy for acute recovery trial (NeuSTART) Cerebrovasc Dis. 2009;28:266–275. doi: 10.1159/000228709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond M, Harrison C, Vicente S, et al. A phase Ib study to assess the safety and tolerability of ruxolitinib in combination with azacitidine in patients with advanced phase myeloproliferative neoplasms (MPN), including myelodysplastic syndromes (MDS) or acute myeloid leukaemia (AML) arising from MPN (the Bloodwise/TAP PHAZAR study on behalf of the UK MPN CSG) Blood 1301649.2017(suppl 1; abstr 634)28733324 [Google Scholar]
- 32.Fleming TR. One-sample multiple testing procedure for phase II clinical trials. Biometrics. 1982;38:143–151. [PubMed] [Google Scholar]
- 33.Gehan EA. The determination of the number of patients required in a preliminary and a follow-up trial of a new chemotherapeutic agent. J Chronic Dis. 1961;13:346–353. doi: 10.1016/0021-9681(61)90060-1. [DOI] [PubMed] [Google Scholar]
- 34.Cheung K, Kaufmann P. Efficiency perspectives on adaptive designs in stroke clinical trials. Stroke. 2011;42:2990–2994. doi: 10.1161/STROKEAHA.111.620765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SM, Backenroth D, Cheung YK, et al. Case example of dose optimization using data from bortezomib dose-finding clinical trials. J Clin Oncol. 2016;34:1395–1401. doi: 10.1200/JCO.2015.66.0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharon E, Polley MY, Bernstein MB, et al. Immunotherapy and radiation therapy: Considerations for successfully combining radiation into the paradigm of immuno-oncology drug development. Radiat Res. 2014;182:252–257. doi: 10.1667/RR13707.1. [DOI] [PubMed] [Google Scholar]
- 37.Lawrence YR, Vikram B, Dignam JJ, et al. NCI-RTOG translational program strategic guidelines for the early-stage development of radiosensitizers. J Natl Cancer Inst. 2013;105:11–24. doi: 10.1093/jnci/djs472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung YK, Chappell R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics. 2000;56:1177–1182. doi: 10.1111/j.0006-341x.2000.01177.x. [DOI] [PubMed] [Google Scholar]
- 39.Furman RR, Martin P, Ruan J, et al. Phase 1 trial of bortezomib plus R-CHOP in previously untreated patients with aggressive non-Hodgkin lymphoma. Cancer. 2010;116:5432–5439. doi: 10.1002/cncr.25509. [DOI] [PubMed] [Google Scholar]
- 40.Andre F, Campone M, O’Regan R, et al. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol. 2010;28:5110–5115. doi: 10.1200/JCO.2009.27.8549. [DOI] [PubMed] [Google Scholar]
- 41.Muler JH, McGinn CJ, Normolle D, et al. Phase I trial using a time-to-event continual reassessment strategy for dose escalation of cisplatin combined with gemcitabine and radiation therapy in pancreatic cancer. J Clin Oncol. 2004;22:238–243. doi: 10.1200/JCO.2004.03.129. [DOI] [PubMed] [Google Scholar]
- 42.Jerusalem G, Fasolo A, Dieras V, et al. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2011;125:447–455. doi: 10.1007/s10549-010-1260-x. [DOI] [PubMed] [Google Scholar]
- 43.Ben-Josef E, Schipper M, Francis IR, et al. A phase I/II trial of intensity modulated radiation (IMRT) dose escalation with concurrent fixed-dose rate gemcitabine (FDR-G) in patients with unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys. 2012;84:1166–1171. doi: 10.1016/j.ijrobp.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. doi: 10.1158/1078-0432.CCR-18-0467. Karam SD, Reddy K, Blatchford PJ, et al: Final report of a phase I trial of olaparib with cetuximab and radiation for heavy smoker patients with locally advanced head and neck cancer. Clin Cancer Res 24:4949-4959, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crew KD, Brown P, Greenlee H, et al. Phase IB randomized, double-blinded, placebo-controlled, dose escalation study of polyphenon E in women with hormone receptor-negative breast cancer. Cancer Prev Res (Phila) 2012;5:1144–1154. doi: 10.1158/1940-6207.CAPR-12-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jakubowiak AJ, Griffith KA, Reece DE, et al. Lenalidomide, bortezomib, pegylated liposomal doxorubicin, and dexamethasone in newly diagnosed multiple myeloma: A phase 1/2 Multiple Myeloma Research Consortium trial. Blood. 2011;118:535–543. doi: 10.1182/blood-2011-02-334755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magenau J, Tobai H, Pawarode A, et al. Clofarabine and busulfan conditioning facilitates engraftment and provides significant antitumor activity in nonremission hematologic malignancies. Blood. 2011;118:4258–4264. doi: 10.1182/blood-2011-06-358010. [DOI] [PubMed] [Google Scholar]
- 48.Jakubowiak AJ, Dytfeld D, Griffith KA, et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood. 2012;120:1801–1809. doi: 10.1182/blood-2012-04-422683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bekele BN, Ji Y, Shen Y, et al. Monitoring late-onset toxicities in phase I trials using predicted risks. Biostatistics. 2008;9:442–457. doi: 10.1093/biostatistics/kxm044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheung Y: Dose Finding by the Continual Reassessment Method. Boca Raton, FL, Chapman & Hall/CRC, 2011. [Google Scholar]
- 51.Polley MY. Practical modifications to the time-to-event continual reassessment method for phase I cancer trials with fast patient accrual and late-onset toxicities. Stat Med. 2011;30:2130–2143. doi: 10.1002/sim.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Slaton JW, Perrotte P, Inoue K, et al: Interferon-alpha-mediated down-regulation of angiogenesis-related genes and therapy of bladder cancer are dependent on optimization of biological dose and schedule. Clin Cancer Res 5:2726-2734, 1999. [PubMed] [Google Scholar]
- 53. Goel R, Hirte H, Shah A, et al: Phase I study of the metalloproteinase inhibitor Bayer 12-9566. Proc Am Soc Clin Oncol 17:217a, 1998 (abstr 840) [Google Scholar]
- 54.Hobbs BP, Barata PC, Kanjanapan Y, et al. Seamless designs: Current practice and considerations for early-phase drug development in oncology. J Natl Cancer Inst. 2019;111:118–128. doi: 10.1093/jnci/djy196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paoletti X, Postel-Vinay S. Phase I-II trial designs: How early should efficacy guide the dose recommendation process? Ann Oncol. 2018;29:540–541. doi: 10.1093/annonc/mdy044. [DOI] [PubMed] [Google Scholar]
- 56.Yan F, Thall PF, Lu KH, et al. Phase I-II clinical trial design: A state-of-the-art paradigm for dose finding. Ann Oncol. 2018;29:694–699. doi: 10.1093/annonc/mdx795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Houede N, Thall PF, Nguyen H, et al. Utility-based optimization of combination therapy using ordinal toxicity and efficacy in phase I/II trials. Biometrics. 2010;66:532–540. doi: 10.1111/j.1541-0420.2009.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, Wang M, Cheung Y. Treatment and dose prioritization in early phase platform trials of targeted cancer therapies. J R Stat Soc Ser C Appl Stat. 2019;68:475–491. [Google Scholar]
- 59.Simon R, Wittes RE, Ellenberg SS. Randomized phase II clinical trials. Cancer Treat Rep. 1985;69:1375–1381. [PubMed] [Google Scholar]
- 60.Thall PF, Simon R, Ellenberg SS, et al. Optimal two-stage designs for clinical trials with binary response. Stat Med. 1988;7:571–579. doi: 10.1002/sim.4780070504. [DOI] [PubMed] [Google Scholar]
- 61.Schaid D, Wieand S, Therneau T. Optimal two-stage screening designs for survival comparisons. Biometrika. 1990;77:507–513. [Google Scholar]
- 62.Cheung YK. Simple sequential boundaries for treatment selection in multi-armed randomized clinical trials with a control. Biometrics. 2008;64:940–949. doi: 10.1111/j.1541-0420.2007.00929.x. [DOI] [PubMed] [Google Scholar]
- 63. doi: 10.1158/1078-0432.CCR-12-1223. Freidlin B, Korn EL. Borrowing information across subgroups in phase II trials: is it useful? Clin Cancer Res 19:1326-1334, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hobbs BP, Landin R. Bayesian basket trial design with exchangeability monitoring. Stat Med. 2018;37:3557–3572. doi: 10.1002/sim.7893. [DOI] [PubMed] [Google Scholar]
- 65. National Center for Advancing Translational Sciences: Trial Innovation Network. https://trialinnovationnetwork.org/
- 66.Gerber DE, Oxnard GR, Govindan R. ALCHEMIST: Bringing genomic discovery and targeted therapies to early-stage lung cancer. Clin Pharmacol Ther. 2015;97:447–450. doi: 10.1002/cpt.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. doi: 10.1158/1078-0432.CCR-18-3544. Cecchini M, Rubin EH, Blumenthal GM, et al: Challenges with novel clinical trial designs: Master protocols. Clin Cancer Res 25:2049-2057, 2019. [DOI] [PubMed] [Google Scholar]