

Abstract
PURPOSE
Next-generation sequencing (NGS) of tumor and germline DNA is foundational for precision oncology, with rapidly expanding diagnostic, prognostic, and therapeutic implications. Although few question the importance of NGS in modern oncology care, the process of gathering primary molecular data, integrating it into electronic health records, and optimally using it as part of a clinical workflow remains far from seamless. Numerous challenges persist around data standards and interoperability, and clinicians frequently face difficulties in managing the growing amount of genomic knowledge required to care for patients and keep up to date.
METHODS
This review provides a descriptive analysis of genomic data workflows for NGS data in clinical oncology and issues that arise from the inconsistent use of standards for sharing data across systems. Potential solutions are described.
RESULTS
NGS technology, especially for somatic genomics, is well established and widely used in routine patient care, quality measurement, and research. Available genomic knowledge bases play an evolving role in patient management but lack harmonization with one another. Questions about their provenance and timeliness of updating remain. Potentially useful standards for sharing genomic data, such as HL7 FHIR and mCODE, remain primarily in the research and/or development stage. Nonetheless, their impact will likely be seen as uptake increases across care settings and laboratories. The specific use case of ASCO CancerLinQ, as a clinicogenomic database, is discussed.
CONCLUSION
Because the electronic health records of today seem ill suited for managing genomic data, other solutions are required, including universal data standards and applications that use application programming interfaces, along with a commitment on the part of sequencing laboratories to consistently provide structured genomic data for clinical use.
INTRODUCTION
Next-generation sequencing (NGS) of tumor and inherited (germline) genomes has revolutionized and refined cancer treatment during the past two decades and is now vital for evaluating therapeutic opportunities in many solid and hematologic malignancies.1 Currently, NGS panels including sets of genes are the most widespread method of rapidly identifying sequence variation in patients with cancer. NGS panels provide information for a variety of purposes, including diagnostics (eg, determination of sarcoma subtype), hereditary risk assessment (eg, Lynch syndrome), prognosis (eg, KRAS mutations in lung adenocarcinoma), and treatment selection (eg, biomarkers for immunotherapy responsiveness, such as tumor mutational burden and microsatellite instability; therapeutic selection for clinically actionable alterations, such as BRAF V600E in melanoma; and biomarkers of resistance, such as loss of B2M for immunotherapy).2,3 As a metric of the significance of NGS in oncology care, 29 of the 43 National Comprehensive Cancer Network clinical practice guidelines denote specific sequence-based biomarkers important for clinical care (Table 1). Of these 29 guidelines for treatment of cancer by site, 12 include both somatic and germline biomarkers, 16 include somatic only, and one includes germline only.
TABLE 1.
Inclusion of Genetic Testing in NCCN Cancer Site Guidelines

Recently, there has been an increased focus on performing NGS on matched normal samples (either adjacent tissue or blood) to compare with tumor biopsies. The use of paired tumor and normal samples supports improved fidelity of variant calling,4 increased sensitivity in low-purity tumor samples,5,6 and unambiguous delineation of germline mutations.4 The identification of pathogenic germline variation not only supports patient and familial risk management but is also embedded in guidelines and reimbursement for certain treatments (eg, poly [ADP-ribose] polymerase inhibitors for ovarian cancer). This is an example of how clinical practice guidelines7 and reimbursement8 can lag behind the evidence.
CONTEXT
Key Objective
The amount of molecular knowledge required for patient care is exploding, but the minimal adoption of genomic data standards in electronic health records compounds the challenges facing oncologists. Using examples from ASCO CancerLinQ, this review covers the current status of next-generation sequencing, describes relevant data standards, highlights the pilot of the genomic SMART-on-FHIR application, and provides a call to action for laboratories and electronic health record vendors to ensure that the chain of data custody for structured genomic content remains unbroken and reaches the clinician.
Knowledge Generated
Several genomic knowledge bases for variant interpretation are available to support clinicians. Nonetheless, transmitting next-generation sequencing results via PDF hinders the use of decision support tools intended for clinical care, including trial selection, and blocks the downstream reuse of genomic data.
Relevance
The consistent use of evolving standards (eg, HL7 FHIR and mCODE) should improve genomic data interoperability and care quality. However, the full integration of genotype and phenotype will require an innovative and socially driven commitment by all oncologic ecosystem participants, beginning with sequencing laboratories, to better leverage technologic solutions.
As the cost of sequencing continues to decrease, the identification of actionable variants improves, and oncogenic pathways become more targetable, whole-exome sequencing and whole-genome sequencing will likely gain more traction over NGS panels.9 For instance, active mutational processes can now be determined from the mutational spectrum of single-patient samples that have undergone whole-exome sequencing, allowing for more personal dissection of the biology of a tumor.10 Tumor signatures predicting response to immune checkpoint inhibitors will likely be refined soon, placing pressure on NGS and biomarker data management to support translation into evidence-based clinical care.11 Sequencing of heterogeneous tumor tissues containing many cell types may eventually be supplemented by single-cell sequencing, and this will magnify data complexity.
NGS AND CLINICAL USE CASES
Routine Care
Molecular testing has become essential to oncology care.12 Survey data from 2017 showed that more than 75% of oncologists used NGS tests to guide cancer treatment. Most commonly, NGS was used in patients with advanced, refractory cancers (consistent with the NGS National Coverage Determination by the Centers for Medicare and Medicaid Services13) and less commonly for patients with rare cancers, cancers of unknown origin, or an initial cancer diagnosis.1 The two most commonly cited clinical purposes of NGS testing were to guide the on-label use of a US Food and Drug Administration (FDA)–approved therapy or to determine clinical trial eligibility. Multiple molecularly targeted agents have now been incorporated into standard patient management and reflected in numerous guidelines.
Clinical Quality Measures
The 2019 Merit-Based Incentive Payment System (MIPS) of the Centers for Medicare and Medicaid Services Quality Payment Program contains 24 general oncology measures, two of which involve genetic testing (KRAS and NRAS in patients with colorectal cancer).14 In addition, a number of clinical quality measures in the ASCO Quality Oncology Practice Initiative reference NGS testing, including non–small-cell lung cancer (NSCLC) measures evaluating the use and turnaround time of molecular testing in patients with adenocarcinoma histology.15 As clinical quality measurement evolves from clinician-focused process measures to more patient-centric outcome measures, it is reasonable to expect that more measures will incorporate results from NGS panels and potentially circulating tumor-free DNA testing to foster appropriate diagnostic testing and therapy selection that affect patient outcomes.
Clinical Trial Matching
Despite the critical need for patients with cancer to participate in clinical trials, only 3% of US patients with cancer actually do so, and 18% of National Clinical Trials Network–sponsored phase II to III adult trials fail to accrue enough patients to meet their statistical end points.16 With eligibility being increasingly predicated on NGS testing,17 several computational clinical trial matching platforms that leverage NGS data have been developed to more efficiently assess eligibility criteria. The sweet spot is to seamlessly integrate patient demographic, clinical, and laboratory data from electronic health records (EHRs) and match patient and trial locations, a major barrier to enrollment.18
BACKGROUND AND DATA CHALLENGES OF INTEGRATING INTO THE CLINICAL WORKFLOW
A comprehensive review of bioinformatic pipelines is beyond the scope of this article; the interested reader is referred to a recent review.19 However, to understand how genomic data move from laboratory to clinical information systems and are used by clinicians requires an analysis of current data challenges.
Presenting Results to Clinicians and the Role of Knowledge Bases
Despite the rapid advancements in NGS technology and bioinformatic pipelines for identifying somatic and germline alterations, major barriers still exist for efficiently implementing interpreted results for use in clinical decision making. Barriers include insufficient clinician knowledge, training, and confidence regarding the use of NGS results.20,21 Although many clinicians may take laboratory-based interpretations at face value, numerous knowledge bases have been created to aid oncologists and clinical researchers in the curation and interpretation of NGS data. These were developed largely because of the recognition that institutional (local) knowledge based on a limited sampling of the cancer population, including that possessed by testing laboratories, would not be sufficient to capture the genomic diversity of any cancer type, common or rare. Perhaps a surprise to clinicians, many commonly tested genes, such as BRCA1 and BRCA2, show considerable disagreement by interpreting laboratories about the pathogenicity of genetic variants.22 One of the earliest resources to address this issue was MyCancerGenome,23 created at Vanderbilt University in 2009. Since that time, there have been numerous entrants into an increasingly crowded field, with the most well-known publicly available resources for variant interpretation being CIViC,24 OncoKB,25and ClinVar,26 the latter of which is the first FDA-recognized public genetic variant database, housing ClinGen expert-curated data.27 Each of these efforts was developed in relative isolation and with slightly different goals, leading to a divergence of data models, evidentiary models, and user experiences. The Variant Interpretation in Cancer Consortium, a driver project of the Global Alliance for Genomics and Health,28 was founded with the mission statement to help unify and harmonize the disparate data sources. The Variant Interpretation in Cancer Consortium now offers a product termed the meta-knowledgebase, which aggregates and displays information from up to eight clinicogenomic knowledge bases (Fig 1).29
FIG 1.
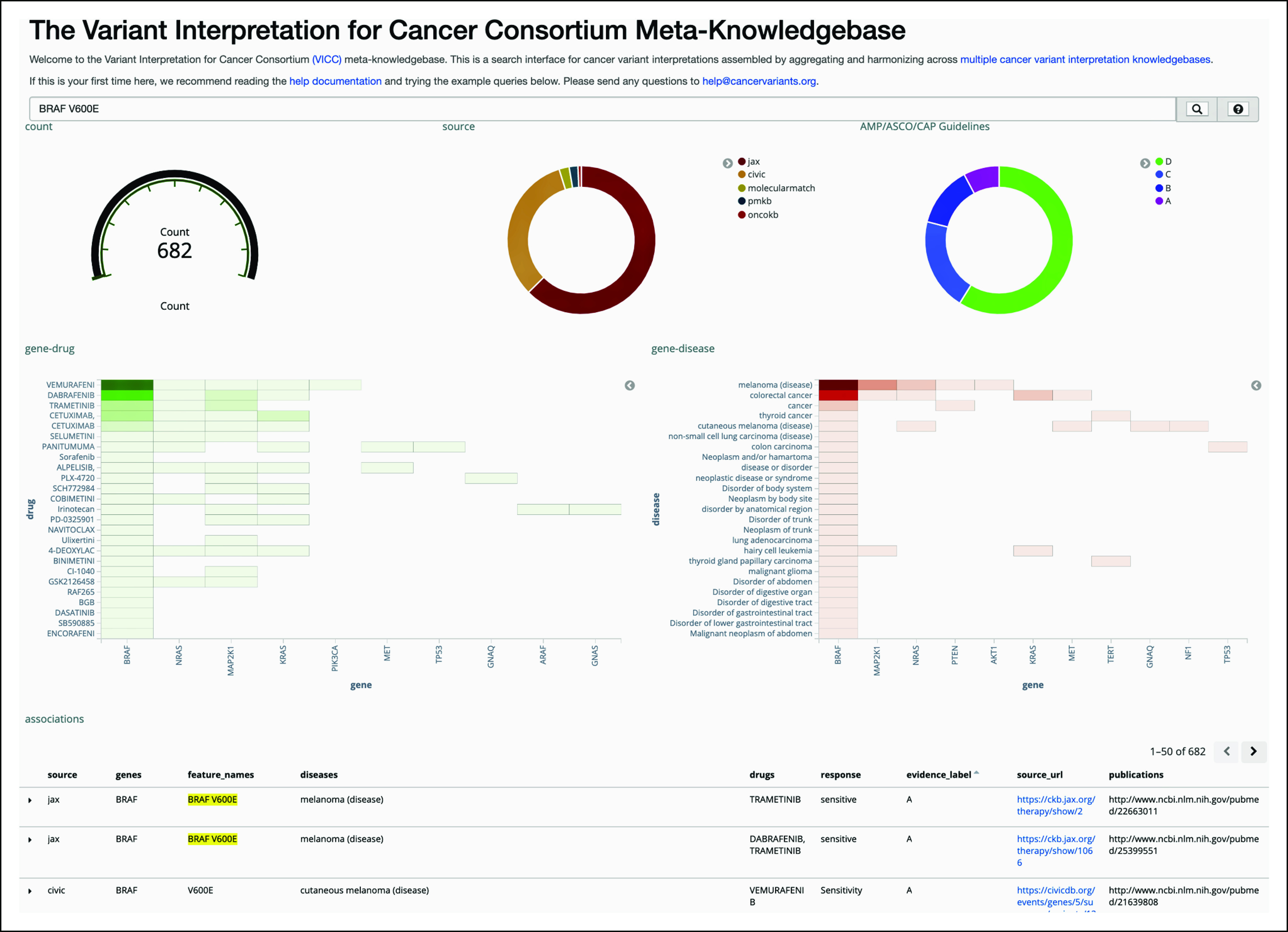
Screenshot of the Variant Interpretation in Cancer Consortium (VICC) meta-knowledgebase user interface.45 Results are shown after searching for BRAF V600E. The top section shows total count of prognostic or predictive assertions related to this gene-variant combination, the breakdown by data source (five are shown), and the breakdown by assertion type per Association for Molecular Pathology (AMP)/ASCO/College of American Pathologists (CAP) guidelines.46 The middle section cross-references drugs and diseases with the gene of interest (first column), as well as potentially related genes (adjacent columns), which are automatically determined; heatmap colors indicate the quantity of evidence for a drug-gene or disease-gene correlation. At the bottom, three of 682 results are shown in tabular format, with active links to the source URLs. Reproduced with permission. The VICC meta-knowledgebase user guide is available online.47
Several challenges associated with knowledge bases must be considered. The first is access and integration. Although the aforementioned knowledge bases are free to users, they are generally not integrated into the clinical workflow.30 The second is trustworthiness and culpability. Can the assertions made by knowledge bases be trusted? If advice is erroneous, the primary responsibility clearly lies with the managing clinician, but can knowledge bases also be held culpable? Furthermore, advice can become outdated; this must be expected to some degree, given the exponential growth of the field and the lag between the announcement of trial results, publication of findings, FDA labeling, and revision of variant interpretations; however, poor recency of data in knowledge bases will diminish trust. Finally, there is the issue of user experience. Many knowledge bases spend much effort on attractive so-called skins and search abilities, but the degree to which clinicians find these interfaces useful has not been extensively studied.
Technological Challenges of Data Integration: Opportunities With FHIR and mCODE
Integration of NGS results into the clinical workflow via EHR interfaces must tackle several technologic challenges. In particular, interpreted genomic results are generally reported in PDF format, which cannot support electronic search, clinical decision support (CDS), or secondary use.31 Moreover, the lack of a common data model for genomic testing impedes all use cases by hampering data storage and interoperability between EHR systems and/or data warehouses, thereby hindering the use of CDS tools intended to assist clinicians in caring for their patients.
These issues have prompted the emergence of initiatives aimed at improving genomic data standards and facilitating genomic data interoperability. One of the broader initiatives is the HL7 (Health Level Seven International) FHIR (Fast Healthcare Interoperability Resources) standard for exchanging health information, with the overall goal of enabling semantic interoperability.32 FHIR can be used as a companion to any EHR vendor technology and has a data model for how health information should be structured that is compatible with other common data models.33 A more oncology-focused initiative is the mCODE (Minimal Common Oncology Data Elements) project, codeveloped by ASCO and other collaborators, which aims to establish a core set of structured data elements, including genomics, with the goal of facilitating cancer data interoperability to improve cancer data quality, clinical care, and cancer research.34,35
POTENTIAL SOLUTIONS: A SMART-ON-FHIR EXAMPLE
Use of Application Programming Interfaces
To provide proof of concept that the structured data elements established through interoperability initiatives could be used to exchange NGS results and integrate genomic information into patient EHRs, two authors (J.R.C. and J.L.W.) obtained sample genomic profiling reports from Foundation Medicine Incorporated (FMI) under the auspices of Sync for Genes.36 These reports were generated from proprietary pipelines used for the FMI FoundationOne and FoundationACT NGS panels. Although customers typically receive a PDF of the findings and interpretations, FMI also sends XML files, based on a custom internal standard used to generate the PDFs, upon request. These XML files include more detailed information than that presented in the PDF, some of which is for research use only, and can be parsed to generate information relevant for FHIR Resources and elements (eg, variant allele fraction, sequencing depth and functional effect for mutations, absolute copy number and copy-number ratio for copy-number events, number of supporting read pairs and identity of both genes involved for rearrangements, and genomic location for all alterations). Briefly, clinical fields such as patient, sample, and physician information could be mapped to Patient, Specimen, and Physician FHIR Resources, respectively. Genomic fields such as alterations (eg, BRAF p.V600E, amplification, rearrangement), therapies, and clinical trials were mapped to elements of the Observation Genetics Resource, with more detailed genetic information (eg, chromosome, start position, end position, variant allele frequency, coverage, and variant type) mapped to the Molecular Sequence Resource. The entire FMI report was represented as a Diagnostic Report Resource, which included references to the Patient, Physician, Specimen, Observation Genetics, and Molecular Sequence Resources. Furthermore, FHIR servers enable easy and scalable exchange of genomic and health information through a RESTful application programming interface (API) service.31,37 By leveraging FHIR standards and the HL7 API server, we were able to successfully integrate FMI NGS results into a test environment at Vanderbilt University Medical Center.
SMART Precision Cancer Medicine
The SMART (Sustainable Medical Apps, Reusable Technology) application platform has been modified to leverage FHIR APIs.38 We developed a SMART application from the perspective of precision oncology: SMART Precision Cancer Medicine (PCM).39 An open-source version of SMART PCM can be found in the SMART App Gallery (https://gallery.smarthealthit.org/apps/category/genomics).
SMART PCM was developed to fit into any clinical oncology workflow with access to genomic data and consists of several visualizations and interactive features to facilitate shared decision making between oncologists and their patients; it remains the only genomic application available on the SMART App Gallery. The modular design (Fig 2) was architected with four major goals: assist in clinical interpretation of variants within context, provide links to external resources, facilitate point-of-care physician-patient conversations, and demonstrate that SMART-on-FHIR technology can be used for a clinicogenomic use case. Current work on the application includes enhancing the visualizations to accommodate high-dimensional NGS panels, structural variants, and signature data (eg, tumor mutational burden) and migrating from static links to API-based queries of knowledge bases. If the ecosystem recommended by Hughes et al30 comes to fruition, we would expect a proliferation of such applications in the future.
FIG 2.
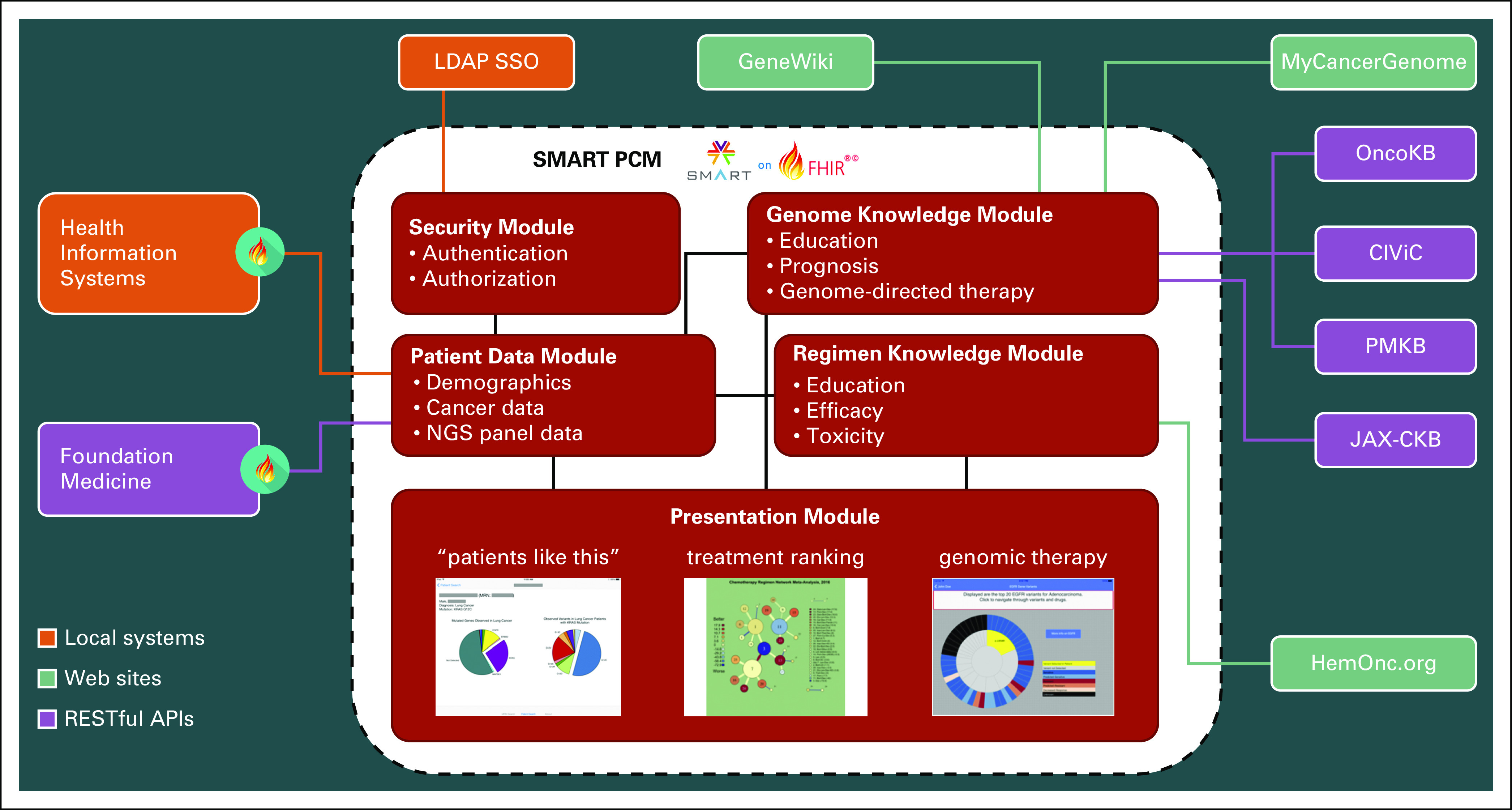
SMART (Sustainable Medical Apps, Reusable Technology) Precision Cancer Medicine (PCM) architecture diagram. The application is designed to be modular and extensible, particularly in the ability to add presentation modules and interfaces to external data sources. External sources with the Fast Healthcare Interoperability Resources (FHIR) logo can expose information using native FHIR; others require some degree of custom mapping. API, application programming interface; CIViC, Clinical Interpretations of Variants in Cancer; JAX-CKB, Jackson Laboratory Clinical Knowledgebase; LDAP SSO, Lightweight Directory Access Protocol Single Sign-On; NGS, next-generation sequencing; PMKB, Precision Medicine Knowledgebase.
CANCERLINQ: OPPORTUNITIES TO DEVELOP A CLINICOGENOMIC DATABASE FOR PATIENT CARE AND DISCOVERY
Background
CancerLinQ is a health technology platform developed and implemented by ASCO that collects and analyzes real-world cancer care data to deliver insights to oncologists, improve the quality of patient care, and drive new research. CancerLinQ aggregates structured and unstructured data from EHRs and other sources via direct feeds and processes the data in a series of cloud-based databases where it is normalized and deidentified for secondary reuse.40
Database
The CancerLinQ database, now representing more than 1.5 million patients, contains vast amounts of detailed longitudinal clinical data, such as demographics, diagnoses, laboratory results, and medications. However, a significant volume of the data is not computable, because many critical concepts (cancer staging, biomarkers, disease progression, and adverse events) reside primarily in text notes and other unstructured documents and not in discrete fields. To supplement the structured data and extract critical information from clinical notes and other sources, CancerLinQ employs technology-assisted manual data curation services delivered by distributed teams of trained clinical abstractors. Although natural language processing technology itself has not yet demonstrated sufficient precision or recall to be used as a standalone solution, it has been successfully embedded in the curation workbench used by the data abstractors, where it functions to surface putative data elements from unstructured text that can then be confirmed or disregarded.
Curation of Genomic Data From NGS Panels in CancerLinQ
As noted, NGS panels are typically brought into the EHR as PDFs or scanned faxes and therefore are not computable. However, since 2017, CancerLinQ has been extracting high-value genomic information via user interface–assisted data abstraction. CancerLinQ has also obtained and processed structured genomic reports in XML format and is evaluating automated processes to scan and extract data from reports with standardized formats.
The magnitude of the genomic data gap in native EHRs is considerable and shows how poorly precision oncology is supported. Only one third of practices in the CancerLinQ database have any structured BRCA1 or BRCA2 gene test results, with results coming predominantly from two practices. Approximately 1.5% of patients with breast or ovarian cancer have natively structured BRCA1 or BRCA2 results, whereas curation has abstracted 5,004 BRCA1 and 4,235 BRCA2 results from 36,346 records. Similarly, for advanced NSCLC, 85.3% of curated patient records had epidermal growth factor receptor tests, but structured epidermal growth factor receptor data were found in native EHRs for only 1.7% of all records for patients with advanced NSCLC.
It is worth noting that although CancerLinQ has developed various technical solutions to structuring genomic data as it is housed in EHRs, all NGS data are originally generated as structured data by molecular diagnostics laboratories. The data capture of the various chemistries that underlie NGS sequencing, as well as the components of bioinformatic pipelines (sequence aligners, variant callers) and even clinical report production, comprises machine-readable, structured genomic data. More properly, curation of NGS reports restructures the data, at a significant cost, along with some erosion of data quality. The technical solutions CancerLinQ has painstakingly developed would not be necessary at all if the originally structured data were provided.
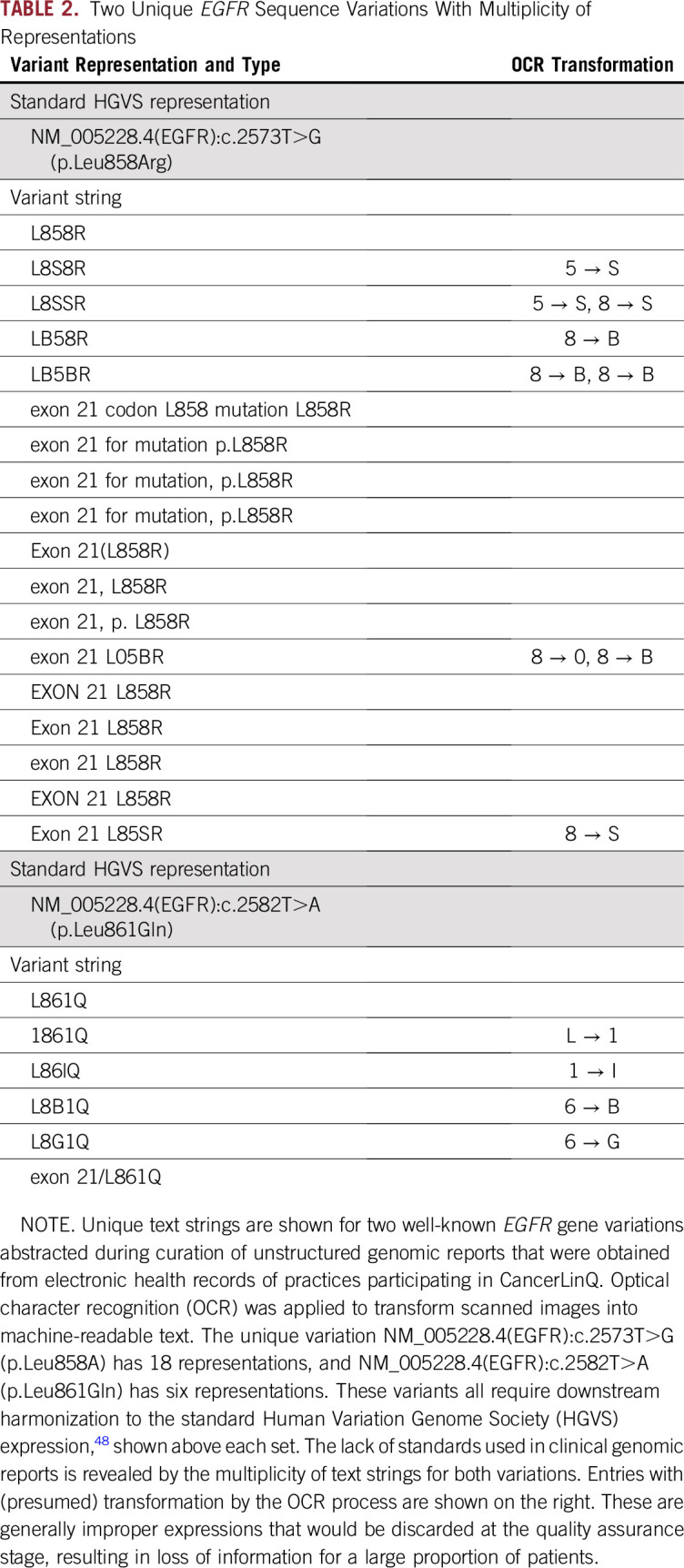
A stark example of how data quality is diminished when optical character recognition, needed to parse genomic reports, is applied, particularly to scanned images (eg, faxed reports) and tables, is listed in Table 2. Downstream, quality-assurance processes remove nonstandard terms, favoring data gaps rather than inaccuracies, but the overall effect is data erosion.
TABLE 2.
Two Unique EGFR Sequence Variations With Multiplicity of Representations
CALL TO ACTION FOR RETAINING STRUCTURED GENOMIC DATA
The utility and importance of NGS testing in oncology care are evident from the incorporation of such testing into 67% of National Comprehensive Cancer Network guidelines as well as payment incentives (eg, MIPS) and quality programs (eg, Quality Oncology Practice Initiative). Clinical trial eligibility in oncology increasingly incorporates genomic biomarkers, and the FDA has been modernizing its policies and review processes for cancer, resulting in an avalanche of new agents and approved indications for targeted therapies.41
There is no dearth of CDS tools to help translate patient data into quality care, nor is there a lack of technical solutions to transmit, harmonize, curate, store, or otherwise manage data. Rather, operationalizing precision oncology is hampered by insufficient adoption of data standards and by failure to share data in computable formats. These problems are not primarily technical, and solving them requires a will to action, with social incentives and disincentives.
For example, NGS test results that are natively structured are sent to ordering physicians and stored in their EHRs, but in a manner that requires costly and data-lossy curation to restructure back into usable information. Considering that the genomic results are reimbursed through federal and private insurers, but the reporting format impedes their utility to clinicians, we call for all molecular diagnostic laboratories to provide structured data as part of routine reporting at the request of ordering physicians and their practices. This would help laboratories meet the definition of interoperability by the 21st Century Cures Act34 to facilitate health information exchange without special effort on the part of the user while avoiding the Act's prohibition of information blocking.
It is notable that on the basis of curated records, structured BRCA1 or BRCA2 gene test results are extremely under-represented among the 10 EHRs used by the more than 50 CancerLinQ practices. This calls into question whether EHRs are technologically competent to house genomic results and whether EHR vendors in general have a will to support precision oncology. It is possible to integrate genomic and clinical data from APIs externally to native EHRs, but greater cooperation is needed from vendors to export clinical data for CDS use and to provide a seamless experience for the clinician, whose attention is tethered to the EHR.
Adoption of data standards, including HL7 FHIR and mCODE, by all the entities that generate, transmit, and receive health information would also facilitate the implementation of precision oncology. The endless customizability of EHRs is at odds with health information technology interoperability and suboptimally supports automated quality measure reporting as required by MIPS.42,43 The dearth of coding for cancer staging, laboratory data, and medications within EHRs could be rectified by adopting widely used standard vocabularies, which would facilitate the computable representation of patients’ oncology records as envisioned by mCODE. To the extent that the complexity of clinicogenomic data is a barrier to enhancing care using NGS technology, we endorse the implementation of solutions in the social sphere to leverage the technical solutions at hand.
Footnotes
Supported by Award No. F31CA239347 from the National Cancer Institute (NCI), National Institutes of Health (J.R.C.), and by NCI Grant No. U01 CA231840 supporting the further development of Sustainable Medical Apps, Reusable Technology, Precision Medical Oncology initiative (J.L.W.).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Provision of study material or patients: Wendy S. Rubinstein, Robert S. Miller
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Jeremy L. Warner
Stock and Other Ownership Interests: HemOnc.org
Consulting or Advisory Role: Westat
No other potential conflicts of interest were reported.
REFERENCES
- 1.Freedman AN, Klabunde CN, Wiant K, et al. Use of next-generation sequencing tests to guide cancer treatment: results from a nationally representative survey of oncologists in the United States. JCO Precis Oncol . doi: 10.1200/PO.18.00169. 10.1200/po.18.00169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dienstmann R, Dong F, Borger D, et al. Standardized decision support in next generation sequencing reports of somatic cancer variants. Mol Oncol. 2014;8:859–873. doi: 10.1016/j.molonc.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamps R, Brandão RD, Bosch BJ, et al. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci. 2017;18:E308. doi: 10.3390/ijms18020308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7:283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology policy statement update: Genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33:3660–3667. doi: 10.1200/JCO.2015.63.0996. [DOI] [PubMed] [Google Scholar]
- 8.Karow J. Tumor-only sequencing may misguide therapy but many labs omit matched control. https://www.genomeweb.com/cancer/tumor-only-sequencing-may-misguide-therapy-many-labs-omit-matched-control
- 9.Damodaran S, Berger MF, Roychowdhury S. Clinical tumor sequencing: Opportunities and challenges for precision cancer medicine. Am Soc Clin Oncol Educ Book. 2015;2015:e175–e182. doi: 10.14694/EdBook_AM.2015.35.e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saigi M, Alburquerque-Bejar JJ, Sanchez-Cespedes M. Determinants of immunological evasion and immunocheckpoint inhibition response in non-small cell lung cancer: The genetic front. Oncogene. 2019;38:5921–5932. doi: 10.1038/s41388-019-0855-x. [DOI] [PubMed] [Google Scholar]
- 12.MacConaill LE. Existing and emerging technologies for tumor genomic profiling. J Clin Oncol. 2013;31:1815–1824. doi: 10.1200/JCO.2012.46.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Center for Medicare and Medicaid Services CMS finalizes coverage of next generation sequencing tests, ensuring enhanced access for cancer patients. https://www.cms.gov/newsroom/press-releases/cms-finalizes-coverage-next-generation-sequencing-tests-ensuring-enhanced-access-cancer-patients
- 14. Center for Medicare and Medicaid Services: Quality Payment Program: MIPS overview—Explore measures and activities. https://qpp.cms.gov/mips/explore-measures/quality-measures?specialtyMeasureSet=Oncology.
- 15. American Society of Clinical Oncology: Quality Oncology Practice Initiative 2019 Reporting Tracks. https://practice.asco.org/sites/default/files/drupalfiles/QOPI-2019-Round-1-Reporting-Tracks-Public-Posting.pdf.
- 16.Bennette CS, Ramsey SD, McDermott CL, et al. Predicting low accrual in the National Cancer Institute’s Cooperative Group clinical trials. J Natl Cancer Inst. 2015;108:djv324. doi: 10.1093/jnci/djv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Lee HJ, Zeng J, et al. Extracting genetic alteration information for personalized cancer therapy from ClinicalTrials.gov. J Am Med Inform Assoc. 2016;23:750–757. doi: 10.1093/jamia/ocw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sahoo SS, Tao S, Parchman A, et al. Trial prospector: Matching patients with cancer research studies using an automated and scalable approach. Cancer Inform. 2014;13:157–166. doi: 10.4137/CIN.S19454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao P, Zhang R, Li J: Comprehensive elaboration of database resources utilized in next-generation sequencing-based tumor somatic mutation detection. Biochim Biophys Acta Rev Cancer. 1872:122-137, 2019. [DOI] [PubMed]
- 20.Gray SW, Hicks-Courant K, Cronin A, et al. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32:1317–1323. doi: 10.1200/JCO.2013.52.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson L-M, Valdez JM, Quinn EA, et al. Integrating next-generation sequencing into pediatric oncology practice: An assessment of physician confidence and understanding of clinical genomics. Cancer. 2017;123:2352–2359. doi: 10.1002/cncr.30581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manolio TA, Fowler DM, Starita LM, et al. Bedside back to bench: Building bridges between basic and clinical genomic research. Cell. 2017;169:6–12. doi: 10.1016/j.cell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanderbilt-Ingram Cancer Center My Cancer Genome. https://www.mycancergenome.org
- 24.Griffith M, Spies NC, Krysiak K, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49:170–174. doi: 10.1038/ng.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol . doi: 10.1200/PO.17.00011. 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landrum MJ, Lee JM, Benson M, et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.US Food and Drug Administration Genetic database recognition decision summary for ClinGen expert curated human variant data. https://www.fda.gov/media/119313/download
- 28.Lawler M, Siu LL, Rehm HL, et al. All the world’s a stage: Facilitating discovery science and improved cancer care through the Global Alliance for Genomics and Health. Cancer Discov. 2015;5:1133–1136. doi: 10.1158/2159-8290.CD-15-0821. [DOI] [PubMed] [Google Scholar]
- 29. doi: 10.1038/s41588-020-0603-8. Wagner AH, Walsh B, Mayfield G, et al: A harmonized meta-knowledgebase of clinical interpretations of cancer genomic variants. https://www.biorxiv.org/content/10.1101/366856v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes KS, Ambinder EP, Hess GP, et al. Identifying health information technology needs of oncologists to facilitate the adoption of genomic medicine: Recommendations from the 2016 American Society of Clinical Oncology Omics and Precision Oncology Workshop. J Clin Oncol. 2017;35:3153–3159. doi: 10.1200/JCO.2017.74.1744. [DOI] [PubMed] [Google Scholar]
- 31.Warner JL, Jain SK, Levy MA. Integrating cancer genomic data into electronic health records. Genome Med. 2016;8:113. doi: 10.1186/s13073-016-0371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arvanitis TN. Semantic interoperability in healthcare. Stud Health Technol Inform. 2014;202:5–8. [PubMed] [Google Scholar]
- 33.Rosenbloom ST, Carroll RJ, Warner JL, et al. Representing knowledge consistently across health systems. Yearb Med Inform. 2017;26:139–147. doi: 10.15265/IY-2017-018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubinstein WS. CancerLinQ: Cutting the Gordian knot of interoperability. J Oncol Pract. 2019;15:3–6. doi: 10.1200/JOP.18.00612. [DOI] [PubMed] [Google Scholar]
- 35. mCODE: Minimal Common Oncology Data Elements. https://mcodeinitiative.org/
- 36. Alterovitz G, Brown J, Chan M, et al: Sync for Genes. https://www.healthit.gov/sites/default/files/sync_for_genes_report_november_2017.pdf. [Google Scholar]
- 37.Hussain MA, Langer SG, Kohli M. Learning HL7 FHIR using the HAPI FHIR server and its use in medical imaging with the SIIM Dataset. J Digit Imaging. 2018;31:334–340. doi: 10.1007/s10278-018-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandl KD, Mandel JC, Murphy SN, et al. The SMART Platform: Early experience enabling substitutable applications for electronic health records. J Am Med Inform Assoc. 2012;19:597–603. doi: 10.1136/amiajnl-2011-000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warner JL, Rioth MJ, Mandl KD, et al. SMART precision cancer medicine: A FHIR-based app to provide genomic information at the point of care. J Am Med Inform Assoc. 2016;23:701–710. doi: 10.1093/jamia/ocw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller RS, Wong JL. Using oncology real-world evidence for quality improvement and discovery: The case for ASCO’s CancerLinQ. Future Oncol. 2018;14:5–8. doi: 10.2217/fon-2017-0521. [DOI] [PubMed] [Google Scholar]
- 41. Gottlieb S: Remarks to the American Society of Clinical Oncology (ASCO) Annual Meeting: June 2, 2018. https://www.fda.gov/news-events/speeches-fda-officials/remarks-american-society-clinical-oncology-asco-annual-meeting-06022018.
- 42. Bernstam E, Warner J, Krauss J, et al: Quantifying interoperability: An analysis of oncology practice electronic health record data variability. J Clin Oncol 37, 2019 (suppl; abstr e18080)
- 43. Schorer A, Koskimaki J, Miller R, et al: Electronic but overly eclectic: Disciplined EHR data management is needed to automate MIPS reporting. J Clin Oncol 37, 2019 (suppl; abstr e18074)
- 44. National Comprehensive Cancer Network: NCCN Guidelines for Treatment of Cancer by Site. https://www.nccn.org/professionals/physician_gls/default.aspx#site.
- 45. Variant Interpretation for Cancer Consortium: Variant Interpretation for Cancer Consortium Meta-Knowledgebase. https://search.cancervariants.org/#BRAF%20V600E.
- 46. doi: 10.1016/j.jmoldx.2016.10.002. Li M, Datto M, Duncavage E: Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19:4-23, 2017. [DOI] [PMC free article] [PubMed]
- 47. Variant Interpretation for Cancer Consortium: The VICC meta-knowledgebase. http://docs.cancervariants.org/en/latest/
- 48. Human Variation Genome Society: Sequence variant nomenclature. https://varnomen.hgvs.org.