

Abstract
Purpose
Desmoplastic small round blue-cell tumors (DSRCTs) are sarcomas that contain the t(11;22) (p13;q12) translocation EWS-WT1 fusion protein. Because this is a rare tumor type, prospective clinical trials in DSRCT are challenging. Patients are treated in a manner similar to those with Ewing sarcoma; however, differences in prognosis and clinical presentation suggest fundamental differences in biology and potentially different therapeutic implications. This study aimed to characterize the molecular characteristics of DSRCT tumors to explore unique therapeutic options for this extremely rare and aggressive cancer type.
Methods
Thirty-five DSRCT tumors were assessed using next-generation sequencing, protein expression (immunohistochemistry), and gene amplification (chromogenic in situ hybridization or fluorescence in situ hybridization). Three patients had tumor mutational load, which was calculated as somatic nonsynonymous missense mutations sequenced with a 592-gene panel. Gene expression data were obtained for an additional seven DSRCT tumors. Molecular alterations were compared with 88 Ewing sarcomas.
Results
The most common alterations that distinguished DSRCTs from Ewing sarcoma included higher androgen receptor (AR), TUBB3, epidermal growth factor receptor, and TOPO2A expression. Independent analysis by RNA sequencing confirmed higher AR expression from an independent data set of EWS-WT1 fusion–positive DSRCTs compared with Ewing sarcoma and a pan-cancer analysis. DSRCTs had somatic mutations that were identified in TP53 and FOXO3, averaged five mutations per megabase, and no programmed death-ligand 1 expression was detected in any DSRCT samples.
Conclusion
The current analysis provides the first comparative analysis, to our knowledge, of molecular aberrations that distinguish DSRCT from Ewing sarcoma. High AR expression seems to be a defining event in these malignancies, and additional investigation of the responsiveness of AR inhibitors in this disease is encouraged.
INTRODUCTION
Desmoplastic small round blue-cell tumors (DSRCTs) originate from a cell with multilineage potential and are defined by a molecular hallmark with EWS-WT1 reciprocal translocation.1 DSRCTs are rare, highly aggressive mesenchymal tumors, with only 200 to 450 affected patients with the disease described to date.1,2 Ewing sarcoma and DSRCTs are treated in a similar manner in the clinic; however, DSRCTs have been represented in limited numbers in sarcoma studies. The clinical presentations of these diseases are different, with important comparisons being the site of disease presentation and poor prognosis in DSRCT groups.
Despite aggressive therapy, median survival ranges from 17 to 25 months,2,3 and the 5-year survival rate remains around 15%3 with higher survival reported among those who underwent removal of at least 90% of the tumor with an absence of extraperitoneal metastasis.4 Almost all of these tumors contain the t(11;22) (p13;q12) translocation that fuses EWS with WT1 leading to production of a chimeric protein with transcriptional regulatory activity. It is likely that EWS-WT1 functions as a transcription factor via WT1 targets.2,5,6 The tumor has a predilection for developing in the abdominal and pelvic cavity of young adult men in 88% to 97% of cases,2,5 whereas Ewing sarcoma commonly occurs in the axial skeleton.7 Despite similar histologic morphology, differences in disease presentation and prognosis likely reflect the impact of different biologic factors, including differences in the transcriptional effects of the EWS-WT1 fusion relative to the EWS-FLI1 fusion observed in Ewing sarcoma.
Although there is no standard clinical management strategy for DSRCT, treatment usually includes neoadjuvant high-dose cyclophosphamide, doxorubicin, and vincristine, alternating with ifosfamide and etoposide chemotherapy combined with aggressively attempted R0 resection.4,8,9 Recent accelerated US Food and Drug Administration (FDA) approval in October 2016 of the compound olaratumab, a platelet-derived growth factor receptor A inhibitor, was based on a highly significant improvement in overall survival of 26.5 months in soft tissue sarcomas of histologic subtypes for which an anthracycline-containing regimen may be appropriate. However, only one patient in the phase II olaratumab study had round blue-cell sarcoma, and that patient was negative for the EWS translocation.10 Patients with DSRCTs are poorly represented in clinical trials as a result of the rare nature of the disease. A list of ongoing clinical trials in this disease is provided in the Data Supplement.
The aim of the current study was to understand the molecular characteristics of DSRCTs by analyzing 35 patients with molecularly and immunohistochemically profiled tumors and to compare these with Ewing sarcoma to explore therapeutic options and potential oncogenic vulnerabilities for this extremely rare and aggressive cancer type. We also analyzed additional transcriptomic gene expression data in this disease. To our knowledge, the assembled cohort is the largest analysis of DSRCTs with molecular data to date.
METHODS
Immunohistochemistry and Molecular Sequencing
Thirty-five DSRCT tumors were assessed with immunohistochemistry (IHC) and in situ hybridization, and we performed next-generation sequencing on full slides of formalin-fixed, paraffin-embedded tumor samples (Caris Life Sciences, Phoenix, AZ). DNA from formalin-fixed, paraffin-embedded samples was sequenced using the NextSeq (592 genes selected on the basis of the COSMIC database; Agilent Sure Select XT; Illumina, San Diego, CA) and MiSeq (47 genes; TruSeq), to evaluate mutation and gene amplification. The full list of markers surveyed is available.11 Twenty-four tumors were sequenced with the 45-gene panel, and 11 tumors were sequenced with the 592-gene panel. Tumor mutational load was calculated as somatic nonsynonymous missense mutations sequenced with a 592-gene panel. Molecular alterations were compared with 88 Ewing sarcomas. AR27 antibody was used for androgen receptor (AR) expression, with 1+ (10%) and 2+ (30%) used as cutoffs. The primary antibody used against programmed death-ligand 1 (PD-L1) was SP142 (Spring Biosciences, Pleasanton, CA). Every biomarker that was assessed with IHC was compared with a predetermined cutoff, percent staining, and staining intensity. Full details of the scoring of staining intensity and percent staining is provided in the Data Supplement. Additional data for AR and epidermal growth factor receptor (EGFR) IHC in other tumors, including prostate cancer, lung cancer, breast cancer, kidney cancer, endometrial cancer, and glioblastoma, were obtained from a larger cohort of 127,000 pan-cancer tumor samples analyzed.
We used χ2 tests for comparison, and statistical significance was determined as P < .05. All tests were optimized and validated and met the standards and requirements of the Clinical Laboratory Improvement Amendments/College of American Pathologists and the International Organization for Standardization. This retrospective analysis used previously collected, de-identified data and was deemed to be exempt by the Western Regional Review Board, the institutional review board of record for Caris Life Sciences.
Gene Set Enrichment Analysis
Enrichment analysis shown in Figure 1 was obtained using the transcriptional signatures and data sets described below and single sample gene set enrichment analysis (ssGSEA).12 The ssGSEA approach allows for computing the enrichment score of a gene set in individual samples without phenotype labels and represents the degree to which the genes in a particular gene set are coordinately upregulated or downregulated within each sample. In this manner, ssGSEA projects a single sample gene expression profile from the space of single genes onto the space of gene sets.
Fig 1.
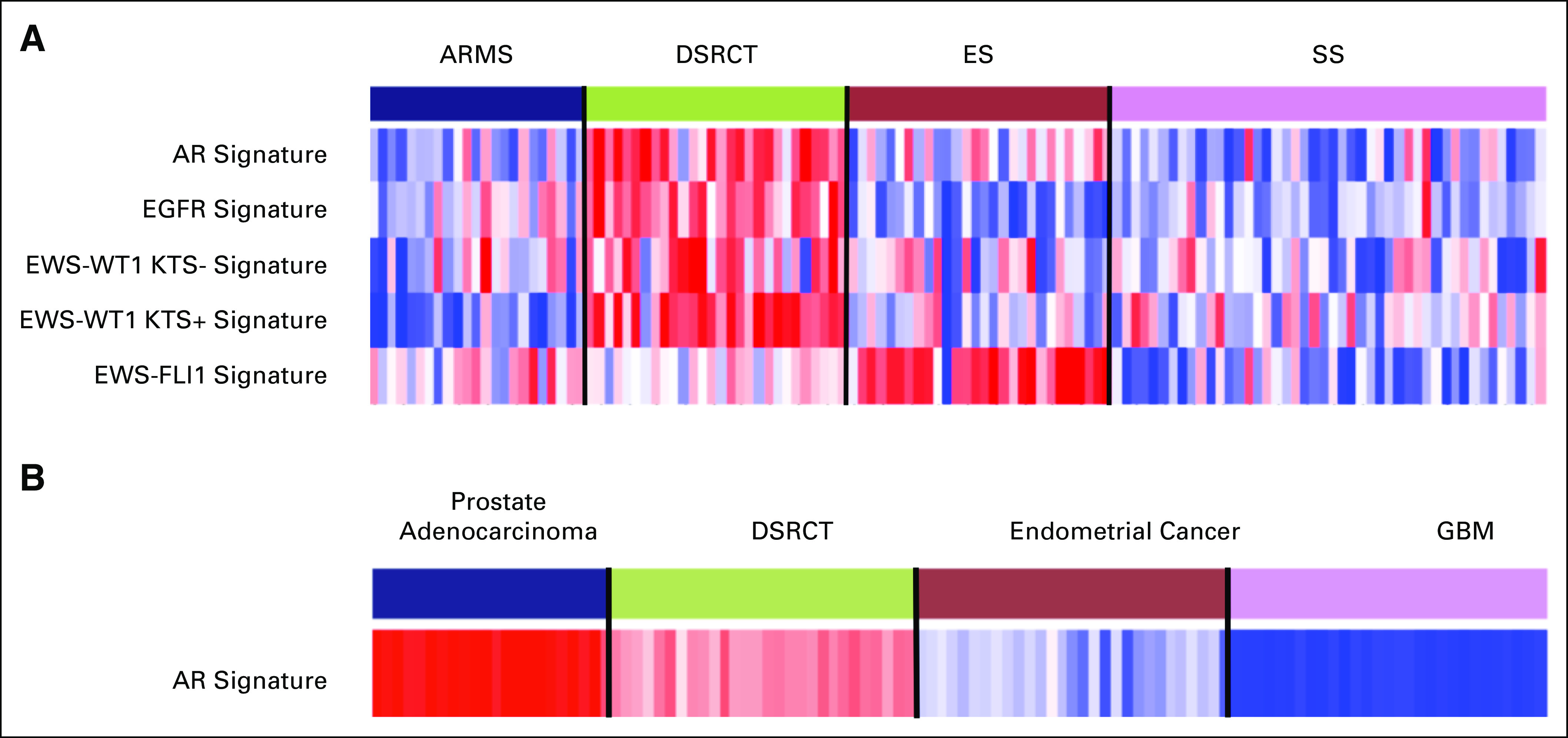
(A) Enrichment scores (single-sample gene set enrichment analysis) of selected transcriptional signatures across alveolar rhabdomyosarcoma, desmoplastic small round blue cell tumors, Ewing sarcoma, and synovial sarcoma. Desmoplastic small round blue-cell tumor (DSRCT) sarcomas are clearly overenriched in androgen receptor (AR), epidermal growth factor receptor (EGFR), and EWS-WT1 (KTS positive/negative) signatures compared with those of other sarcomas. Variant fusion isoforms of KTS are generated as a result of alternative mRNA splicing. Some of these cases express full-length WT1 or have variant transcripts (KTS positive), which results in atypical staining patterns. KTS-positive variant has transcriptional targets that are different than those of the KTS-negative isoform. Ewing sarcomas are enriched in the EWS-FLI1 signature as expected. (B) Enrichment scores of the AR signature in DSRCTs and three others cancer types—prostate, endometrial, and glioblastoma multiforme (GBM). The highest level of enrichment of the AR signature takes place in prostate cancer. DSRCT sarcomas display increased levels of AR enrichment compared with endometrial and GBM cancers. ARMS, alveolar rhabdomyosarcoma; ES, Ewing sarcoma; SS, synovial sarcoma.
The signature for AR is the HALLMARK_ANDROGEN_RESPONSE gene set from the Molecular Signatures Database hallmark subcollection.13 The EGFR signature corresponds to the EGFR_UP.V1_UP/DN gene sets from the Molecular Signatures Database subcollection C6.14 The EWS-WT1 (KTS positive/negative) signatures were generated from the GEO data set GSE53301.15 The EWS-FLI1 signature was made from the 20 genes listed in Figure 1 from Mendiola et al.16
Transcriptional Data Sets
The multiple sarcoma data set shown in Figure 1A is a subset of the data set from Filion et al.17 Sarcoma samples in Figure 1B are the DSRCT subset from Filion et al.17 Prostate samples, shown in Figure 1B, consisted of 22 primary prostate cancer samples from the GEO data set GSE32269.18 Endometrial samples are 30 samples from the endometrial cancer data set from Raeder et al.19 Glioblastoma multiforme samples consisted of a subset of 30 samples from the glioblastoma multiforme data set from Verhaak et al.20
RESULTS
Demographic data from the 35 patients whose tumors underwent clinical molecular testing are provided in the Data Supplement. The average age of patients with DSRCTs was 30.4 years. (range, 7 to 69 years), and the average age of patients with Ewing sarcoma was 28 years (range, 8 to 62 years). Thirty (86%) of 35 patients with DSRCTs were male, which is consistent with prior reports,5 compared with only 38 (57%) of 50 of the patients with Ewing sarcoma tumors (P = .003). A distribution of anatomic sites from which the DSRCTs and Ewing sarcoma tumors were resected or biopsied is shown in the Data Supplement. Most patients with DSRCTs had abdominal and pelvic disease, whereas most patients with Ewing sarcoma had lung and bone involvement, which is consistent with the known distribution pattern of these tumors.21
In the 35 DSRCTs, increased expression of TOP2A was observed in 63% of cases, TOPO1 in 63%, PTEN in 62%, AR in 59%, and EGFR in 42% of tumors. Increased expression of TUBB3 was observed in 56% of tumors, MGMT in 55%, TS in 52%, RRM1 in 43%, and ERCC1 in 24% (Fig 2A). Comparing DSRCTs with Ewing sarcoma tumors, we found that both AR (59% v 3%; P = 1.7E10) and TUBB3 (56% v 29%; P = .03) were expressed in significantly more DSRCTs (Fig 2B). No significant differences were observed between the two tumor types for the other IHC markers. A full list of all of the patients with IHC staining is provided in the Data Supplement, which includes a description of staining thresholds.
Fig 2.
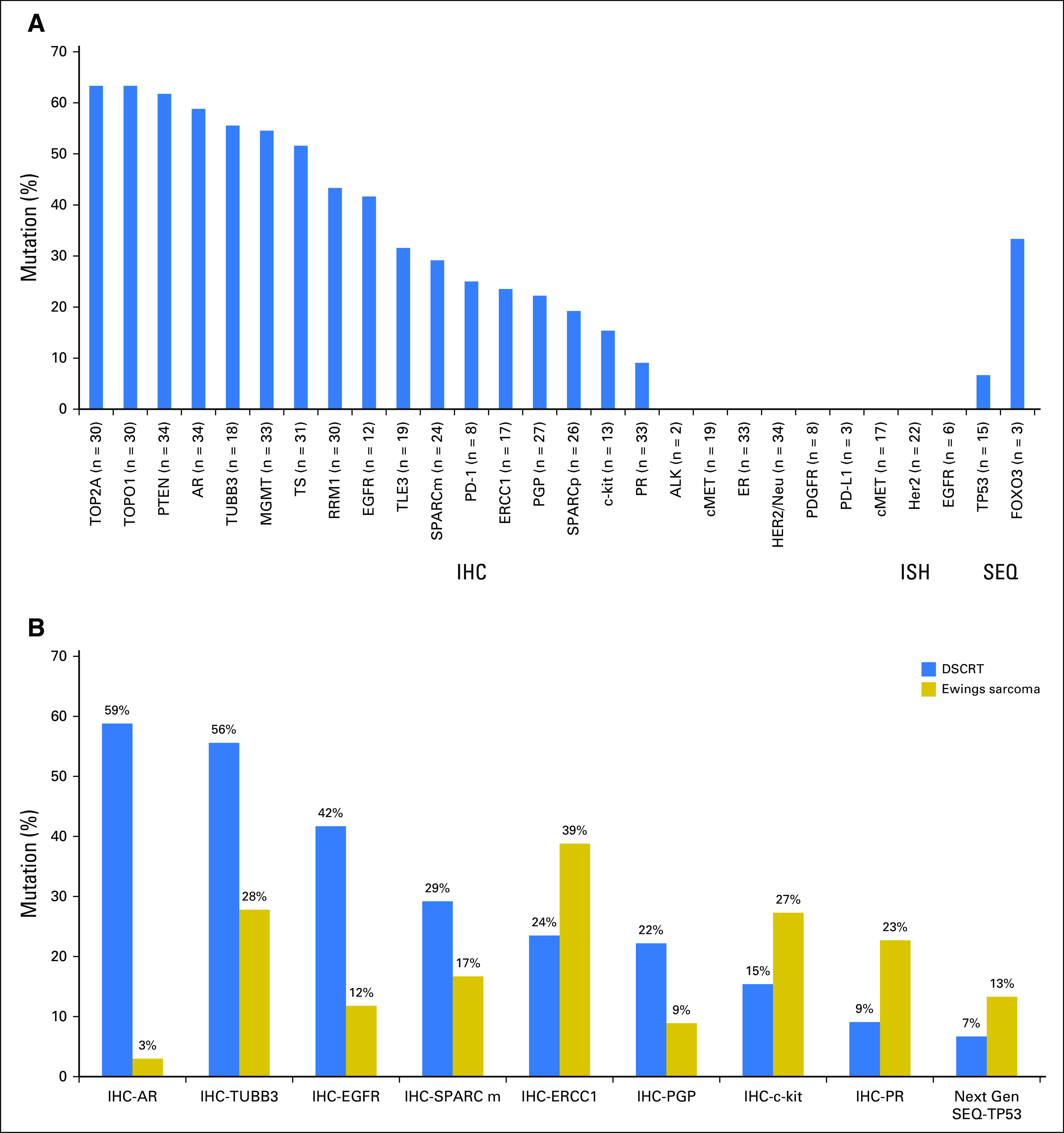
(A) Thirty-five desmoplastic small round blue-cell tumors (DSRCTs) went through molecular profiling using a commercial, multiplatform profiling service (Caris Life Sciences). Specific tests performed included sequencing (next-generation sequencing [SEQ]), protein expression (immunohistochemistry [IHC]), and gene amplification (chromogenic in situ hybridization or fluorescence in situ hybridization [ISH]). Twelve tumors were sequenced with the 45-gene panel, and three tumors were sequenced with the 592-gene panel. The only pathogenic or presumed pathogenic mutations observed were one TP53 mutation (G245S) and one FOXO3 mutation (L382fs). The three tumors with the 592-gene panel carried tumor mutational loads of 4, 6, and 8 mutations per megabase. (B) Comparison of selected biomarker features of DSRCTs (N = 35) and Ewing sarcoma tumors (N = 88). Shown are biomarkers with incidences that are > 50% different between the two tumor types. Connective lines indicate statistical significance by Fisher exact test (P < .05).
Given that both the AR and EGFR pathways can be effectively targeted with FDA-approved drugs, we sought to further evaluate the activity of these pathways in DSRCTs. We obtained gene expression profiles for 28 DSRCTs as well as profiles for Ewing sarcoma, alveolar rhabdomyosarcomas, and synovial sarcoma for comparison with other primitive sarcomas.17 Performing ssGSEA, we found that the majority of DSRCTs was enriched for the AR signature, which indicated that these tumors coordinately upregulated genes that are known to be highly expressed when the AR pathway is activated (Fig 1A). Similarly, the EGFR signature was also enriched in the majority of DSRCTs. None of the other sarcomas displayed consistent AR or EGFR enrichment. As expected, Ewing sarcoma tumors were enriched for the EWS-FLI1 signature, and DSRCTs were enriched for the EWS-WT1 signature, which is consistent with the translocation event pathognomonic for each tumor.
To evaluate the significance of AR pathway activation in DSRCTs, we made comparisons with tumors that are known to be driven by the AR pathway. Using ssGSEA, we found that the degree of enrichment for the AR signature in DSRCTs was less than that of prostate adenocarcinoma, but was greater than that of endometrial cancer or glioblastoma, two tumor types that have low AR expression in our data set (9% [n = 57 tumors] in the glioblastoma cohort, and 22% [n = 1,034 tumors] in the endometrial cancer cohort. Compared with 59% AR expression in DSRCT, IHC results indicated that AR expression was most frequent in prostate cancer (94% [n = 1,340 tumors]); however, DSRCT was greater than any other tumor type, including breast cancer (50% [n = 1,962 tumors]), kidney cancer (30% [n = 40 tumors]), and the pan-cancer analysis, which demonstrated < 30% expression (n = 127,000). A pan-cancer analysis of EGFR expression demonstrated a percentage of expression (53% [n > 20,000]) that was similar to the 42% observed in DSRCT. Furthermore, TUBB3 expression (51% [n = 66,000]) and TOP2A (69% [n = 77,000]) was similar to that observed in DSRCT.
PD-L1 expression (SP142) was not detected in tumors tested (zero of four), whereas programmed death 1 (PD-1) staining on tumor-infiltrating lymphocytes (TILs) was found in 25% of tumors tested (two of eight). PD-L1 expression on tumor cells in Ewing sarcoma was absent (zero of 10), and PD-1 expression on TILs was observed in 32% (six of 19) of Ewing sarcoma tumors (Fig 2). The three tumors with the 592-gene panel carried a tumor mutational load of 4, 6, and 8 mutations per megabase. Tumor mutational load was calculated in 11 Ewing sarcoma tumors, and mean tumor mutational load was five mutations per megabase (range, three to eight mutations).
Next-generation sequencing revealed a synonymous TP53 G245G mutation and a FOXO3 mutation (L382fs) in DSCRT, whereas six TP53 mutations (13%), two APC mutations (L1129S and I1307K), one BRCA1 (c.301+1G > A), and one CTNNB1 (T41A) mutation were identified in Ewing sarcoma. There was no copy number amplification identified in DSRCT samples for cMET, HER2 (human epidermal growth factor receptor 2), and EGFR with in situ hybridization (Data Supplement).
DISCUSSION
Our understanding of the biology of DSRCTs and their distinction from other sarcomas is evolving. Ewing sarcoma–like tumors and desmoplastic small-cell tumors can be difficult to differentiate morphologically or by light microscopy alone, and differences across disease may be linked to unique transcriptional regulation that is influenced by different fusion partners of the EWS gene.22-24 From our targeted analysis, apart from the canonical EWS-WT1 fusion, some major differences between DSRCT and Ewing sarcoma reflect those between AR and EGFR expression.
Significantly higher AR expression in DSRCTs compared with Ewing sarcoma tumors suggest the possibility of a mechanistic link between EWS-WT1 activation and androgen receptor expression. Various EWS-WT1 proteins regulate the promoter activity of EGR-1, a zinc finger transcription factor that contributes to AR pathway activation.25,26 EGR-1 knockdown by small interfering RNA inhibited activity of the AR signaling pathway.27 Evidence for AR activation in DSRCTs was observed by IHC as well as the coordinate regulation of the AR-responsive transcriptome. The degree of AR enrichment in DSRCTs was higher than that of many other tumors, and additional work is needed to understand the clinical sensitivity of these tumors to AR inhibition.
FDA-approved novel AR inhibitors, including enzalutamide, and CYP17A1 inhibitors, including abiraterone, will need to be explored and provide a potential opportunity for continued drug expansion in this space. Fine et al28 have previously demonstrated by IHC that heavily pretreated patients have significant AR expression in 37% (10 of 27) of DSRCTs. Three of the six patients who were treated with combined androgen blockade attained a clinical benefit for 3 to 6 months. The three patients who had benefit from complete androgen blockade (CAB) had normal testosterone levels before CAB, whereas the three who did not experience a response had baseline castrate levels of testosterone.28
In vitro studies that evaluated the functional status of AR using Western blot analysis and the stimulation of growth by dihydrotestosterone in MTT calorimetric assays in 27 patient tumors suggested that AR in DSRCTs is functional, and inhibition of basal and testosterone-stimulated growth by flutamide could be achieved.28
A phase IB study in prostate cancer has combined treatment with enzalutamide and docetaxel chemotherapy and has resulted in a ≥ 50% decline from baseline in prostate-specific antigen in 19 of 20 patients, and a phase II study is ongoing in prostate cancer (ClinicalTrials.gov identifier: NCT02685267).29 Given the poor outcomes found with conventional therapy, the role of combinatorial therapy with AR inhibitors alone or in combination with chemotherapy in DSRTC deserves additional clinical investigation.
Tumor mutational load may affect response rates to immunotherapy in melanoma and non–small-cell lung cancers.30 Unfortunately, single-agent anti–PD-1 antibodies, to date, have had limited efficacy across sarcomas, and an ongoing phase II study (SARC028) is evaluating the role of pembrolizumab across various sarcoma histologies (ClinicalTrials.gov identifier: NCT02301039).31 None of the patients in our cohort had identifiable tumoral PD-L1 expression by SP142 antibody testing, and the significance of PD-1–positive TILs is unclear at this time.
In conclusion, there are distinguishing molecular characteristics between DSRCTs and Ewing sarcoma. AR expression may facilitate novel clinical trial designs and drug development. Additional work will be focused on defining the prevalence of AR expression in this disease in larger cohorts and the functional significance of targeting the AR pathway in this malignancy.
Appendix
Transcriptional Signatures
The signature for androgen receptor (AR) is the HALLMARK_ANDROGEN_RESPONSE gene set from the Molecular Signatures Database hallmark subcollection (msigdb.org).16 The epidermal growth factor receptor signature corresponds to the EGFR_UP.V1_UP/DN gene sets from Molecular Signatures Database subcollection C6 (msigdb.org).14 The EWS-WT1 (KTS positive/negative) signatures were generated from the GEO data set GSE53301.15 The EWS-FLI1 signature was made from the 20 genes listed in Figure 1 of Mendiola et al (Mendiola M, et al: Int J Cancer 118:1381-1389, 2006).
Transcriptional Data Sets
The multiple sarcoma data set shown in Figure 1A is a subset of the data set from Filion et al.17 Sarcoma samples in Figure 1B are the desmoplastic small round blue-cell tumor subset of Filion et al.17 Prostate samples in Figure 1B consisted of 22 primary prostate cancer samples from the GEO data set GSE32269.18 Endometrial samples are 30 samples from the endometrial cancer data set from Raeder et al.19 Glioblastoma multiforme samples consisted of a subset of 30 samples from the glioblastoma multiforme data set from Verhaak et al.20
Gene Set Enrichment Analysis
Enrichment analysis shown in Figures 1A and 1B was obtained using the transcriptional signatures and data sets described above and single-sample gene set enrichment analysis (ssGSEA).12 The ssGSEA approach allows for computing the enrichment score of a gene set in individual samples without phenotype labels and represents the degree to which the genes in a particular gene set are coordinately upregulated or downregulated within each sample. In this manner, ssGSEA projects a single sample gene expression profile from the space of single genes onto the space of gene sets.
Footnotes
Funded by University of California San Diego Moores Cancer Center Institutional Funds and National Institutes of Health Grants No. U01-CA217885 and P30-CA023100 (P.T.).
AUTHOR CONTRIBUTIONS
Conception and design: Ajaz Bulbul, Pablo Tamayo, Hatim Husain
Financial support: Hatim Husain
Administrative support: Hatim Husain
Provision of study materials or patients: Ajaz Bulbul, Joanne Xiu, Hatim Husain
Collection and assembly of data: All authors
Data analysis and interpretation: Ajaz Bulbul, John Paul Shen, Pablo Tamayo, Hatim Husain
Manuscript writing:All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Ajaz Bulbul
Consulting or Advisory Role: Pfizer, AstraZeneca
Travel, Accommodations, Expenses: Pfizer, AstraZeneca
John Paul Shen
No relationship to disclose
Joanne Xiu
Employment: Caris Life Sciences
Pablo Tamayo
No relationship to disclose
Hatim Husain
Consulting or Advisory Role: AstraZeneca, AbbVie, Foundation Medicine
Speakers' Bureau: AstraZeneca, Merck, Bristol-Myers Squibb
Research Funding: Pfizer (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Merck, Bristol-Myers Squibb, Foundation Medicine, AbbVie
REFERENCES
- 1.Honoré C, Amroun K, Vilcot L, et al. : Abdominal desmoplastic small round cell tumor: Multimodal treatment combining chemotherapy, surgery, and radiotherapy is the best option. Ann Surg Oncol 22:1073-1079, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Dufresne A, Cassier P, Couraud L, et al. : Desmoplastic small round cell tumor: Current management and recent findings. Sarcoma 2012:714986, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lal DR, Su WT, Wolden SL, et al. : Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40:251-255, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Hayes-Jordan A, Anderson PM: The diagnosis and management of desmoplastic small round cell tumor: A review. Curr Opin Oncol 23:385-389, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Lae ME, Roche PC, Jin L, et al. : Desmoplastic small round cell tumor: A clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol 26:823-835, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Kushner BH, LaQuaglia MP, Wollner N, et al. : Desmoplastic small round-cell tumor: Prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14:1526-1531, 1996 [DOI] [PubMed] [Google Scholar]
- 7.Grier HE, Krailo MD, Tarbell NJ, et al. : Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348:694-701, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Farhat F, Culine S, Lhommé C, et al. : Desmoplastic small round cell tumors: Results of a four-drug chemotherapy regimen in five adult patients. Cancer 77:1363-1366, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Hayes-Jordan A, LaQuaglia MP, Modak S: Management of desmoplastic small round cell tumor. Semin Pediatr Surg 25:299-304, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tap WD, Jones RL, Van Tine BA, et al. : Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: An open-label phase 1b and randomised phase 2 trial. Lancet 388:488-497, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caris Molecular Intelligence : Profiling menu. https://www.carismolecularintelligence.com/profiling-menu
- 12.Barbie DA, Tamayo P, Boehm JS, et al. : Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462:108-112, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liberzon A, Birger C, Thorvaldsdóttir H, et al. : The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1:417-425, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creighton CJ, Hilger AM, Murthy S, et al. : Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res 66:3903-3911, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Kang HJ, Park JH, Chen W, et al. : EWS-WT1 oncoprotein activates neuronal reprogramming factor ASCL1 and promotes neural differentiation. Cancer Res 74:4526-4535, 2014 [DOI] [PubMed] [Google Scholar]
- 16.Mendiola M, Carrillo J, García E, et al. : The orphan nuclear receptor DAX1 is up-regulated by the EWS/FLI1 oncoprotein and is highly expressed in Ewing tumors. Int J Cancer 118:1381-1389, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Filion C, Motoi T, Olshen AB, et al. : The EWSR1/NR4A3 fusion protein of extraskeletal myxoid chondrosarcoma activates the PPARG nuclear receptor gene. J Pathol 217:83-93, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai C, Wang H, He HH, et al. : ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest 123:1109-1122, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raeder MB, Birkeland E, Trovik J, et al. : Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS One 8:e54873, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhaak RG, Hoadley KA, Purdom E, et al. : Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98-110, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulbul A, Fahy BN, Xiu J, et al. : Desmoplastic small round blue cell tumor: A review of treatment and potential therapeutic genomic alterations. Sarcoma 2017:1278268, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antonescu CR, Gerald WL, Magid MS, et al. : Molecular variants of the EWS-WT1 gene fusion in desmoplastic small round cell tumor. Diagn Mol Pathol 7:24-28, 1998 [DOI] [PubMed] [Google Scholar]
- 23.Gerald WL, Rosai J, Ladanyi M: Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci USA 92:1028-1032, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Nau MM, Yeh JC, et al. : Molecular heterogeneity and function of EWS-WT1 fusion transcripts in desmoplastic small round cell tumors. Clin Cancer Res 6:3522-3529, 2000 [PubMed] [Google Scholar]
- 25.Sukhatme VP, Cao XM, Chang LC, et al. : A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 53:37-43, 1988 [DOI] [PubMed] [Google Scholar]
- 26.Milbrandt J: A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science 238:797-799, 1987 [DOI] [PubMed] [Google Scholar]
- 27.Yang SZ, Abdulkadir SA: Early growth response gene 1 modulates androgen receptor signaling in prostate carcinoma cells. J Biol Chem 278:39906-39911, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Fine RL, Shah SS, Moulton TA, et al. : Androgen and c-Kit receptors in desmoplastic small round cell tumors resistant to chemotherapy: Novel targets for therapy. Cancer Chemother Pharmacol 59:429-437, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Morris MJ, Rathkopf DE, Novotny W, et al. : Phase Ib study of enzalutamide in combination with docetaxel in men with metastatic castration-resistant prostate cancer. Clin Cancer Res 22:3774-3781, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Champiat S, Ferté C, Lebel-Binay S, et al. : Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. OncoImmunology 3:e27817, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgess MA, Crowley J, Reinke DK, et al. : SARC 028: A phase II study of the anti-PD1 antibody pembrolizumab (P) in patients (Pts) with advanced sarcomas. J Clin Oncol 33, 2015 (suppl; abstr TPS10578) [Google Scholar]