

Abstract
In this precision oncology era, where molecular profiling at the individual patient level becomes increasingly accessible and affordable, more and more clinical trials are now driven by biomarkers, with an overarching objective to optimize and personalize disease management. As compared with the conventional clinical development paradigms, where the key is to evaluate treatment effects in histology-defined populations, the choices of biomarker-driven clinical trial designs and analysis plans require additional considerations that are heavily dependent on the nature of biomarkers (eg, prognostic or predictive, integral or integrated) and the credential of biomarkers’ performance and clinical utility. Most recently, another major paradigm change in biomarker-driven trials is to conduct multi-agent and/or multihistology master protocols or platform trials. These trials, although they may enjoy substantial infrastructure and logistical advantages, also face unique operational and conduct challenges. Here we provide a concise overview of design options for both the setting of single-biomarker/single-disease and the setting of multiple-biomarker/multiple-disease types. We focus on explaining the trial design and practical considerations and rationale of when to use which designs, as well as how to incorporate various adaptive design components to provide additional flexibility, enhance logistical efficiency, and optimize resource allocation. Lessons learned from real trials are also presented for illustration.
INTRODUCTION
In the past 10 to 15 years, cancer clinical trials have experienced some important paradigm changes to embrace the era of precision oncology as defined by various biomarkers. The central hypothesis driving this movement is that by integrating the right biomarker information we can properly select, or at least enrich, trial cohorts for patients who are most likely to benefit from a particular therapy. Multiple factors collectively contribute to this movement—our knowledge about oncogenic pathways has been greatly advanced; high-throughput screening and other drug discovery developments have made a lot more candidate agents available for evaluation; and the rapid development of various omics-based technologies, especially the increasing availability of next-generation genomic sequencing, makes it more feasible and affordable to incorporate biomarker information into clinical trials. All of these call for novel biomarker-driven trial designs to expedite the clinical development of multiple treatment agents and newly defined patient populations.
In this review, we use biomarker to generically describe any characterizations of biologic molecules or diagnostic tests carried out on DNA, RNA, proteins, and metabolites from blood, body fluids, or tissues for diagnosis purposes including disease confirmation, staging, subtyping, and so on. Under this working definition, the presence of some actionable mutation, such as a mutation in epidermal growth factor receptor (EGFR) measured at the protein level, is viewed as a single biomarker; a DNA-based multiplex genotyping that simultaneously determines multiple actionable mutations such as EGFR, KRAS, or EML4-ALK is viewed as multiple biomarkers.
Compared with the traditional paradigm, there are even greater consequences of asking the right questions (or wrong questions) in these biomarker-driven clinical trials. For example, although often there is good biologic rationale to consider biomarker-negative (M-negative) patients unlikely or less likely to benefit from the new (targeted) therapy, the clinical evidence of whether the potential treatment benefit is confined only in biomarker-positive (M-positive) patients may or may not be strong. Moreover, the development of a validated companion biomarker, including establishing a widely accepted partition or cutoff value to determine whether the biomarker is positive or negative, is sometimes lagging behind the development of novel therapeutic agents. Therefore, as we simultaneously evaluate new treatments and identify new patient populations defined by the corresponding biomarkers, it is crucial to properly tailor the trial designs and prioritize research questions on the basis of the development stage of the biomarker and the credentials of its clinical utility.
Context
Key Objective
In the past few decades, clinical trials have experienced a major paradigm shift, aiming to incorporate ever-growing tumor biomarker information and evaluate biomarker-defined patient cohorts, while accelerating the clinical development process simultaneously. What are the key considerations when designing and conducting biomarker-driven clinical trials?
Knowledge Generated
We present an overview of the rationale, trial types, key design elements and features, and practical considerations for commonly used biomarker-driven clinical trials. Examples of trials conducted and the lessons learned are also presented.
Relevance
When properly designed and implemented with adequate resources, biomarker-driven clinical trials may efficiently and effectively generate evidence on biomarker-based personalized disease management. Clinicians and clinical trialists should think thoroughly and critically if and how to design and conduct these trials.
The demand for trial development and conduct has become more daunting than ever, as high-throughput molecular profiling becomes more accessible. Rather than mounting separate trials, an important paradigm shift to expedite clinical development is to establish the so-called master protocol or platform trial—a program of trial development and conduct implemented with an up-front molecular screen and enrollment infrastructure into subtrials across multiple biomarkers and/or multiple disease types—to enhance logistic and regulatory efficiency.
Some important and useful concepts in development of biomarkers for clinical utility are first briefly introduced here and expanded on in the next sections. Throughout this review, we assume biomarkers of interest already have good analytical validity (eg, they can be accurately, reliably, and reproducibly measured).1,2 A biomarker is called prognostic if it is associated with disease prognosis regardless of treatment type and is called predictive when it exerts prognostic influence differentially according to different treatments. It is desirable to have a biomarker (either prognostic or predictive) with good clinical utility (ie, the biomarker can reliably prompt clinical actions that benefit patients). According to Dancey et al,3 biomarkers are integral when they are inherent in the study design from the onset and must be performed in real time to establish eligibility, identify the correct stratum for stratified enrollment, or assign treatment. In other words, to make a biomarker usable for clinical trial setting, an integral biomarker should be fully developed and validated (eg, the cutoff that dichotomizes the continuous marker measurement is fixed), with a rapid assay turnaround time. Alternatively, biomarkers are considered integrated if they are used to test specific hypotheses with defined objectives and statistical analysis plans in the study. Fast assay turnaround time is desirable but not required for integrated biomarkers.
To design more efficient trials and overcome the aforementioned challenges, the idea of adaptive designs has been promoted, and aspects of this approach play an important role in virtually all existing biomarker-driven trials. Although modern oncology trial design has promoted adaptive designs as a new aspect of clinical trials, we note that adaptive elements have played a long-standing role in oncology clinical trials, dating back to early multistage design concepts4,5 and continuing in the rich development of group sequential monitoring methods that permit early efficacy or futility stopping.6 Also, both traditional and newer phase I oncology trial designs are by their nature adaptive to accumulating data. We will discuss the rationale and features of established as well as newer adaptive design elements as they arise in respective biomarker-driven trial designs.
Finally, a new nomenclature that generally describes trial constructs has been introduced, attempting to describe heuristically the structure, particularly when multiple biomarkers and/or treatment agents are considered. The term basket trial refers to a trial with an agent tested among multiple disease types sharing a common molecular feature or target identified by biomarker(s). In contrast, an umbrella trial describes the case where, for a common disease entity, multiple agents may be investigated in conjunction with specific molecular targets and biomarkers. Either of these designs may be implemented under a master protocol or platform trial, as an expedient method of execution relative to multiple distinct trials. We note that all of these terms lack precision, and thus the specific study features will provide more detail as to the structure.
This article aims to provide a concise and up-to-date overview on the current status of biomarker-driven clinical trials, with a focus on the underlying design considerations, rationale, and lessons learned. We organize these discussions in terms of the biomarker’s role (integral v integrated), number of biomarkers involved (one v multiple), and trial objective (confirmatory v discovery). Adaptive design elements and selected examples are embedded as we summarize the features of each design (Table 1).
TABLE 1.
Summary of Biomarker-Driven Trial Designs

ADAPTIVE DESIGN FEATURES
As stated above, the notion of decisions toward trial modification on the basis of accumulating information, or adaptive strategies, was already a common feature of oncology trials before biomarkers were a design focus. Interim monitoring, including but not limited to group sequential methods, provides the statistical framework to allow repeated assessments of treatment efficacy and early stopping as soon as the accumulated data already provide adequate evidence to conclude that the experimental regimen is highly likely (efficacy monitoring) or unlikely (futility monitoring) beneficial. These adaptive design elements have been extensively used and appear in virtually all oncology trials nowadays. For example, phase II trials traditionally have implemented a single-arm, noncomparative design, with a planned interim futility analysis, such as Simon’s two-stage design,7 to minimize patient exposure to ineffective regimens. In the late-phase setting, integrated randomized phase II/III design8 is a useful adaptive design that combines both screening (phase II component) and confirmatory (phase III component) in a single trial. Such designs allow phase II patient data to be included in the principal phase III trial analysis to improve the overall logistic efficiency. As the data based on the phase II component are used to provisionally test the study hypothesis of the phase III component, integrated phase II/III designs can effectively be viewed as phase III studies with rather aggressive (ie, likely to stop) interim futility analyses. If more than one experimental regimen is of interest in the phase II component, a plan of selecting treatment arms can also be incorporated when making the go/no-go decision from phase II to phase III.
More formally speaking, adaptive designs are those that allow prospectively planned modifications in both the statistical and scientific aspects of study designs, on the basis of accumulating data while the trials are still in progress.9 Adaptations to the statistical aspects of study designs arise when the primary estimand (eg, the target of estimation) addressing the scientific question of interest10 remains unchanged; examples include group sequential designs,6 sample size adaptation,11 and, more recently (although the concept dates back several decades), outcome/response-adaptive randomization.12-14 Adaptations to the scientific aspects of study designs occur when the primary estimand does change (eg, enriching patient population, or selecting new treatment arms or end points while the trial is still ongoing). Among these adaptive design features, group sequential theory–based interim analysis and treatment assignment modification (such as add or drop arms) have been used most frequently, whereas other adaptive design features have not been widely adopted in practice. We also note that although the term adaptive design may be frequently considered synonymous with outcome adaptive randomization via Bayesian methods (as, for example, in the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination [BATTLE] trials15), trials with these aforementioned design features should also be considered adaptive.
In biomarker-driven trials, as we seek to simultaneously understand the biomarker-treatment relationship, both types of adaptations may be desirable to enhance logistical efficiency and optimize resource allocation, while maintaining balance with respect to ethical considerations.
DESIGNS WITH SINGLE INTEGRAL BIOMARKER
An enrichment design, which only enrolls M-positive patients (Fig 1A), can be considered when there is strong rationale and evidence suggesting the putative treatment effect of the novel agent is confined within the M-positive subpopulation only,16 because it does not permit a treatment effect evaluation among M-negative patients at all or permit evaluating whether the biomarker is predictive. For discovery purposes, both nonrandomized and randomized designs may be considered. Single-arm enrichment designs may be appropriate when tumor response (tumor shrinkage) provides a meaningful measure of clinical benefit without a comparator. If the new agent is expected to have little effect on tumor shrinkage, or must be combined with an active agent, randomized phase II screening designs17 with end points such as progression-free survival may be considered and provide valuable information on whether a confirmatory, randomized phase III enrichment design should be performed. Of note, although enrichment designs should be efficient (a small sample size) because we anticipate a large effect size, if the prevalence of M-positive patients is low, many patients must be screened to obtain the necessary sample size; if the prevalence is too low, the trial may simply be infeasible.
FIG 1.
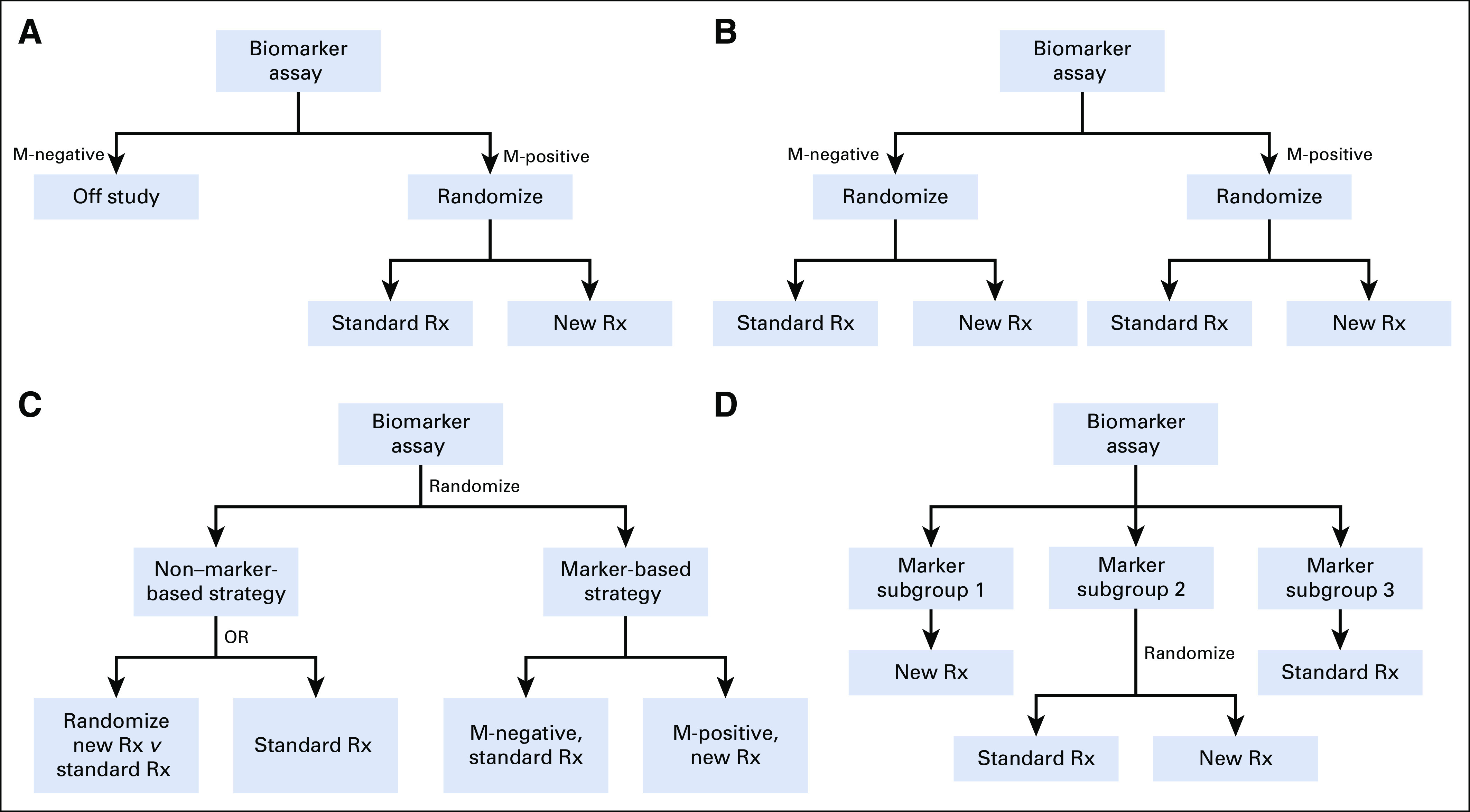
Schemas for single integral biomarker-driven trial designs. (A) Enrichment design. (B) Biomarker-stratified design. (C) Biomarker-strategy design. (D) Biomarker-directed design. M-negative, biomarker-negative; M-positive, biomarker-positive; Rx, treatment.
When there is strong evidence suggesting the biomarker is predictive with respect to the experimental regimen (ie, M-positive patients benefit), but it remains unclear whether the new treatment may also have a clinically meaningful (but likely smaller) benefit for M-negative patients, the so-called biomarker-stratified design, which randomly assigns all patients with a valid marker result (M-positive and M-negative) as a stratification factor, can be considered (Fig 1B). One can also view this as an umbrella trial (defined in a later section) that contains two separate RCTs for M-positive and M-negative subgroups. The key design consideration for confirmatory trials is if and how to prioritize the multiple hypotheses within the M-positive subgroup, the M-negative subgroup, or the overall population and properly power for each separately as applicable. To control the overall type I error due to multiple comparisons, various analysis strategies have been proposed to reflect different prioritizations and preserve the power of the most interesting questions.18,19 Here are some examples:
If we assume the new treatment is unlikely to be beneficial in the M-negative subgroup unless it works first in the M-positive subgroup, a sequential, α-recycling strategy can be considered by first testing the M-positive subgroup at significance level α and then testing the M-negative subgroup at the same α level if and only if the first test is statistically significant.20 An alternative approach to reflect such prioritization is to split the overall α unequally with a Bonferroni correction (eg, α1 = 0.04 and α2 = 0.01 for M-positive and M-negative, respectively).
If the effect in the overall population is of secondary interest, the aforementioned sequential procedure needs to be modified, because the treatment effect of the overall population can still seem to be clinically meaningful even when the new treatment only works in the M-positive subpopulation but not in the M-negative subpopulation at all. To mitigate the potentially misleading conclusion that treatment works in all comers (M-positive and M-negative), the Marker Sequential Test design21 was proposed, which first tests the M-positive subgroup at a reduced significance level α1 (< α): if the test yields a statistically significant result, the M-negative subgroup will be tested at level α, whereas the overall population will be tested at α − α1 if the test among the M-negative subgroup is not significant.
If there is no convincing evidence suggesting a particular biomarker is predictive, a fallback strategy can be used, which first tests the overall population at α1 (< α): if the result is significant, one can claim that the treatment is effective in all patients; if it is not significant, then the M-positive population must meet α − α1 for significance.22
Although biomarker-stratified designs are typically used in the context of a confirmatory phase III setting, randomized phase II screening designs may still be considered if the overarching goal is to efficiently inform whether to conduct a randomized phase III enrichment design. When the primary interest is to evaluate whether the biomarker-based treatment assignment strategy is more effective than non–biomarker-based treatment assignment strategy, one may consider a biomarker-strategy design, which randomly assigns all patients (M-positive and M-negative) to receive treatments either on the basis of or independent of biomarker status (Fig 1C). This design may also be used to evaluate whether the biomarker is predictive with some efficiency loss,16,23 because there can be a significant portion of patients with the same biomarker status receiving the same treatments in both arms, reducing the treatment effect size that can realistically be specified.
Biomarker-directed design may be considered when it is desirable to have an integral biomarker evaluation for all patients, and there is compelling existing evidence suggesting a particular biomarker-defined subgroup should receive a certain regimen with satisfactory efficacy and safety profiles (Fig 1D). In this case, like enrichment designs, a biomarker-directed design only randomizes the biomarker-defined subgroup where the biomarker’s clinical utility in directing treatment decisions remains unclear; meanwhile the other biomarker-defined subgroups are treated deterministically. This design therefore is suitable for evaluating the clinical utility of not only an integral predictive biomarker but also a prognostic biomarker. For example, in TAILORx (Program for the Assessment of Clinical Cancer Tests [PACCT-1]: Trial Assigning Individualized Options for Treatment),24 patients with breast cancer treated with tamoxifen were classified as low, intermediate, and high risk on the basis of a 21-gene recurrence score (Oncotype DX, Genomic Health, Redwood City, CA). Patients with intermediate risk were randomly assigned to receive hormonal therapy with or without chemotherapy, whereas low-risk patients and high-risk patients always received only hormonal therapy or hormonal therapy with chemotherapy, respectively.
In these confirmatory integral biomarker-driven trials, interim monitoring, especially futility monitoring, plays an even more critical role to help prioritize the limited resources by dropping subgroups with unpromising or unresponsive treatment benefit and/or dropping ineffective experimental regimens if more than one is being investigated. For example, for biomarker-stratified designs, futility monitoring can be easily and flexibly incorporated for M-positive, M-negative, and overall population, such that if the treatment benefit is unlikely to be observed in M-negative subgroup, the trial can terminate accrual to M-negative and only accrue M-positive patients.23 One straightforward extension is to accrue M-negative and M-positive (all comers) initially and only continue to accrue biomarker-defined subgroups where promising treatment effects are observed, using proper multiplicity adjustments.25 This concept has been generalized more broadly as the adaptive enrichment design, where all comers are accrued initially, and the eligibility criteria may change adaptively on the basis of planned interim analysis results and subgroups defined by one or more biomarkers.26,27 Alternatively, one can start with M-positive patients only and expand to the overall population if it is suggested that the treatment effect may be not be confined within M-positive subgroup alone.28
DESIGNS WITH SINGLE INTEGRATED BIOMARKER
When an integrated biomarker is analytically validated, but its clinical utility is not fully developed by the time of trial initiation, or the biomarker cannot be obtained with fast turnaround time, it is desirable to have proper trial design and analysis plans to provide a valid treatment effect evaluation for biomarker-based subgroups. For a biomarker classifier that is based on a multiplex assay of genomic, proteomic, and transcriptomic data, the adaptive signature design29 was proposed to evaluate whether the experimental regimen is effective in the overall population or a subset of patients only while developing the biomarker classifier simultaneously. This design modifies the fallback analysis strategy for biomarker-stratified designs in a learn-and-confirm fashion. If the treatment comparison for all patients is not significant in the overall population at α1 (< α), we will either split all patients into training and testing subsets or use K-fold cross-validation30 to develop the biomarker and then evaluate efficacy in identified subgroups. Of note, the cross-validated approach has also been shown to substantially improve the power of identifying the M-positive subgroup that benefits from the new treatment. By transforming the multiple candidate genes to a binary classifier, the approach does not suffer too much with respect to type I error control as the dimension of genes increases. When a biomarker or gene signature can be quantified on a continuous or ordinal scale, we can consider the biomarker-adaptive threshold design31 using a similar strategy to identify and validate an optimal cutoff point that separates M-positive and M-negative subgroups. The use of bootstrap resampling for estimation and inference of the threshold, although accounting for the multiplicity issue due to combining the tests for subgroup M-positive and overall population, has been shown to preserve the power to detect a global treatment effect while developing the biomarker. Comparing with the conventional approaches, these methods have been shown to substantially improve the likelihood of detecting the M-positive subgroup when differential treatment effect does exist, especially when the prevalence of M-positive is low. A more comprehensive review has been performed by Renfro et al.32
BASKET TRIALS FOR INTEGRAL BIOMARKER(S)
A basket trial, in its simplest form, studies a single targeted therapy among patients characterized by a corresponding biomarker in the context of multiple disease types or histology. Basket trial designs may be practically viewed as a collection of enrichment designs across different disease types or histology (Fig 2A). For example, after the approval of vemurafenib for BRAFV600 mutation-positive metastatic melanoma, a nonrandomized phase II basket trial of vemurafenib for multiple nonmelanoma cancers with BRAFV600 mutation was conducted,33 with objective response as the primary end point.
FIG 2.
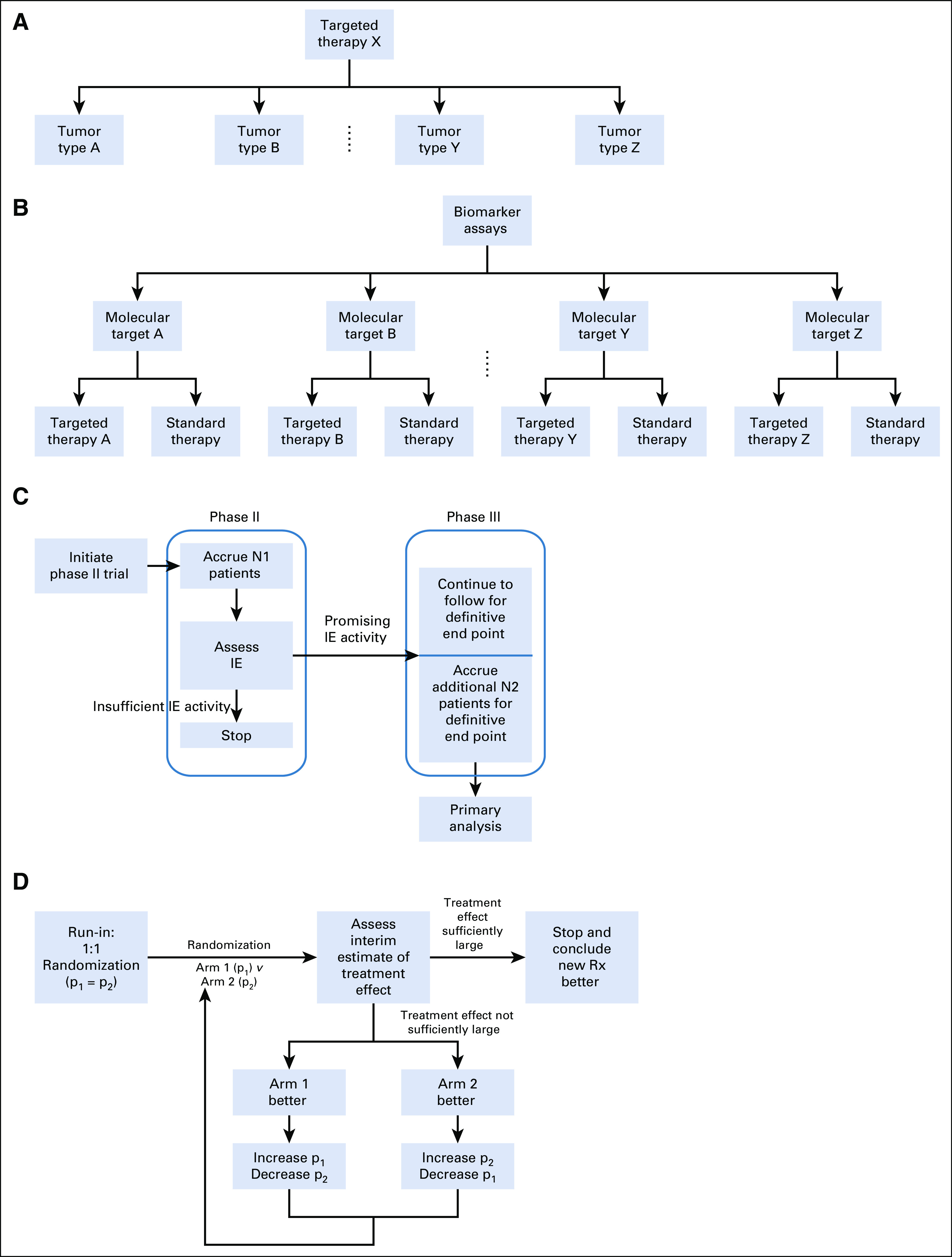
Schemas for master protocols and adaptive design elements. (A) Basket trial. (B) Umbrella trial. (C) Seamless phase II/III design. (D) Outcome-adaptive randomization. IE, intermediate end point.
From a trial conduct perspective, basket trials can be more efficient than multiple histology-specific enrichment trials conducted separately and are convenient to carry out because the biomarker can (although not necessarily) be assessed locally at participating sites as part of the eligibility criteria screen. The latter is an important feature and makes it different from umbrella trials with respect to trial conduct logistics. Furthermore, this trial design can be quite appealing to patients because it conveniently provides access to the experimental therapy across multiple disease types, including in settings where our understanding of the biomarker-treatment relationship is relatively limited. Consequently, basket designs most often serve for discovery purposes (ie, early phase II, pilot efficacy only), as we hope to investigate whether we can extrapolate the findings and gain understanding of biomarker-drug interaction within a particular disease type to all relevant disease types on the basis of the biomarker.
From a scientific perspective, the underlying rationale of conducting a basket trial is that the biomarker’s presence may independently predict responses attributed to the corresponding targeted therapy, regardless of histology or disease type.34 If this truly holds, it would be reasonable to redefine cancer in a histology-agnostic fashion, suggesting an exciting new approach to therapy development. For example, in May 2017, the US Food and Drug Administration approved pembrolizumab for any patients with microsatellite instability–high/deficient DNA mismatch repair regardless of histology.35 This approval was the first-ever histology-agnostic indication and was partly based on a small, proof-of-concept basket trial.36
Because basket trials may be viewed as a collection of enrichment trials, they also inherit all of the advantages and disadvantages, with the same practical considerations as elaborated earlier. One unique challenge facing basket trials is the balance between feasibility and the exchangeability (eg, histology agnostic) hypothesis, given the potential heterogeneity across different disease types. That is, if and when can we assume the molecular profiling is sufficient to replace histology and pool patients together with the same biomarker? For many biomarkers, prevalence is so low that it may be infeasible to accrue enough patients and analyze by each disease type. Meanwhile, pooling all patients with the same biomarker can be questionable if not at all unrealistic, because the approach implies we can completely ignore the prognosis heterogeneity across different histology and assume disease subtype is not prognostic at all. In the case of vemurafenib for patients with BRAFV600 mutations, response to treatment was high when the primary site was melanoma but low when the primary site was colorectal cancer.33 A practical compromise is to combine some disease types for which prevalence is significantly lower than others. In Le et al,36 patients with microsatellite instability–high/deficient DNA mismatch repair were accrued and analyzed by colorectal and noncolorectal cohorts separately, because the prevalence in colorectal cancer is notably higher than other disease types.
Several novel adaptive designs have been developed to avoid separate analyses and properly share response information across disease types. One approach is based on preplanned interim analyses to determine the next steps.37 For example, if there is adequate evidence suggesting some histology-specific cohorts have similar and promising activities, these cohorts will be aggregated to allow more efficient statistical inference with fewer patients; otherwise, histology-specific cohorts with exceptionally favorable response will remain separate, and cohorts with low responses will be terminated. The other approach is based on statistical modeling and Bayesian inference,38,39 which explicitly allows information sharing across different histology-specific cohorts and permits early stopping for some cohorts naturally on the basis of posterior probability of histology-specific response rates. Nonetheless, it was argued that, for sample sizes typically used in phase II trials and a reasonable number of cohorts/histology types under investigation, these designs that are meant to share information may not be as useful as one would hope, unless there is a strong rationale and evidence indicating uniform responses across cohorts.40
Another important consideration when designing discovery basket trials is whether randomization should be used or not. The nonrandomized basket trial may be clearly preferred because of its feasibility and close connection to the conventional single-arm phase II design traditionally used in early-stage development. However, nonrandomized basket design practically mandates objective response to be the only choice of the primary end point, because it is generally considered the only interpretable efficacy end point without a comparator. Even with this end point, concerns may still arise regarding the relevance of historical control in at least some histology cohorts, because the biomarker likely defines a new disease subtype for which there is little or no historical information on prognosis.41 Furthermore, if the experimental regimen is to be administered in combination with other active regimens, how to isolate the impacts of background treatments and properly interpret the experimental regimen’s role can be challenging. Multi-agent, nonrandomized basket trials also have been proposed, which are essentially a collection of single-agent basket trials. In this case, because a centralized molecular screening platform is typically used, one may also view them as umbrella trials.
From a pure statistical perspective, one could argue that basket trials may have multiplicity issues, because we simultaneously evaluate multiple histology-specific cohorts. Pragmatically speaking, this may not be that problematic, because we are more tolerant of a higher type I error for discovery purposes; the same issue also exists if we conduct separate trials for each cohort, and more practical concerns such as accrual feasibility often outweigh the type I error control. Nonetheless, avoidance of false-positive signals as well as bias are concerns worth addressing as these trial designs evolve.
UMBRELLA TRIALS FOR MULTIPLE INTEGRAL BIOMARKERS
A typical umbrella trial evaluates multiple experimental regimens within a single disease histology. An up-front, centralized molecular screening platform and a multiplex assay are used to simultaneously obtain the biomarkers that determine eligibility and treatment. Patients with biomarkers of interest are allocated into mutually exclusive marker-specific subtrials, which use either nonrandomized or randomized enrichment designs (Fig 2B). Patients whose molecular profiles are not part of these markers of interest can be grouped as an unmatched cohort and evaluated separately or treated off protocol.
If the biomarkers of interest have good clinical utility, confirmatory-intent umbrella trials may be considered, where for each marker-specific cohort, randomized phase II or integrated phase II/III designs8 are used. A randomized phase II/III trial example is ALCHEMIST (Adjuvant Lung Cancer Enrichment Marker Identification and Sequencing Trial) for resectable non–small-cell lung cancer (NSCLC),42,43 which consists of three treatment subtrials, ALCHEMIST-EGFR for patients with EGFR mutation (ClinicalTrials.gov identifier: NCT02193282), ALCHEMIST-ALK for patients with ALK rearrangements (ClinicalTrials.gov identifier: NCT02201992), and ANVIL (ClinicalTrials.gov identifier: NCT02595944) for unmatched patients (eg, squamous histology, or nonsquamous histology and neither EGFR mutation nor ALK rearrangements). An observation cohort is also available for unmatched patients who refuse to participate in ANVIL. Another prominent example is Lung-MAP (ClinicalTrials.gov identifier: NCT02154490),44 which was initially for squamous lung cancer but recently was amended for all types of advanced NSCLC. In all examples, statistical considerations, including sample size justification and interim analyses for efficacy and futility, are largely driven by needs within each of the subtrials. Using sequential development features such as phase II/III designs and interim analyses that permit adaptation to findings, these trials adaptively provide the necessary flexibility for the umbrella trial objectives as a whole (Fig 2C).
When there are multiple candidate regimens that are of interest equally for some or all biomarker cohorts, umbrella trials also can be designed solely for discovery objectives. For example, in the BATTLE-1 (ClinicalTrials.gov identifier: NCT00411632, NCT00409968, NCT00410059, NCT00410189, NCT00411632, NCT00411671) trial, patients with chemotherapy-refractory NSCLC were assayed for four candidate biomarkers to be allocated to a total of five marker strata (including one nonmatched) and then randomly assigned to one of four drug regimens.15 In NRG-LU003 (ClinicalTrials.gov identifier: NCT03737994), an umbrella trial of previously treated patients with ALK-positive lung cancer, a total of 10 marker cohorts (including an unmatched cohort) and up to seven new-generation ALK inhibitors are to be evaluated. In both cases, the primary interests are to explore the antitumor activities and identify predictive biomarkers that are promising enough to guide patient assignment. However, as the total number of possible drug-marker strata increases, the likelihood to accrue enough patients to have a reasonably accurate estimate of efficacy for each drug-biomarker stratum decreases. By exploiting the fact that biomarker allocation is not informative for treatment assignments, BATTLE-1 considered a learn-as-go approach by explicitly using a Bayesian hierarchical probit model for information sharing,45 along with outcome-adaptive randomization,12,46 which allocates more patients to drug-marker strata that are more likely to have exceptional activities, to potentially provide individuals with more efficacious treatments and improve estimation precision (Fig 2D). In NRG-LU003, for each biomarker the investigators are able to determine and prioritize the experimental regimens of interest on the basis of preclinical data, which substantially reduces the total number of drug-biomarker strata to be evaluated. In addition, within each drug-marker stratum, a Simon’s two-stage design is used to flexibly retire those ineffective strata as soon as possible. Freidlin and Korn40 suggested that under typical phase II trial design settings, both approaches may require similar resources.
One implication of conducting umbrella trials is that an explicit rule governing how to match biomarkers and candidate regimens needs to be specified prospectively. Therefore, umbrella trials also provide a unique opportunity to evaluate whether a rule-based policy that matches biomarkers and drugs is effective at all and, if so, to what extent across all genomic characterizations. To make such evaluation interpretable, the rule-based assignment policy should be as stable as possible during the trial conduct and ideally cover a wide range of biomarker-drug combinations.47 Another unique consideration with umbrella trials is that a patient could be eligible for multiple biomarker cohort allocation because the tumor contains multiple biomarkers. How to address this needs to be prospectively specified, either deterministically (eg, one biomarker overrides others) or randomly (eg, with a probability inversely proportional to prevalence).
MASTER PROTOCOLS AND PLATFORM TRIALS
A common feature for both basket and umbrella trials is that within each biomarker-defined cohort a nonrandomized or randomized enrichment design is implemented. With the emergent use of molecular profiling, basket trials and umbrella trials, as well as extensions discussed here, can be collectively called master protocols or platform trials. These trials consist of multiple enrichment subtrials defined by molecular profiles and use a centralized screening platform and common data collection infrastructure. More importantly, such a protocol provides substantial flexibility in terms of discontinuing unpromising investigations, carrying forward favorable early results to definitive testing in a phase II/III framework, and introducing new subtrials as targets and agents are identified in a perpetual manner, using the aforementioned adaptive design methods for single-biomarker settings.48 In addition, the infrastructure advantages of conducting master protocols, such as centralized and streamlined trial conduct (enrollment, informed consent), data collection, governance (institutional review board, data and safety monitoring committee), and quality assurance (clinical monitoring, imaging reading), are substantial as compared with conducting individual trials separately. Meanwhile, the new paradigm presents unique challenges, and here we describe the development history of two master protocols to highlight some of these challenges when conducting these logistically demanding trials.
The NCI-MATCH (National Cancer Institute Molecular Analysis for Therapy Choice ClinicalTrials.gov identifier: NCT02465060) trial is a multiple targeted-therapy basket trial designed to evaluate whether biomarkers may exist in advanced solid tumors and lymphoma that are refractory to standard first-line therapy. When it was activated in 2015, it started with 10 parallel biomarker-based cohorts to evaluate eight different targeted therapies. In May 2016, a preplanned feasibility interim analysis revealed that only 9% of patients had actionable mutations that could be matched with any of the multiple targeted therapies under investigation. The lower-than-expected matching success rate led to a major amendment to improve the overall matching rate by adding additional marker-specific cohorts.49 In June 2017, when the original screening target was met (N = 6,000), it was found that the common molecular subtypes were rarer than expected.50 The study therefore underwent another major amendment by collapsing multiple subtype cohorts and relaxing the screening process. To date, the study remains open to accrual, with a total of 19 cohorts to evaluate 13 drugs.51
Lung-MAP was originally designed in 2014 for advanced squamous NSCLC, consisting of four targeted therapy subgroups and one nonmatched subgroup, each using a phase II/III seamless design, with progression-free survival and overall survival as primary end points, respectively.52 Since 2015, the treatment landscape of advanced NSCLC has changed tremendously because of a series of approvals in immunotherapies especially for squamous and nonsquamous NSCLC by the US Food and Drug Administration, which fundamentally changed the standard of care and the study control arms. After multiple minor amendments for adding and closing biomarker-specific subtrials, in 2018 the study was completely revamped by expanding eligibility to all histology of advanced NSCLC, using a new screening protocol and introducing new biomarker-defined subtrials.53
In summary, although conceptually offering efficiency and flexibility, master protocols are more prone to various factors that are unknown (eg, low biomarker prevalence) or cannot be foreseen (eg, changing treatment landscape) at the trial initiation. These trials also come with substantial logistical complications, especially when several sponsors are involved who may have conflicting proprietary interests and regulatory concerns.
DISCUSSION
Biomarker-driven clinical trials allow us to investigate patient heterogeneity on the basis of molecular profiling, which consequently introduces new opportunities and challenges. Comparing with the conventional paradigm, these trials require even more thorough planning and comprehensive evaluation on the overarching objectives (discovery or confirmatory), the credential of biomarker’s clinical utility, choice of adaptive design and analysis plans, knowledge of cancer biology, existing data from preclinical and early clinical trials, prevalence of each subtype population, logistic readiness to conduct immortal clinical trials, and so on. The success of any biomarker-driven trials therefore certainly relies on an even closer collaboration among all involved in advancing cancer care, including clinical investigators, statisticians, sponsors, regulators, drug and assay developers, and patient advocates.
Our discussions have been limited to phase II and III trial designs for discovery and confirmatory purposes. There are ample opportunities to incorporate dose selection when incorporating biomarkers in the early stages of clinical development. In addition, we are not able to cover aspects evaluating the analytical validity of a companion biomarker or diagnostic test whose development is just as critical as drug development.54 For example, any putative treatment effect will be diluted if the assay has low specificity or low sensitivity for resistance variants, which in turn will negatively affect the treatment evaluation.
ACKNOWLEDGMENT
We thank Ying Lu, PhD, the Associate Editor of the journal, and two anonymous referees whose comments constructively improved the article.
Footnotes
Supported in part by National Institutes of Health Grants No. U10-CA180822 and P30-CA006973.
AUTHOR CONTRIBUTIONS
Conception and design: Chen Hu
Financial support: All authors
Administrative support: Chen Hu
Provision of study material or patients: Chen Hu
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Chen Hu
Consulting or Advisory Role: Merck Sharp & Dohme
James J. Dignam
Consulting or Advisory Role: Merck, Celgene, Northwest Biotherapeutics
No other potential conflicts of interest were reported.
REFERENCES
- 1.McShane LM, Hayes DF. Publication of tumor marker research results: The necessity for complete and transparent reporting. J Clin Oncol. 2012;30:4223–4232. doi: 10.1200/JCO.2012.42.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pennello GA. Analytical and clinical evaluation of biomarkers assays: When are biomarkers ready for prime time? Clin Trials. 2013;10:666–676. doi: 10.1177/1740774513497541. [DOI] [PubMed] [Google Scholar]
- 3.Dancey JE, Dobbin KK, Groshen S, et al. Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin Cancer Res. 2010;16:1745–1755. doi: 10.1158/1078-0432.CCR-09-2167. [DOI] [PubMed] [Google Scholar]
- 4.Fleming TR. One-sample multiple testing procedure for phase II clinical trials. Biometrics. 1982;38:143–151. [PubMed] [Google Scholar]
- 5.Gehan EA. The determination of the number of patients required in a preliminary and a follow-up trial of a new chemotherapeutic agent. J Chronic Dis. 1961;13:346–353. doi: 10.1016/0021-9681(61)90060-1. [DOI] [PubMed] [Google Scholar]
- 6. Jennison C, Turnbull BW: Group sequential methods with applications to clinical trials. https://www.crcpress.com/Group-Sequential-Methods-with-Applications-to-Clinical-Trials/Jennison-Turnbull/p/book/9780849303166.
- 7.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 8.Korn EL, Freidlin B, Abrams JS, et al. Design issues in randomized phase II/III trials. J Clin Oncol. 2012;30:667–671. doi: 10.1200/JCO.2011.38.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. US Food and Drug Administration: Adaptive designs for clinical trials of drugs and biologics. https://www.fda.gov/media/78495/download.
- 10.Little RJ, D’Agostino R, Cohen ML, et al. The prevention and treatment of missing data in clinical trials. N Engl J Med. 2012;367:1355–1360. doi: 10.1056/NEJMsr1203730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang-Stein C, Anderson K, Gallo P, et al. Sample size reestimation: A review and recommendations. Drug Inf J. 2006;40:475–484. [Google Scholar]
- 12.Berry DA, Eick SG. Adaptive assignment versus balanced randomization in clinical trials: A decision analysis. Stat Med. 1995;14:231–246. doi: 10.1002/sim.4780140302. [DOI] [PubMed] [Google Scholar]
- 13.Korn EL, Freidlin B. Outcome--adaptive randomization: Is it useful? J Clin Oncol. 2011;29:771–776. doi: 10.1200/JCO.2010.31.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JJ, Chen N, Yin G. Worth adapting? Revisiting the usefulness of outcome-adaptive randomization. Clin Cancer Res. 2012;18:4498–4507. doi: 10.1158/1078-0432.CCR-11-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: Personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandrekar SJ, Sargent DJ. Clinical trial designs for predictive biomarker validation: Theoretical considerations and practical challenges. J Clin Oncol. 2009;27:4027–4034. doi: 10.1200/JCO.2009.22.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubinstein LV, Korn EL, Freidlin B, et al. Design issues of randomized phase II trials and a proposal for phase II screening trials. J Clin Oncol. 2005;23:7199–7206. doi: 10.1200/JCO.2005.01.149. [DOI] [PubMed] [Google Scholar]
- 18.Freidlin B, Korn EL. Biomarker enrichment strategies: Matching trial design to biomarker credentials. Nat Rev Clin Oncol. 2014;11:81–90. doi: 10.1038/nrclinonc.2013.218. [DOI] [PubMed] [Google Scholar]
- 19.Freidlin B, Sun Z, Gray R, et al. Phase III clinical trials that integrate treatment and biomarker evaluation. J Clin Oncol. 2013;31:3158–3161. doi: 10.1200/JCO.2012.48.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon R, Wang SJ. Use of genomic signatures in therapeutics development in oncology and other diseases. Pharmacogenomics J. 2006;6:166–173. doi: 10.1038/sj.tpj.6500349. [DOI] [PubMed] [Google Scholar]
- 21.Freidlin B, Korn EL, Gray R. Marker sequential test (MaST) design. Clin Trials. 2014;11:19–27. doi: 10.1177/1740774513503739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simon R. The use of genomics in clinical trial design. Clin Cancer Res. 2008;14:5984–5993. doi: 10.1158/1078-0432.CCR-07-4531. [DOI] [PubMed] [Google Scholar]
- 23.Freidlin B, McShane LM, Korn EL. Randomized clinical trials with biomarkers: Design issues. J Natl Cancer Inst. 2010;102:152–160. doi: 10.1093/jnci/djp477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sparano JA, Gray RJ, Makower DF, et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N Engl J Med. 2018;379:111–121. doi: 10.1056/NEJMoa1804710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang SJ, O’Neill RT, Hung HM. Approaches to evaluation of treatment effect in randomized clinical trials with genomic subset. Pharm Stat. 2007;6:227–244. doi: 10.1002/pst.300. [DOI] [PubMed] [Google Scholar]
- 26.Wang SJ, Hung HM, O’Neill RT. Adaptive patient enrichment designs in therapeutic trials. Biom J. 2009;51:358–374. doi: 10.1002/bimj.200900003. [DOI] [PubMed] [Google Scholar]
- 27.Simon N, Simon R. Adaptive enrichment designs for clinical trials. Biostatistics. 2013;14:613–625. doi: 10.1093/biostatistics/kxt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu A, Liu C, Li Q, et al. A threshold sample-enrichment approach in a clinical trial with heterogeneous subpopulations. Clin Trials. 2010;7:537–545. doi: 10.1177/1740774510378695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freidlin B, Simon R. Adaptive signature design: An adaptive clinical trial design for generating and prospectively testing a gene expression signature for sensitive patients. Clin Cancer Res. 2005;11:7872–7878. doi: 10.1158/1078-0432.CCR-05-0605. [DOI] [PubMed] [Google Scholar]
- 30.Freidlin B, Jiang W, Simon R. The cross-validated adaptive signature design. Clin Cancer Res. 2010;16:691–698. doi: 10.1158/1078-0432.CCR-09-1357. [DOI] [PubMed] [Google Scholar]
- 31.Jiang W, Freidlin B, Simon R. Biomarker-adaptive threshold design: A procedure for evaluating treatment with possible biomarker-defined subset effect. J Natl Cancer Inst. 2007;99:1036–1043. doi: 10.1093/jnci/djm022. [DOI] [PubMed] [Google Scholar]
- 32.Renfro LA, Mallick H, An MW, et al. Clinical trial designs incorporating predictive biomarkers. Cancer Treat Rev. 2016;43:74–82. doi: 10.1016/j.ctrv.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon R. Genomic alteration–driven clinical trial designs in oncology. Ann Intern Med. 2016;165:270–278. doi: 10.7326/M15-2413. [DOI] [PubMed] [Google Scholar]
- 35. US Food and Drug Administration: FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560040.htm.
- 36.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leblanc M, Rankin C, Crowley J. Multiple histology phase II trials. Clin Cancer Res. 2009;15:4256–4262. doi: 10.1158/1078-0432.CCR-08-2069. [DOI] [PubMed] [Google Scholar]
- 38.Thall PF, Wathen JK, Bekele BN, et al. Hierarchical Bayesian approaches to phase II trials in diseases with multiple subtypes. Stat Med. 2003;22:763–780. doi: 10.1002/sim.1399. [DOI] [PubMed] [Google Scholar]
- 39. Simon R, Geyer S, Subramanian J, et al: The Bayesian basket design for genomic variant-driven phase II trials. Semin Oncol 43:13-18, 2016. [DOI] [PubMed] [Google Scholar]
- 40.Freidlin B, Korn EL. Borrowing information across subgroups in phase II trials: Is it useful? Clin Cancer Res. 2013;19:1326–1334. doi: 10.1158/1078-0432.CCR-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ratain MJ, Sargent DJ. Optimising the design of phase II oncology trials: The importance of randomisation. Eur J Cancer. 2009;45:275–280. doi: 10.1016/j.ejca.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 42.Le-Rademacher J, Dahlberg S, Lee JJ, et al. Biomarker clinical trials in lung cancer: Design, logistics, challenges, and practical considerations. J Thorac Oncol. 2018;13:1625–1637. doi: 10.1016/j.jtho.2018.08.2019. [DOI] [PubMed] [Google Scholar]
- 43.Govindan R, Mandrekar SJ, Gerber DE, et al. ALCHEMIST trials: A golden opportunity to transform outcomes in early-stage non–small cell lung cancer. Clin Cancer Res. 2015;21:5439–5444. doi: 10.1158/1078-0432.CCR-15-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. National Cancer Institute: Lung-MAP: Master protocol for lung cancer. https://www.cancer.gov/types/lung/research/lung-map.
- 45.Lee JJ, Xuemin Gu, Suyu Liu Bayesian adaptive randomization designs for targeted agent development. Clin Trials. 2010;7:584–596. doi: 10.1177/1740774510373120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou X, Liu S, Kim ES, et al. Bayesian adaptive design for targeted therapy development in lung cancer--a step toward personalized medicine. Clin Trials. 2008;5:181–193. doi: 10.1177/1740774508091815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon R, Polley E. Clinical trials for precision oncology using next-generation sequencing. Per Med. 2013;10:485–495. doi: 10.2217/pme.13.36. [DOI] [PubMed] [Google Scholar]
- 48.Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377:62–70. doi: 10.1056/NEJMra1510062. [DOI] [PubMed] [Google Scholar]
- 49. National Cancer Institute: NCI-MATCH: A status report and future directions. https://www.cancer.gov/news-events/cancer-currents-blog/2016/nci-match-update.
- 50. Harris L, Chen A, O’Dwyer P, et al: Abstract B080: Update on the NCI-Molecular Analysis for Therapy Choice (NCI-MATCH/EAY131) precision medicine trial. 2018.
- 51. National Cancer Institute: NCI-MATCH Trial (Molecular Analysis for Therapy Choice). https://www.cancer.gov/about-cancer/treatment/clinical-trials/nci-supported/nci-match.
- 52. doi: 10.3978/j.issn.2304-3865.2015.09.02. Ferrarotto R, Redman MW, Gandara DR, et al: Lung-MAP: Framework, overview, and design principles. Chin Clin Oncol 4:36, 2015. [DOI] [PubMed] [Google Scholar]
- 53. Hohman R, Lawton W: Lung-MAP precision medicine trial expands to include more patients. https://www.lung-map.org/media/press/lung-map-precision-medicine-trial-expands-include-more-patients.
- 54.Lih CJ, Sims DJ, Harrington RD, et al. Analytical validation and application of a targeted next-generation sequencing mutation-detection assay for use in treatment assignment in the NCI-MPACT trial. J Mol Diagn. 2016;18:51–67. doi: 10.1016/j.jmoldx.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 56.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. doi: 10.1200/JCO.2009.27.6055. Peeters M, Jay Price TJ, Cervantes A: Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28:4706-4713, 2010. [DOI] [PubMed] [Google Scholar]
- 58.Herbst RS, Redman MW, Kim ES, et al. Cetuximab plus carboplatin and paclitaxel with or without bevacizumab versus carboplatin and paclitaxel with or without bevacizumab in advanced NSCLC (SWOG S0819): A randomised, phase 3 study. Lancet Oncol. 2018;19:101–114. doi: 10.1016/S1470-2045(17)30694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31:4105–4114. doi: 10.1200/JCO.2012.47.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bradley JD, Paulus R, Komaki R, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): A randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015;16:187–199. doi: 10.1016/S1470-2045(14)71207-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vansteenkiste JF, Cho BC, Vanakesa T, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17:822–835. doi: 10.1016/S1470-2045(16)00099-1. [DOI] [PubMed] [Google Scholar]
- 62.Cobo M, Isla D, Massuti B, et al. Customizing cisplatin based on quantitative excision repair cross-complementing 1 mRNA expression: A phase III trial in non-small-cell lung cancer. J Clin Oncol. 2007;25:2747–2754. doi: 10.1200/JCO.2006.09.7915. [DOI] [PubMed] [Google Scholar]