

Abstract
PURPOSE
To compare the prevalence of germline mutations in metastatic hormone-sensitive prostate cancer (mHSPC) and metastatic castrate-resistant prostate cancer (mCRPC) and assess the impact of mutations on progression to castration resistance and overall survival.
METHODS
Targeted sequencing of germline DNA from 704 men (221 at the time of mHSPC and 483 at the time of mCRPC) enrolled in two advanced prostate cancer registries at Mayo Clinic between 2003 and 2013 was performed for 21 predisposition genes. Frequencies of pathogenic mutations were compared in patients and reference controls to identify genes enriched in metastatic prostate cancer. Multivariable Cox proportional hazards regression was used to identify predictors of progression to mCRPC and overall survival.
RESULTS
Sixty-eight germline mutations in 12 genes were identified in 66 men (9.4%). Mutations in ATM, BRCA2, CHEK2, FANCM, and TP53 were significantly enriched (odds ratio greater than 2.0) in the metastatic cohorts compared with reference controls. The frequency of germline mutations was similar for patients with mHSPC and mCRPC (11.8% v 8.3%; P = .16). The median time to progression from mHSPC to mCRPC was 23.1 and 32.5 months for patients with and without mutations, respectively (P = .96). Although older age at diagnosis, Gleason score greater than 7, elevated alkaline phosphatase level, and high volume of disease were associated with shorter duration of progression to mCRPC and poor overall survival, mutation status was not (progression to mCRPC hazard ratio, 0.81; 95% CI, 0.61 to 1.09; P = .17; overall survival hazard ratio, 1.00; 95% CI, 0.75 to 1.34; P = .98).
CONCLUSION
Similarly elevated rates of germline predisposition gene mutations in mHSPC and mCRPC suggest that germline genetic testing may help to guide medical management for all patients with advanced metastatic prostate cancer. Mutation status was not associated with shorter progression to mCRPC or poor overall survival.
INTRODUCTION
Prostate cancer is a leading cause of cancer in men in the United States and worldwide.1,2 Approximately 30% of patients experience a relapse with advanced disease after curative-intent local therapy for prostate cancer,3,4 and between 5% and 7% of patients are diagnosed initially with advanced stage. Androgen deprivation therapy (ADT), alone or in combination with other therapeutic agents, is the mainstay treatment in newly diagnosed metastatic disease. However, the majority of patients with metastatic hormone-sensitive prostate cancer (mHSPC) eventually stop responding to ADT and progress to metastatic castrate-resistant prostate cancer (mCRPC). Several factors have been implicated in the progression from mHSPC to mCRPC,5-8 but the role of mutations in cancer predisposition genes has not been adequately explored.
Inherited mutations in cancer predisposition genes, such as BRCA1/2 and other DNA repair genes, are detected in approximately 4% of patients with localized prostate cancer and less than 1% of the general population.9 In comparison, 7% to 17% of men with mCRPC carry deleterious germline mutations in cancer predisposition genes,9-13 which suggests a contributory role of these mutations in the progression from localized prostate cancer to mCRPC. However, the frequency of germline mutations in patients with mHSPC is not known, and whether mutation carrier status can predict rapid progression from hormonal sensitivity to castration resistance is unclear.
Novel drug combinations with ADT in patients with mHSPC already have demonstrated efficacy in slowing progression to mCRPC and death,14-16 but currently, interventions are not based on the presence of specific genomic aberrations or mutations. In the mCRPC state, the targeting of mutations in DNA repair genes with poly (ADP-ribose) polymerase (PARP) inhibitors, such as olaparib or platinum-based chemotherapy,17-19 has demonstrated clinical benefit. Because the influence of DNA repair gene defects in mHSPC on disease progression has not been determined, the use of PARP inhibitors or carboplatin in this subcohort has not been explored. In this study, we undertook a comprehensive assessment to study the contributions of DNA repair gene defects in mHSPC and mCRPC and the implications for clinical outcomes.
Context
Key Objective
To compare the prevalence of germline mutations in metastatic hormone-sensitive prostate cancer (mHSPC) and metastatic castrate-resistant prostate cancer (mCRPC) and assess the impact of mutations on progression to castration resistance and overall survival.
Knowledge Generated
In this analysis of 704 patients with metastatic prostate cancer enrolled in a prospective registry, germline mutations were observed in 9.4% of men, 11.8% of men with mHSPC, and 8.3% of men with mCRPC. Mutations in ATM, BRCA2, CHEK2, FANCM, and TP53 were enriched significantly in the metastatic cohorts compared with reference controls from disease-specific and population genetic studies. No significant difference was found in time to progression from the hormone-sensitive state to the castrate-resistant state or in overall survival between mutation carriers and noncarriers.
Relevance
These findings have significant implications for informing prognosis and designing future clinical trials with targeted agents in patients with mHSPC and mCRPC.
METHODS
Patient Selection
Patients with advanced prostate cancer were derived from two prospective registries at Mayo Clinic. The Prostate Cancer Specialized Program of Research Excellence Clinical Research database enrolled patients with advanced prostate cancer between 2003 and 2008. Patients who experienced ADT failure were eligible to enroll in this registry. A second registry enrolled patients with mHSPC and mCRPC between September 2009 and January 2013. Details of both cohorts have been published previously.20-23 A total of 826 patients with either mHSPC or mCRPC enrolled in either registry were evaluated for inclusion in this study. Patients without a germline DNA sample or available clinical data were excluded from germline sequencing.
Patients were stratified into mHSPC or mCRPC enrollment cohorts on the basis of hormone sensitivity or castration resistance at the time of enrollment. The date of starting ADT or orchiectomy for mHSPC was considered as the date of mHSPC diagnosis. mHSPC was defined as the development of metastasis as seen on bone imaging scans or computed tomography scans of the abdomen and pelvis, with or without a rising prostate-specific antigen (PSA), and no previous ADT in the past 2 years. mCRPC was defined as the occurrence of either two serial PSA rises during continuous ADT measured at least 1 week apart with an absolute PSA level greater than 2 ng/mL or detection of imaging-based progressive metastasis, whichever came first. High volume of metastasis was defined as the presence of visceral metastases or four or more bone lesions with one or more beyond the vertebral bodies and pelvis per the E3805 study.14 Data on initial diagnosis of localized cancer, including date of diagnosis, T stage, N stage, and Gleason score, were collected by review of medical records at the time of enrollment. For the mCRPC cohort, information on date of mHSPC diagnosis, PSA and alkaline phosphatase (ALP) levels, and volume of disease at mHSPC diagnosis were collected by medical record review at the time of enrollment. All patients provided informed consent and a blood sample for genetic testing at the time of enrollment. All patients were prospectively followed until death. The last date of follow-up for data analysis was November 16, 2018. This study was approved by the institutional review board at Mayo Clinic.
Sequencing and Bioinformatics Analysis
Germline DNA from peripheral blood mononuclear cells was enriched for the coding regions and consensus splice sites of 21 cancer predisposition genes (ATM, BARD1, BRCA1, BRCA2, BRIP1, CHEK2, FANCC, FANCM, MRE11A, MLH1, MSH2, MSH6, NBN, PALB2, PTEN, RAD50, RAD51C, RAD51D, STK11, TP53, and XRCC2) using a custom amplicon-based QIAseq panel (QIAGEN, Hilden, Germany).
Pooled libraries (n = 728) were subjected to paired-end 150-base pair sequencing in a single lane of a HiSeq 4000 (Illumina, San Diego, CA), which corresponds to a median coverage of 200×. Reads were trimmed with Cutadapt version 1.1024 and aligned with BWA-MEM.25 Sequence realignment, recalibration, haplotype calling, and depth of coverage were conducted using Genome Analysis Toolkit version 3.4-46.26 Large genomic rearrangements were detected with PatternCNV version 1.1.3.27 Annotations were provided through the BioR toolkit,28 which leverages dbNSFP version 3.0,29 ClinVar,30 Clinical Annotation of Variants,31 and population frequencies from Genome Aggregation Database (gnomAD)32 and Exome Aggregation Consortium33 non–The Cancer Genome Atlas controls. Mutations were viewed with VCF-Miner.34
Interpretation of Variants
The pathogenicity of germline variants was determined using a framework consistent with established American College of Medical Genetics and Genomics and Association for Molecular Pathology guidelines.35 Only patients with likely pathogenic or pathogenic mutations were classified as mutation carriers. Missense mutations in the CHEK2 gene and other low-penetrance alterations were excluded from analyses. The details of variant classification are described in the Data Supplement.
Statistical Analysis
Statistical analysis was performed using R version 3.5.1 (https://cran.r-project.org) software. Fisher’s exact test was used to evaluate associations between categorical variables, and Mann-Whitney U test was used for comparing continuous variables. In case-control association analysis, mutation frequencies by gene were compared with mutation frequencies for non-Finnish whites in gnomAD.32 Large genomic rearrangements in all genes and gnomAD filter non-PASS variants were excluded from case-control association studies. Odds ratios (ORs) and 95% CIs for the strength of association with prostate cancer were estimated by Fisher’s exact test.
Gene-specific mutation enrichment in the mHSPC enrollment cohorts relative to the mCRPC cohort were assessed by logistic regression adjusted for age at mHSPC diagnosis and reported as ORs with 95% CIs. Time to progression to mCRPC and death were analyzed using Kaplan-Meier curves. The log-rank test was used for comparisons between subgroups. Multivariable analysis for time to progression to mCRPC and overall survival was performed using a Cox proportional hazards regression model that included age, local versus advanced disease at initial presentation, Gleason score, T and N stage at initial diagnosis, PSA and ALP level at mHSPC diagnosis, volume of metastasis, and mutation status as covariates. Seventeen patients who did not receive ADT despite a diagnosis of metastatic prostate cancer were excluded from analysis of progression from hormone-sensitive stage to castration resistance but were included in the overall survival analysis. All tests were two-sided, and P < .05 was considered to indicate statistical significance.
RESULTS
Of 728 samples subjected to germline sequencing, 17 failed coverage, and six were identified as duplicates; one patient was excluded because of a diagnosis of neuroendocrine prostate cancer (Data Supplement). Of the 704 patients included in the final analysis, 221 (31.4%) were enrolled in the mHSPC cohort and 483 (68.6%) in the mCRPC cohort. The median age at diagnosis of metastatic disease was 68.8 years for the mHSPC cohort and 67.0 years for the mCRPC cohort. The median duration of follow-up from mHSPC diagnosis was 71 months for the entire cohort, 53 months for the mHSPC cohort, and 77 months for the mCRPC cohort. Baseline characteristics of the mHSPC and mCRPC cohorts are listed in Table 1.
TABLE 1.
Demographic and Clinical Characteristics of the Metastatic Prostate Cancer Cohorts

Frequency of Germline Cancer Predisposition Gene Mutations
Among the 21 genes evaluated in the final analysis, 68 germline mutations were identified in 66 men (9.4%). Mutations were observed in 12 genes, including BRCA2 (2%), CHEK2 (2%), ATM (1.8%), FANCM (1%), TP53 (0.6%), BRCA1 (0.4%), BRIP1 (0.4%), RAD50 (0.4%), FANCC (0.3%), NBN (0.3%), PALB2 (0.3%), and BARD1 (0.1%; Data Supplement). No mutations were observed in MRE11A, MLH1, MSH2, MSH6, PTEN, RAD51C, RAD51D, STK11, and XRCC2. ATM, BRCA2, and CHEK2 were the most frequently mutated genes in both cohorts (Data Supplement). Twelve of the 14 CHEK2 mutations were c.1100delC. Large genomic rearrangements accounted for seven (10.3%) of 68 mutations. Deletion of exon 8 in FANCM (three patients) was the most commonly observed genomic rearrangement, followed by deletion of exon 6 in BRIP1 (two patients; Data Supplement).
The overall frequencies of mutations in patients with mHSPC and mCRPC were not significantly different (11.8% v 8.3%; P = .16). Similarly, no difference in gene-specific mutation frequency was observed in the mHSPC and mCRPC cohorts, although numbers were limited (Data Supplement). No difference was found in the frequency of germline mutations by volume of disease at metastasis (11.8% for high volume v 8.8% for low volume; P = .25) or by stage at initial diagnosis (9.4% for both local and de novo metastatic groups; P = 1.00).
Associations Between Cancer Predisposition Genes and Metastatic Prostate Cancer
Allele frequencies of the mutated genes from the overall cohort of 704 patients were compared with allele frequencies in public reference controls from gnomAD. ATM, BRCA2, CHEK2, FANCM, and TP53 mutations were significantly enriched in patients with prostate cancer relative to controls, with ORs ranging from 2.8 for CHEK2 to 15.9 for TP53 (Table 2). In the mHSPC cohort, ATM, BRCA2, CHEK2, and FANCM mutations were significantly enriched compared with reference controls (Data Supplement), whereas ATM, BRCA2, CHEK2, and TP53 mutations were significantly enriched in the mCRPC cohort (Data Supplement).
TABLE 2.
Enrichment of Cancer Predisposition Genes Compared With Reference Controls in the Entire Cohort*

Impact of Germline Mutations on Progression to Castration Resistance
No significant difference was found in the baseline characteristics of patients with and without mutations for the entire cohort or for the mHSPC and mCRPC enrollment cohorts (Table 1). For the entire cohort, the median time to progression from mHSPC to mCRPC was 23.1 months for mutation carriers and 32.5 months for noncarriers (P = .96; Fig 1A). When the mHSPC and mCRPC cohorts were analyzed separately, a significant difference in the rate of progression from mHSPC to mCRPC was not observed between mutation carriers and noncarriers in either cohort (Figs 1B and 1C).
FIG 1.
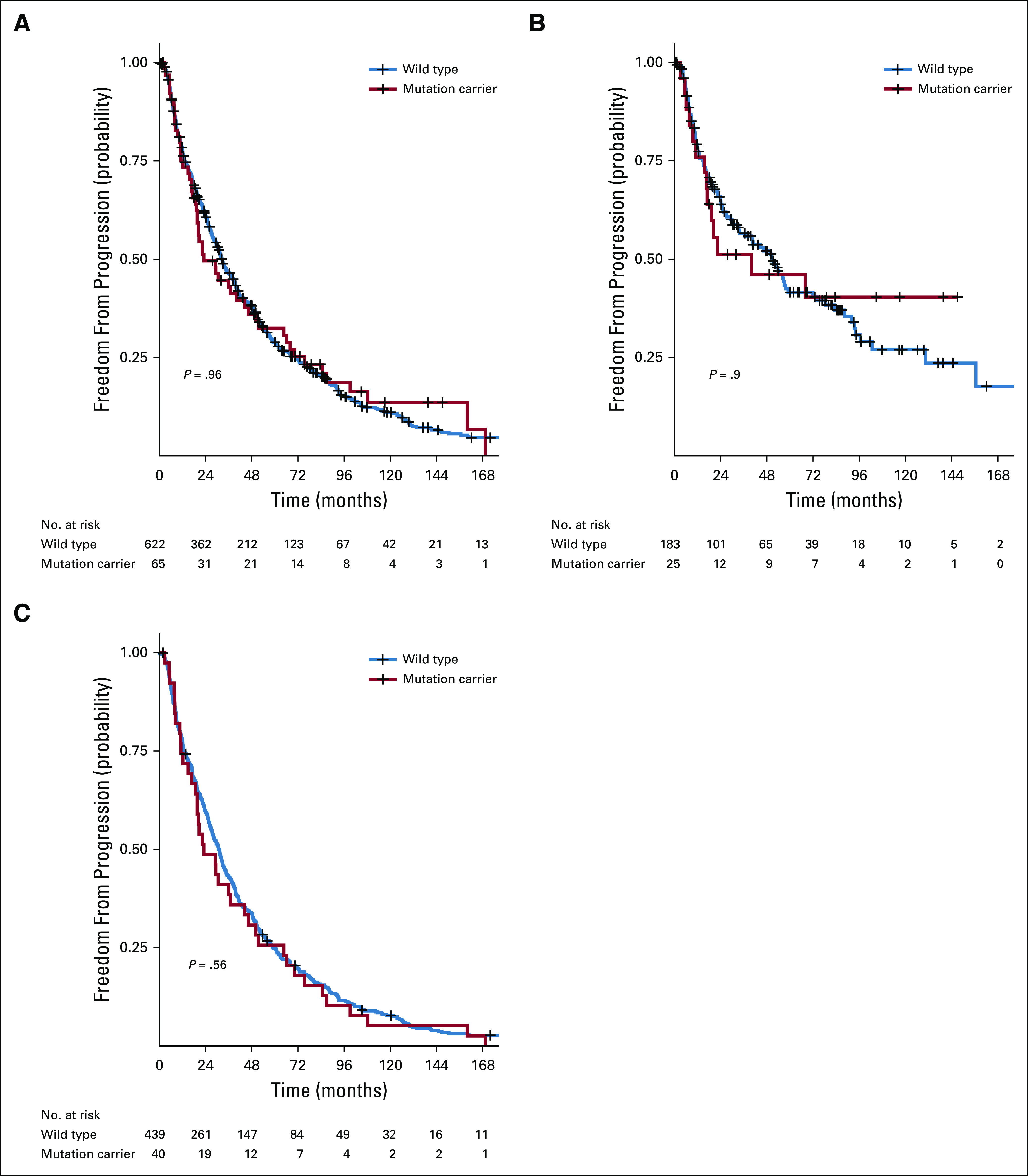
Comparison of the rate of progression from metastatic hormone-sensitive prostate cancer (mHSPC) to metastatic castrate-resistant prostate cancer (mCRPC) between mutation carriers and noncarriers. (A) Rate of mHSPC to mCRPC progression conditioned on mutation status for the entire cohort. (B) Rate of mHSPC to mCRPC progression conditioned on mutation status for the mHSPC cohort. (C) Rate of mHSPC to mCRPC progression conditioned on mutation status for the mCRPC cohort.
In univariable Cox proportional hazards regression analysis, older age at diagnosis of mHSPC, metastatic disease at initial presentation, Gleason score greater than 7, higher PSA level, elevated ALP level, and high volume of metastatic disease were significant predictors of mHSPC to mCRPC progression (Table 3). In multivariable Cox proportional hazards regression analysis, older age at diagnosis, Gleason score greater than 7, elevated ALP level, and high volume of metastasis remained significant predictors of progression to castration resistance (Table 3). Mutation status was not associated with progression to castration resistance in a univariable (hazard ratio [HR], 0.99; 95% CI, 0.75 to 1.32; P = .96) or multivariable model (HR, 0.81; 95% CI, 0.61 to 1.09; P = .17). On further analysis by enrollment cohort, mutation status was not associated with progression to castration resistance in multivariable models of the mHSPC or mCRPC cohorts (Data Supplement).
TABLE 3.
Univariable and Multivariable Cox Proportional Hazards Regression Model for Predictors of Progression From mHSPC to mCRPC for the Entire Cohort

Impact of Germline Mutations on Overall Survival
During the follow-up period, 528 patients (75.0%) died: 108 (48.9%) from the mHSPC enrollment cohort and 420 (87.0%) from the mCRPC enrollment cohort. No difference in overall survival was found from diagnosis of mHSPC between patients with and without mutations (median, 66 v 84 months; P = .14; Fig 2A). When the mHSPC and mCRPC enrollment cohorts were analyzed separately, no statistically significant difference in overall survival was observed (Figs 2B and 2C). In multivariable hazard regression analysis for overall survival, older age at diagnosis, Gleason score greater than 7, elevated ALP level, higher PSA level, and high volume of metastasis were significant predictors of poor overall survival (Table 4), whereas mutation status was not (HR, 1.00; 95% CI, 0.75 to 1.34; P = .98). On further subset analysis by enrollment cohort, mutation status was not associated with overall survival in multivariable analysis of the mHSPC or mCRPC cohorts (Data Supplement).
FIG 2.
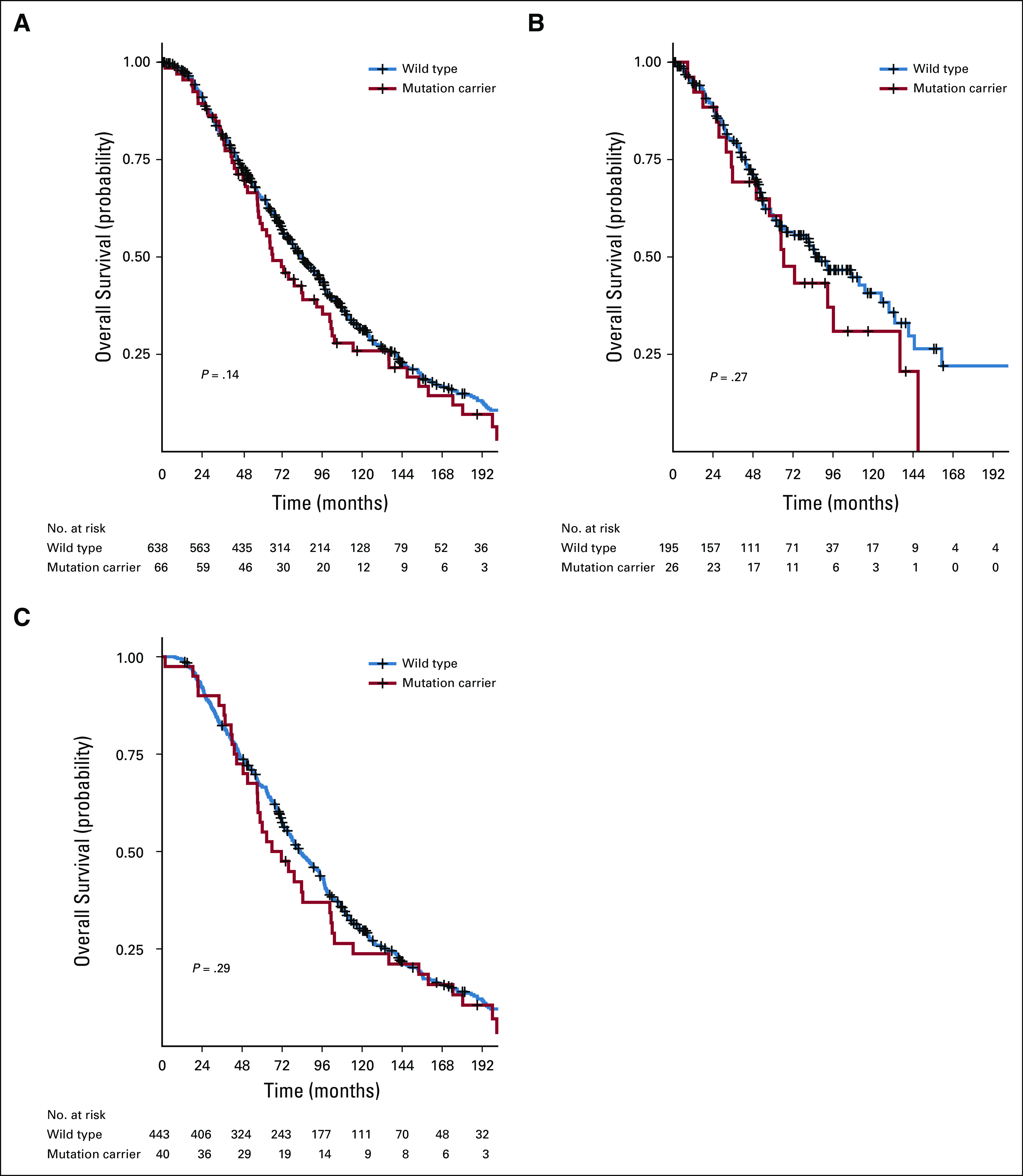
Comparison of overall survival between mutation carriers and noncarriers. (A) Overall survival between mutation carriers and noncarriers for the entire cohort relative to the first diagnosis of metastatic disease. (B) Overall survival between mutation carriers and noncarriers for the metastatic hormone-sensitive prostate cancer cohort relative to the first diagnosis of metastatic disease. (C) Overall survival between mutation carriers and noncarriers for the metastatic castrate-resistant prostate cancer cohort relative to the first diagnosis of metastatic disease.
TABLE 4.
Univariable and Multivariable Cox Proportional Hazards Regression Model for Predictors of Overall Survival for the Entire Cohort

DISCUSSION
This study showed that more than 10% of patients in the mHSPC state of advanced prostate cancer harbor a germline mutation in one of the predisposition genes associated with the cellular response to DNA damage and that the frequency of germline mutations is not significantly different between patients with mHSPC and mCRPC. Although prior studies have reported on the frequency of germline mutations in metastatic prostate cancer, these were mostly focused on patients with mCRPC.9,11-13,36 Our findings support the current National Comprehensive Cancer Network guidelines on further genetic risk evaluation in patients with metastatic prostate cancer irrespective of whether they are hormone sensitive or castration resistant.37
The three most commonly mutated genes in this study were ATM, BRCA2, and CHEK2, which is consistent with other studies.9,11-13,38 Twelve of the 14 CHEK2 alterations noted in our study were the common 1100delC variant. The 1100delC variant has been associated previously with a twofold higher risk for prostate cancer,39,40 which is consistent with the findings in the current study. This variant also has been noted to be more frequent in patients with lethal prostate cancer compared with low-risk prostate cancer.38 Whether other variants in CHEK2 confer similar prostate cancer risk is unclear. Although the frequencies of ATM and CHEK2 mutations were similar to prior studies, the frequency of BRCA2 mutations was lower compared with studies by Pritchard et al,9 Annala et al,11 and Castro et al36 but similar to a study by Antonarakis et al.12 The reasons behind the differences in the frequency of BRCA2 mutations across these studies are unclear, but selection of patients enriched in family history of cancer along with unstable frequency estimates because of small numbers of mutation carriers in some of the studies may have played a role. For instance, in the study by Pritchard et al, more than 50% of patients had a first-degree relative with cancer, whereas in the study by Castro et al, those with BRCA2 mutations had the greatest association with a family history of cancer. Although to our knowledge the current study is the largest in terms of sample size among all reported studies, we did not evaluate family history of cancer, which is a limitation when comparing participating populations across different studies. In addition, some variation existed in the genes evaluated in these studies.
In all, five genes were significantly associated with a high risk (OR greater than 2) for metastatic prostate cancer (ATM, BRCA2, CHEK2, FANCM, and TP53) compared with the general population in this study. In a similar comparison, Pritchard et al9 found that six genes (ATM, BRCA1, BRCA2, CHEK2, GEN1, and RAD51D) were significantly enriched in mCRPC. We did not detect any mutations in RAD51D, did not sequence GEN1, and detected only three BRCA1 mutations. The association of FANCM with prostate cancer risk, on the other hand, is a novel finding of the current study. FANCM is considered a member of the Fanconi anemia (FA) family of DNA repair genes (Online Mendelian Inheritance in Man 609644), a component of the upstream FA pathway that mediates the assembly of the FA core complex, and a regulator of the S-phase checkpoint upon interstrand crosslink-induced DNA damage.41 FANCM has been associated with an increased risk of breast cancer.42-45 In the current study, three of the seven mutations noted in FANCM were due to the deletion of exon 8, which has not been reported previously to be associated with prostate cancer risk, and hence, needs to be validated in future studies. The association between germline TP53 mutations and prostate cancer risk is also a novel finding of this study but needs to be interpreted with caution because of the small number of mutation carriers and the potential for age- and/or therapy-related clonal hematopoiesis events, which can be seen in up to one quarter of TP53 mutation carriers.46 We did not attempt to confirm the Mendelian inheritance of these TP53 mutations through analysis of DNA from secondary tissue sources,46 which is a limitation of this and other germline TP53 studies. The studies by Pritchard et al9 and Castro et al36 did not evaluate germline TP53 mutation status. Based on the results of our study, additional studies should investigate associations between germline TP53 mutations and risk of prostate cancer while also accounting for clonal hematopoiesis events.
Germline mutation carriers with localized prostate cancer have been reported to have a higher rate of progression to metastatic disease and an overall poorer prognosis.47-52 This is also supported by significant enrichment of mutation carriers among patients with metastatic prostate cancer compared with patients with localized prostate cancer.9 Although mutation carriers are more likely to progress from localized prostate cancer to metastatic prostate cancer, our results suggest that the rate of progression to castration resistance or death is not affected by mutation carrier status. The biologic mechanisms of metastasis and androgen resistance are distinct from each other.53,54 Genomic analysis of metastatic prostate tumors has demonstrated that mutation or amplification of the androgen receptor gene is the most enriched alteration in mCRPC compared with mHSPC tumors, and mutations in DNA repair genes are less common.55
In recent years, several clinical trials evaluated the addition of other therapeutic agents to ADT in early mHSPC stage,14,15,56-58 with the goal of slowing down progression to mCRPC and improving overall survival. At present, targeted therapies for patients with DNA repair defects, such as PARP inhibitors, have been studied only in the mCRPC stage.17 Our study suggests that future clinical trials with early addition of PARP inhibitors or similar drugs to ADT in the mHSPC stage may be feasible. This number of potentially eligible patients will be even higher when patients with somatic mutations in DNA repair genes are taken into account. These somatic alterations in DNA repair genes can arise during different stages of disease progression as a result of acquired resistance to therapy or as part of tumor evolution59 even in the absence of germline mutations. Our study did not evaluate somatic mutations in DNA repair genes in corresponding tumor material, but such patients are known to respond to PARP inhibitors.17 In addition, PARP inhibitors may potentially replace ADT in a select group of patients with mHSPC because of superior adverse effect profiles, as is being evaluated in an ongoing phase II clinical trial of PARP inhibitors instead of ADT in patients with mHSPC.60
Because this study was performed using prospective registries from a single institution, it has several strengths compared with similar prior studies. However, a number of limitations exist. First, we only sequenced 21 clinically relevant DNA repair genes. It is possible that some of the patients in the wild-type group may harbor mutations in a gene not evaluated in the study. Second, although this study used prospective registries, we included some retrospective data before enrollment for analysis. Third, we only analyzed patients who consented to participate in the registries. Hence, a selection bias cannot be ruled out. In addition, we did not account for treatment effect, although the majority of patients were enrolled before the publication of studies that established preferential use of docetaxel14 or abiraterone in the mHSPC setting15,58 and were unlikely to receive these treatments outside a clinical trial. However, patients may have received these treatments at progression to castration resistance, which can potentially affect the overall survival analysis. Finally, we did not take into account somatic mutations in DNA repair genes, which can independently affect outcomes.
This study demonstrated that approximately 10% of patients with metastatic prostate cancer harbor germline mutations in cancer predisposition genes, and it identified mutations in five genes (ATM, BRCA2, CHEK2, FANCM, and TP53) that are enriched in patients with advanced prostate cancer. Although mutations were common at the mHSPC and mCRPC stages, these were not specifically associated with progression to mCRPC or death. These findings have significant implications for future studies that evaluate novel therapeutic agents that target mutation carriers in the mHSPC stage.
Footnotes
Supported by National Institutes of Health Grants CA192393, CA176785, and CA225262 (F.J.C.) and CA212097 (M.K.); Mayo Clinic Center for Individualized Medicine; and the Department of Laboratory Medicine and Pathology.
AUTHOR CONTRIBUTIONS
Conception and design: Siddhartha Yadav, Steven N. Hart, Fergus J. Couch, Manish Kohli
Financial support: Fergus J. Couch
Administrative support: Fergus J. Couch
Provision of study material or patients: Fergus J. Couch
Collection and assembly of data: Siddhartha Yadav, Chunling Hu, David Hillman, Fergus J. Couch, Manish Kohli
Data analysis and interpretation: Siddhartha Yadav, Steven N. Hart, Chunling Hu, Kun Y. Lee, Rohan Gnanaolivu, Jie Na, Eric C. Polley, Fergus J. Couch, Manish Kohli
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Eric C. Polley
Research Funding: GRAIL
Fergus J. Couch
Consulting or Advisory Role: AstraZeneca
Research Funding: GRAIL
Travel, Accommodations, Expenses: GRAIL, QIAGEN
Other Relationship: Ambry Genetics
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Pound CR, Partin AW, Eisenberger MA, et al. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281:1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 4.Amling CL, Blute ML, Bergstralh EJ, et al. Long-term hazard of progression after radical prostatectomy for clinically localized prostate cancer: Continued risk of biochemical failure after 5 years. J Urol. 2000;164:101–105. [PubMed] [Google Scholar]
- 5.Sharma J, Gray KP, Harshman LC, et al. Elevated IL-8, TNF-α, and MCP-1 in men with metastatic prostate cancer starting androgen-deprivation therapy (ADT) are associated with shorter time to castration-resistance and overall survival. Prostate. 2014;74:820–828. doi: 10.1002/pros.22788. [DOI] [PubMed] [Google Scholar]
- 6.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501–5511. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma J, Gray KP, Evan C, et al. Elevated insulin-like growth factor binding protein-1 (IGFBP-1) in men with metastatic prostate cancer starting androgen deprivation therapy (ADT) is associated with shorter time to castration resistance and overall survival. Prostate. 2014;74:225–234. doi: 10.1002/pros.22744. [DOI] [PubMed] [Google Scholar]
- 8.Keto CJ, Aronson WJ, Terris MK, et al. Obesity is associated with castration-resistant disease and metastasis in men treated with androgen deprivation therapy after radical prostatectomy: Results from the SEARCH database. BJU Int. 2012;110:492–498. doi: 10.1111/j.1464-410X.2011.10754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hart SN, Ellingson MS, Schahl K, et al. Determining the frequency of pathogenic germline variants from exome sequencing in patients with castrate-resistant prostate cancer. BMJ Open. 2016;6:e010332. doi: 10.1136/bmjopen-2015-010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Annala M, Struss WJ, Warner EW, et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur Urol. 2017;72:34–42. doi: 10.1016/j.eururo.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Antonarakis ES, Lu C, Luber B, et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur Urol. 2018;74:218–225. doi: 10.1016/j.eururo.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. doi: 10.1016/j.cell.2015.05.001. Robinson D, Van Allen EM, Wu YM, et al: Integrative clinical genomics of advanced prostate cancer. Cell 161:1215-1228, 2015 [Erratum: Cell 162:454, 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373:737–746. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. 2017;377:352–360. doi: 10.1056/NEJMoa1704174. [DOI] [PubMed] [Google Scholar]
- 16.Kohli M, Tindall DJ. New developments in the medical management of prostate cancer. Mayo Clin Proc. 2010;85:77–86. doi: 10.4065/mcp.2009.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng HH, Pritchard CC, Boyd T, et al. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur Urol. 2016;69:992–995. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pomerantz MM, Spisák S, Jia L, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer. 2017;123:3532–3539. doi: 10.1002/cncr.30808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohli M, Riska SM, Mahoney DW, et al. Germline predictors of androgen deprivation therapy response in advanced prostate cancer. Mayo Clin Proc. 2012;87:240–246. doi: 10.1016/j.mayocp.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X, Yuan T, Liang M, et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur Urol. 2015;67:33–41. doi: 10.1016/j.eururo.2014.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia S, Kohli M, Du M, et al. Plasma genetic and genomic abnormalities predict treatment response and clinical outcome in advanced prostate cancer. Oncotarget. 2015;6:16411–16421. doi: 10.18632/oncotarget.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia Y, Huang CC, Dittmar R, et al. Copy number variations in urine cell free DNA as biomarkers in advanced prostate cancer. Oncotarget. 2016;7:35818–35831. doi: 10.18632/oncotarget.9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin M: Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10-12, 2011. [Google Scholar]
- 25. Li H: Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, 2013. https://arxiv.org/pdf/1303.3997.pdf.
- 26.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, Evans JM, Bhagwate AV, et al. PatternCNV: A versatile tool for detecting copy number changes from exome sequencing data. Bioinformatics. 2014;30:2678–2680. doi: 10.1093/bioinformatics/btu363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kocher JP, Quest DJ, Duffy P, et al. The Biological Reference Repository (BioR): A rapid and flexible system for genomics annotation. Bioinformatics. 2014;30:1920–1922. doi: 10.1093/bioinformatics/btu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Wu C, Li C, et al. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrum MJ, Lee JM, Benson M, et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Münz M, Ruark E, Renwick A, et al. CSN and CAVA: Variant annotation tools for rapid, robust next-generation sequencing analysis in the clinical setting. Genome Med. 2015;7:76. doi: 10.1186/s13073-015-0195-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Broad Institute: gnomAD. http://gnomad.broadinstitute.org.
- 33. Broad Institute: ExAC Browser (Beta): Exome Aggregation Consortium. http://exac.broadinstitute.org.
- 34.Hart SN, Duffy P, Quest DJ, et al. VCF-Miner: GUI-based application for mining variants and annotations stored in VCF files. Brief Bioinform. 2016;17:346–351. doi: 10.1093/bib/bbv051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castro E, Romero-Laorden N, Del Pozo A, et al. PROREPAIR-B: A prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37:490–503. doi: 10.1200/JCO.18.00358. [DOI] [PubMed] [Google Scholar]
- 37. doi: 10.6004/jnccn.2017.0003. National Comprehensive Cancer Network: NCCN guidelines version 2.2019: Genetic/familial high-risk assessment: Breast and ovarian. https://www.nccn.org/professionals/physician_gls/default.aspx. [DOI] [PubMed]
- 38.Wu Y, Yu H, Zheng SL, et al. A comprehensive evaluation of CHEK2 germline mutations in men with prostate cancer. Prostate. 2018;78:607–615. doi: 10.1002/pros.23505. [DOI] [PubMed] [Google Scholar]
- 39.Hale V, Weischer M, Park JY. CHEK2 (∗) 1100delC mutation and risk of prostate cancer. Prostate Cancer. 2014;2014:294575. doi: 10.1155/2014/294575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Näslund-Koch C, Nordestgaard BG, Bojesen SE. Increased risk for other cancers in addition to breast cancer for CHEK2*1100delC heterozygotes estimated from the Copenhagen General Population Study. J Clin Oncol. 2016;34:1208–1216. doi: 10.1200/JCO.2015.63.3594. [DOI] [PubMed] [Google Scholar]
- 41.Collis SJ, Ciccia A, Deans AJ, et al. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell. 2008;32:313–324. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 42.Peterlongo P, Catucci I, Colombo M, et al. FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum Mol Genet. 2015;24:5345–5355. doi: 10.1093/hmg/ddv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neidhardt G, Hauke J, Ramser J, et al. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol. 2017;3:1245–1248. doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiiski JI, Tervasmäki A, Pelttari LM, et al. FANCM mutation c.5791C>T is a risk factor for triple-negative breast cancer in the Finnish population. Breast Cancer Res Treat. 2017;166:217–226. doi: 10.1007/s10549-017-4388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiiski JI, Pelttari LM, Khan S, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc Natl Acad Sci U S A. 2014;111:15172–15177. doi: 10.1073/pnas.1407909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weitzel JN, Chao EC, Nehoray B, et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med. 2018;20:809–816. doi: 10.1038/gim.2017.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Na R, Zheng SL, Han M, et al. Germline mutations in ATM and BRCA1/2 Distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71:740–747. doi: 10.1016/j.eururo.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gleicher S, Kauffman EC, Kotula L, et al. Implications of high rates of metastatic prostate cancer in BRCA2 mutation carriers. Prostate. 2016;76:1135–1145. doi: 10.1002/pros.23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akbari MR, Wallis CJ, Toi A, et al. The impact of a BRCA2 mutation on mortality from screen-detected prostate cancer. Br J Cancer. 2014;111:1238–1240. doi: 10.1038/bjc.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–1757. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edwards SM, Evans DG, Hope Q, et al. Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br J Cancer. 2010;103:918–924. doi: 10.1038/sj.bjc.6605822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castro E, Goh C, Leongamornlert D, et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol. 2015;68:186–193. doi: 10.1016/j.eururo.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 53.Clarke NW, Hart CA, Brown MD. Molecular mechanisms of metastasis in prostate cancer. Asian J Androl. 2009;11:57–67. doi: 10.1038/aja.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakazawa M, Paller C, Kyprianou N. Mechanisms of therapeutic resistance in prostate cancer. Curr Oncol Rep. 2017;19:13. doi: 10.1007/s11912-017-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. doi: 10.1200/PO.17.00029. Abida W, Armenia J, Gopalan A, et al: Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kyriakopoulos CE, Chen YH, Carducci MA, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer: Long-term survival analysis of the randomized phase III E3805 CHAARTED trial. J Clin Oncol. 2018;36:1080–1087. doi: 10.1200/JCO.2017.75.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tombal B, Borre M, Rathenborg P, et al. Enzalutamide monotherapy in hormone-naive prostate cancer: Primary analysis of an open-label, single-arm, phase 2 study. Lancet Oncol. 2014;15:592–600. doi: 10.1016/S1470-2045(14)70129-9. [DOI] [PubMed] [Google Scholar]
- 58.James ND, de Bono JS, Spears MR, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338–351. doi: 10.1056/NEJMoa1702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. doi: 10.1038/s41588-018-0078-z. Armenia J, Wankowicz SAM, Liu D, et al: The long tail of oncogenic drivers in prostate cancer. Nat Genet 50:645-651, 2018 [Erratum Nat Genet 51:1194, 2019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. doi: 10.1093/oncolo/oyae120. Markowski MC, Wang H, Sullivan R, et al: Phase II trial of rucaparib (without ADT) in patients with metastatic hormone-sensitive prostate cancer harboring germline DNA repair gene mutations (TRIUMPH). J Clin Oncol 36, 2018 (suppl; abstr TPS5095) [DOI] [PMC free article] [PubMed] [Google Scholar]