

INTRODUCTION
Fluoropyrimidine drugs, both fluorouracil (FU) and its prodrug capecitabine, are widely used in the treatment of solid tumors such as breast, colorectal, and gastric cancers.1 Over 2 million patients newly diagnosed with cancer are treated each year with fluoropyrimidines.2 Between 10% and 40% of these patients develop severe, sometimes life-threatening toxicities, which may include mucositis, neutropenia, nausea, severe diarrhea, vomiting, stomatitis, and hand-foot syndrome.2 These toxicities can be caused by genetic variants in DPYD, the gene that encodes for dihydropyrimidine dehydrogenase (DPD), the rate-limiting enzyme responsible for FU catabolism.1,2
Clinical DPYD genotypic testing typically includes the following known toxicity-associated DPYD variants: c.1905+1G>A (*2A, rs3918290), c.1679T>G (*13, rs55886062), c.2846A>T (rs67376798), and c.1129-5923T>G (rs75017182). Guidelines for using these results to guide fluoropyrimidine therapy have been published by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group.2,3 However, targeted genotyping is limited to testing known toxicity-associated DPYD variants and may not include novel or rare variants, which can also be deleterious to DPD function and contribute to severe toxicity.4-9
Here, we present a case of a patient with a rare variant of unknown significance in DPYD who had a severe, life-threatening capecitabine toxicity. The variant was predicted to be deleterious using a recently reported in silico tool (DPYD-Varifier),10 and impaired function was confirmed in vitro.
METHODS
This study was approved by the Indiana University Institutional Review Board. Informed consent was obtained from the patient to have patient-derived specimens and medical records used for research and reporting. Patient germline DNA was obtained from whole blood and used for whole-genome sequencing (WGS), targeted DPYD genotyping, and Sanger sequencing. Detailed methods can be found in the Appendix for WGS and Sanger sequencing. Targeted genotyping was performed in the following Clinical Laboratory Improvement Amendments–certified laboratories: ARUP Laboratories (Salt Lake City, UT) and the Indiana University Pharmacogenomics Laboratory (Indianapolis, IN).
Integrated Genomics Viewer Version 2.4.10 (Broad Institute, Cambridge, MA)11 was used to visualize WGS data, and Ingenuity Variant Analysis (Qiagen, Germantown, MD) was used for variant identification and annotation. DPYD-Varifier10 was used to evaluate the functional impact of p.R235Q on DPD function. The effect of p.R235Q on DPD enzyme activity was determined in vitro as previously described.10
CASE REPORT
A 59-year-old Indian woman was diagnosed with metastatic colon cancer and started on a neoadjuvant treatment regimen of capecitabine (3 500-mg tablets orally twice a day), oxaliplatin, and bevacizumab (a treatment time line is presented in Fig 1). After 9 days of treatment, the patient developed grade 4 mucositis at the ileum and was hospitalized, at which time all chemotherapy was stopped. Hospital records indicate the presence of febrile neutropenia at admission; however, WBC counts were not available to report. While hospitalized, the patient was found to have an ileal obstruction. In all, the patient was hospitalized for 3 weeks. Because of the severity of toxicity, the patient was believed to be DPD deficient, and commercial testing for 3 toxicity-associated DPYD variants (c.1905+1G>A, c.1679T>G, and c.2846A>T) was performed. The patient was found to be wild-type for all 3 variants.
FIG 1.
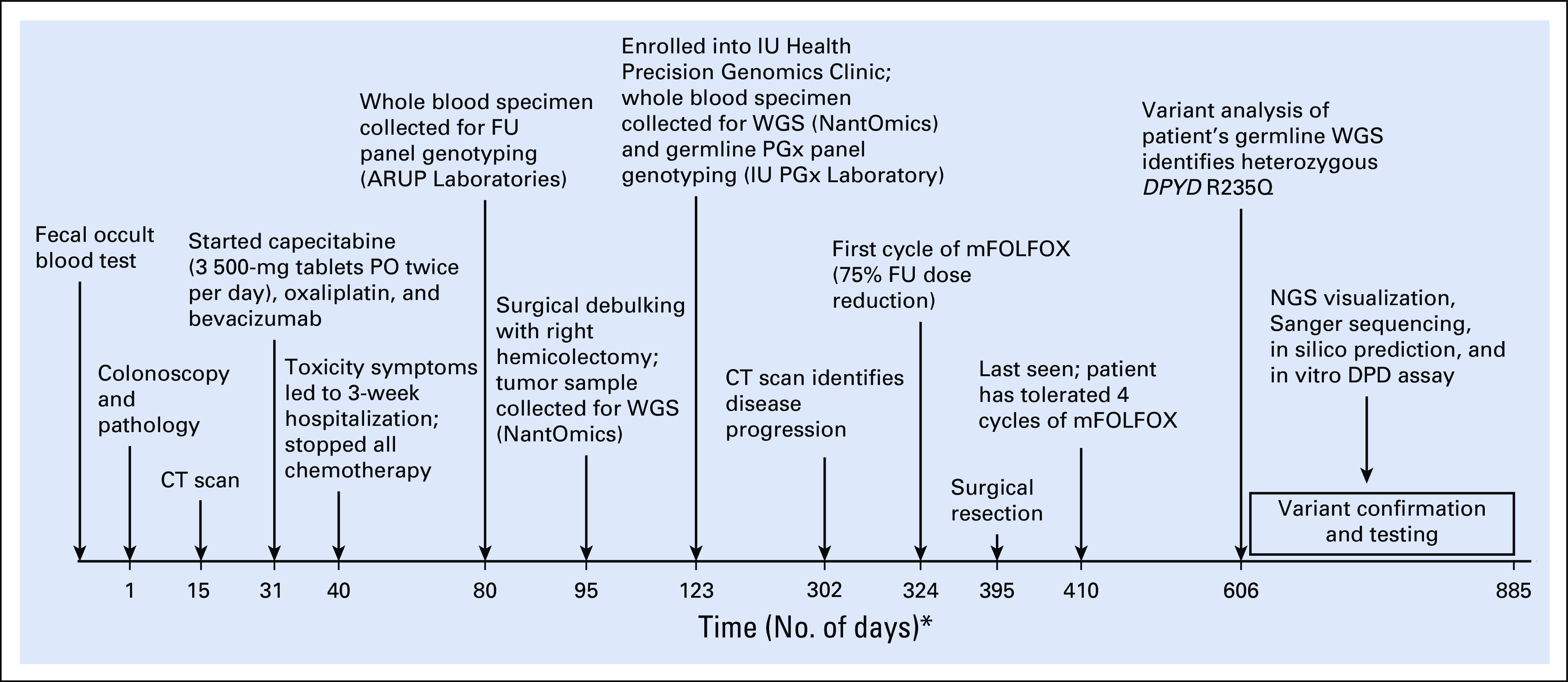
Time line of patient’s case report. This diagram shows the time line of key events in the diagnosis, treatment, and laboratory testing of DPYD variants. The horizontal line represents the number of days from initial diagnosis to the patient’s last encounter after modified infusional fluorouracil, leucovorin, and oxaliplatin (mFOLFOX) treatment. (*) Not drawn to scale. CT, computed tomography; DPD, dihydropyrimidine dehydrogenase; FU, fluorouracil; IU, Indiana University; NGS, next-generation sequencing; PGx, pharmacogenomics; PO, oral; WGS, whole-genome sequencing.
After surgical debulking, the patient enrolled into the Indiana University Health Precision Genomics Clinic for genome-guided cancer therapy (Fig 1). WGS was performed on germline and tumor DNA. As part of care, noncarrier status for c.1905+1G>A was confirmed. The patient remained off chemotherapy until computed tomography scan revealed disease progression. As a result of the previously observed toxicity, the patient was started on a modified leucovorin, FU, and oxaliplatin (mFOLFOX) regimen with a specific 75% FU dose reduction. The patient has tolerated 4 cycles of mFOLFOX therapy with no reported toxicity to date.
On the basis of the extreme observed toxicity, results of targeted DPYD genotyping, and tolerance to the reduced mFOLFOX regimen, we hypothesized that the patient might carry a deleterious DPYD variant that was not covered by the testing panel. Review of the germline WGS data confirmed noncarrier status for c.1905+1G>A, c.1679T>G, c.2846A>T, and c.1129-5923T>G. However, the patient carried a heterozygous rare missense variant in DPYD, rs755416212 (NM_000110.3:c.704G>A; NP_000101.2:p.Arg235Gln; referred to as p.R235Q; Fig 2A). Genotype was confirmed using Sanger sequencing (Fig 2B). Data in the Genome Aggregation Database and Exome Aggregation Consortium database indicate that this variant has a minor allele frequency of 0.00001.
FIG 2.
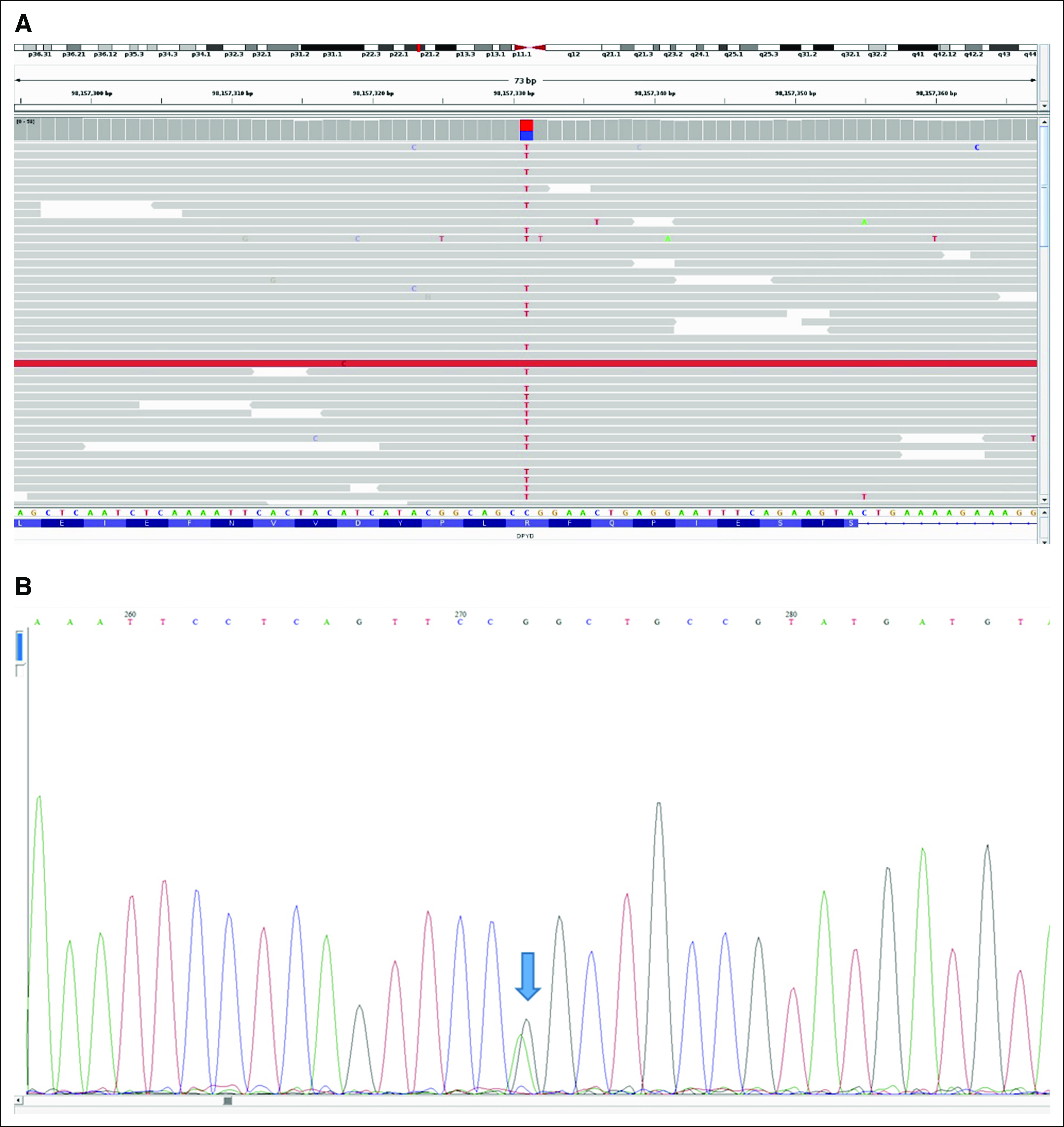
Presence of the heterozygous DPYD p.R235Q (c.704G>A) variant in the patient’s germline genome. (A) Whole-genome sequencing reads identified the heterozygous single-nucleotide variant (C>T on the + DNA strand depicted) in DPYD. Sequencing reads matching the reference sequence are shown in gray. Data were displayed using Integrated Genomics Viewer. (B) Sanger sequencing confirmed the presence of the heterozygous single-nucleotide variant (G>A on the − coding strand) in DPYD as identified by the arrow.
DPYD-Varifier, a gene-specific in silico variant classifier,6,10 predicted that p.R235Q was deleterious to DPD function. This prediction was validated using a previously reported in vitro assay for measuring variant function,10 which showed that the p.R235Q variant significantly reduced DPD activity by 88% compared with wild-type DPD (P = 6.26 × 10−7; Fig 3). We conclude that p.R235Q likely contributed to the patient’s severe, life-threatening capecitabine toxicity.
FIG 3.

Functional assessment of p.R235Q DPYD variant. The in vitro enzyme activity of recombinantly expressed *2A (c.1905+1G>A), *13 (c.1679T>G, p.I560S), p.D949V (c.2846A>T), p.R235Q (c.704G>A), and p.R235W (c.703C>T) variants was compared with reference (wild-type [WT]) dihydropyrimidine dehydrogenase (DPD). For each variant, the mean of 4 independent biologic replicate experiments is presented as a horizontal bar ± standard deviation (SD). Each biologic replicate was measured in triplicate (ie, 3 technical replicates each).
DISCUSSION
This case study illustrates the limitation of using targeted DPYD genotyping and the benefit of using sequence-based approaches to identify individuals at risk for severe fluoropyrimidine toxicity. The 4 known toxicity-associated DPYD variants (c.1905+1G>A, c.1679T>G, c.2846A>T, and c.1129-5923T>G) have been well characterized in the White population and have recommendations for genotype-guided dosing.2,3 However, several studies have identified novel rare DPYD variants associated with fluoropyrimidine toxicity using sequencing.4-9 Our study illustrates that sequencing may be beneficial for detecting unique, common, and rare variants, especially in individuals of non-White ancestry for whom there is limited pharmacogenetic knowledge.
The p.R235Q missense variant was previously observed in a patient presenting with hematuria who was diagnosed with DPD deficiency.12 The patient was compound heterozygous for the p.R235Q missense and p.C79X nonsense variants. DPYD analysis of the parents revealed that the father carried p.C79X and the mother carried p.R235Q. DPD activity in the mother was approximately 50% below the population average, as measured ex vivo using peripheral-blood mononuclear cells. This observation corroborates our in vitro finding of impaired DPD function of the p.R235Q variant. To our knowledge, this is the first report of an association between DPYD p.R235Q and capecitabine toxicity. Collectively, these data support the clinical actionability of p.R235Q.
Interestingly, p.R235Q’s effect was similar to the previously reported p.R235W variant (Fig 3), which had a significant 86% decrease in DPD activity compared with wild-type DPD (P = 6.44 × 10−7).13 This variant was found in a patient with DPD deficiency like the p.R235Q variant.14 Furthermore, one study examining the role of amino acid 235 on DPD function reported that additional missense mutations (p.R235A and p.R235K) deplete DPD activity in vitro.15 Amino acid 235 is well conserved and important for flavin adenine dinucleotide (FAD) binding.15,16 Deleterious variants in DPD have been found enriched at conserved residues and in close proximity to important domains such as the FAD domain.10 Altogether, our findings and these studies indicate this amino acid position does not tolerate missense changes and should be considered clinically actionable.
The p.R235W variant has been incorporated into CPIC’s guidelines for DPYD genotype-guided dosing with an assigned haploid activity score of 0 as a nonfunctional allele.2 Given the similar function of p.R235Q and p.R235W (Fig 3), we feel it may be appropriate to consider p.R235Q similarly with a haploid activity score of 0. As such, the patient in this study would be considered an intermediate metabolizer with a calculated activity score of 1 according to CPIC guidelines. The recommended FU dose reduction with an activity score of 1 is 50%. The fact that the patient tolerated FU with a 75% specific FU dose reduction with no reported toxicity suggests that CPIC’s recommendation of a 50% FU dose reduction might be sufficient to avoid toxicity while preserving drug efficacy for treatment. However, dose titration with therapeutic drug monitoring to prevent underdosing is recommended when available.
The patient was also tested for TYMS germline polymorphisms (rs11280056, rs45445694, and rs34743033) as part of the targeted genotyping panel. The patient was homozygous for the 6 base pair (bp) deletion (rs11280056) in the 3′ untranslated region (UTR; DEL/DEL), which is predictive for low TYMS expression.17,18 The patient also had 3 copies (3R) of the 28-bp tandem repeat in the 5′ UTR promoter enhancer region (rs45445694) with the absence of a variant (rs34743033, G>C) within the second tandem repeat of the 3R allele. Altogether, the 3RG/3RG genotype is associated with increased TYMS expression.19,20 The clinical association and utility of these TYMS polymorphisms with fluoropyrimidine toxicity are unclear.21-25 A recent meta-analysis found no significant association with severe GI or hematologic toxicities for TYMS (rs45445694 and rs11280056) or ENOSF1 (rs2612091) variants.26 On the basis of the literature, the TYMS DEL/DEL genotype was unlikely to contribute to the patient’s severe toxicity.
In conclusion, this case demonstrates the potential benefits of sequence-based DPYD genotyping to fluoropyrimidine dose individualization. One challenge to using sequence-level data is the classification of novel and understudied variants. We overcame this challenge using DPYD-Varifier, an in silico classifier, to predict whether the variant was deleterious to DPD function. The prediction was then validated by assessing the variant’s impact on DPD activity in vitro. Collectively, this approach of using genomic sequencing along with DPYD-Varifier and functional testing can help to identify rare toxicity-linked DPYD variants and improve genotype-guided therapy in patients treated with fluoropyrimidines.
ACKNOWLEDGMENT
We thank Surendra Buddi for bioinformatic support.
APPENDIX
Whole-Genome Sequencing
Germline and somatic whole-genome sequencing (WGS) was performed by NantOmics (Culver City, CA), a Clinical Laboratory Improvement Amendments–certified laboratory, as previously described (Rabizadeh S, et al: Oncotarget 9:19223-19232, 2018). Sequencing depth for all DPYD exons was ≥ 30× by WGS.
Sanger Sequencing
The patient’s DNA served as the template to amplify the region containing the p.R235Q variant by polymerase chain reaction (PCR). PCR was performed using Platinum SuperFi PCR Mastermix (Thermofisher Scientific, Waltham, MA) using the following primers: 5′-GCATCTTTCTGCTTCTGCCTGAT-3′ (forward 1), 5′-GTATTGAAATTGCTTTTGGCCAGTT-3′ (reverse 1), 5′-TGTCCTCATGCATATCTTGTGTG-3′ (forward 2), and 5′-TCCTTTCTTTTTGAGCAGTACACA-3′ (reverse 2). Primers were obtained from Integrated DNA Technologies (Coralville, IA). Amplification conditions were 98°C for 30 seconds, 35 cycles at 98°C for 10 seconds, 62.1°C for 10 seconds, and 72°C for 30 seconds. Final extension was at 72°C for 5 minutes. Individual PCR products from both PCR reactions were purified using the MinElute PCR purification kit (Qiagen, Germantown, MD) according to the user manual. DNA concentration was measured using the Qubit dsDNA BR assay kit according to the user manual (Invitrogen, Waltham, MA). Samples were prepared and Sanger sequenced by ACGT (Germantown, MD) using each PCR product as the template with 1 of the 4 primers individually described earlier.
Presented in part at the American Society for Clinical Pharmacology and Therapeutics Conference, Houston, TX, March 18-21, 2020.
Supported by National Institutes of Health (NIH) Grant No. R35GM131812 (T.C.S.), NIH Grant No. P30CA15083 (R.B.D.), Vera Bradley Foundation (R.C.L., M.R., B.P.S.), and the Indiana University Grand Challenge Precision Health Initiative (M.R., B.P.S., T.C.S.).
The Indiana University School of Medicine Pharmacogenomics Laboratory is a fee-for-service clinical laboratory that offers clinical pharmacogenetic testing (V.M.P.).
AUTHOR CONTRIBUTIONS
Conception and design: Reynold C. Ly, Victoria M. Pratt, Bryan P. Schneider, Todd C. Skaar
Financial support: Todd C. Skaar
Provision of study materials or patients: Milan Radovich
Collection and assembly of data: Reynold C. Ly, Remington E. Schmidt, Milan Radovich
Data analysis and interpretation: Reynold C. Ly, Remington E. Schmidt, Patrick J. Kiel, Victoria M. Pratt, Milan Radovich, Steven M. Offer, Robert B. Diasio, Todd C. Skaar
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Patrick J. Kiel
Employment: Amgen
Speakers' Bureau: Takeda, Celgene, Genentech
Victoria M. Pratt
Stock and Other Ownership Interests: Quest Diagnostics, LabCorp
Other Relationship: Avalon Healthcare
Uncompensated Relationships: Veritas Genetics
Milan Radovich
Stock and Other Ownership Interests: LifeOmic, Macrogenics, Immunomedics, ArQule, Tyme Technologies
Travel, Accommodations, Expenses: LifeOmic
Todd C. Skaar
Honoraria: Tabula Rasa Healthcare
Travel, Accommodations, Expenses: Tabula Rasa Healthcare
Other Relationship: National Institutes of Health
No other potential conflicts of interest were reported.
REFERENCES
- 1.Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018;19:1459–1467. doi: 10.1016/S1470-2045(18)30686-7. [DOI] [PubMed] [Google Scholar]
- 2.Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103:210–216. doi: 10.1002/cpt.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunenburg CATC, van der Wouden CH, Nijenhuis M, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2020;28:508–517. doi: 10.1038/s41431-019-0540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del Re M, Quaquarini E, Sottotetti F, et al. Uncommon dihydropyrimidine dehydrogenase mutations and toxicity by fluoropyrimidines: A lethal case with a new variant. Pharmacogenomics. 2016;17:5–9. doi: 10.2217/pgs.15.146. [DOI] [PubMed] [Google Scholar]
- 5.van Kuilenburg AB, Meijer J, Maurer D, et al. Severe fluoropyrimidine toxicity due to novel and rare DPYD missense mutations, deletion and genomic amplification affecting DPD activity and mRNA splicing. Biochim Biophys Acta Mol Basis Dis. 2017;1863:721–730. doi: 10.1016/j.bbadis.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 6.Shrestha S, Tapper EE, Trogstad-Isaacson CS, et al. Dose modification for safe treatment of a compound complex heterozygous DPYD variant carrier with 5-fluorouracil. JCO Precis Oncol. doi: 10.1200/PO.18.00179. 10.1200/PO.18.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong CC, Lam CW, Lam KO, et al. A novel DPYD variant associated with severe toxicity of fluoropyrimidines: Role of pre-emptive DPYD genotype screening. Front Oncol. 2018;8:279. doi: 10.3389/fonc.2018.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bukhari N, Azam F, Alfawaz M, et al. Identifying a novel DPYD polymorphism associated with severe toxicity to 5-FU chemotherapy in a Saudi patient. Case Rep Genet. 2019;2019:5150725. doi: 10.1155/2019/5150725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmirotta R, Lovero D, Delacour H, et al. Rare dihydropyrimidine dehydrogenase variants and toxicity by fluoropyrimidines: A case report. Front Oncol. 2019;9:139. doi: 10.3389/fonc.2019.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shrestha S, Zhang C, Jerde CR, et al. Gene-specific variant classifier (DPYD-varifier) to identify deleterious alleles of dihydropyrimidine dehydrogenase. Clin Pharmacol Ther. 2018;104:709–718. doi: 10.1002/cpt.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Kuilenburg AB, Meijer J, Dobritzsch D, et al. Identification of two novel mutations C79X and R235Q in the dihydropyrimidine dehydrogenase gene in a patient presenting with hematuria. Nucleosides Nucleotides Nucleic Acids. 2008;27:809–815. doi: 10.1080/15257770802146247. [DOI] [PubMed] [Google Scholar]
- 13.Offer SM, Fossum CC, Wegner NJ, et al. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014;74:2545–2554. doi: 10.1158/0008-5472.CAN-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vreken P, Van Kuilenburg AB, Meinsma R, et al. Dihydropyrimidine dehydrogenase (DPD) deficiency: Identification and expression of missense mutations C29R, R886H and R235W. Hum Genet. 1997;101:333–338. doi: 10.1007/s004390050637. [DOI] [PubMed] [Google Scholar]
- 15.Lohkamp B, Voevodskaya N, Lindqvist Y, et al. Insights into the mechanism of dihydropyrimidine dehydrogenase from site-directed mutagenesis targeting the active site loop and redox cofactor coordination. Biochim Biophys Acta. 2010;1804:2198–2206. doi: 10.1016/j.bbapap.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Weidensee S, Goettig P, Bertone M, et al. A mild phenotype of dihydropyrimidine dehydrogenase deficiency and developmental retardation associated with a missense mutation affecting cofactor binding. Clin Biochem. 2011;44:722–724. doi: 10.1016/j.clinbiochem.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 17.Mandola MV, Stoehlmacher J, Zhang W, et al. A 6 bp polymorphism in the thymidylate synthase gene causes message instability and is associated with decreased intratumoral TS mRNA levels. Pharmacogenetics. 2004;14:319–327. doi: 10.1097/00008571-200405000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Pullmann R, Jr, Abdelmohsen K, Lal A, et al. Differential stability of thymidylate synthase 3′-untranslated region polymorphic variants regulated by AUF1. J Biol Chem. 2006;281:23456–23463. doi: 10.1074/jbc.M600282200. [DOI] [PubMed] [Google Scholar]
- 19.Mandola MV, Stoehlmacher J, Muller-Weeks S, et al. A novel single nucleotide polymorphism within the 5′ tandem repeat polymorphism of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activity. Cancer Res. 2003;63:2898–2904. [PubMed] [Google Scholar]
- 20.Yawata A, Kim SR, Miyajima A, et al. Polymorphic tandem repeat sequences of the thymidylate synthase gene correlates with cellular-based sensitivity to fluoropyrimidine antitumor agents. Cancer Chemother Pharmacol. 2005;56:465–472. doi: 10.1007/s00280-005-1018-z. [DOI] [PubMed] [Google Scholar]
- 21.Lecomte T, Ferraz JM, Zinzindohoué F, et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Cancer Res. 2004;10:5880–5888. doi: 10.1158/1078-0432.CCR-04-0169. [DOI] [PubMed] [Google Scholar]
- 22.Jennings BA, Kwok CS, Willis G, et al. Functional polymorphisms of folate metabolism and response to chemotherapy for colorectal cancer, a systematic review and meta-analysis. Pharmacogenet Genomics. 2012;22:290–304. doi: 10.1097/FPC.0b013e328351875d. [DOI] [PubMed] [Google Scholar]
- 23.Rosmarin D, Palles C, Pagnamenta A, et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut. 2015;64:111–120. doi: 10.1136/gutjnl-2013-306571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meulendijks D, Jacobs BA, Aliev A, et al. Increased risk of severe fluoropyrimidine-associated toxicity in patients carrying a G to C substitution in the first 28-bp tandem repeat of the thymidylate synthase 2R allele. Int J Cancer. 2016;138:245–253. doi: 10.1002/ijc.29694. [DOI] [PubMed] [Google Scholar]
- 25.Rosmarin D, Palles C, Church D, et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: Investigation in the QUASAR2 study, systematic review, and meta-analysis. J Clin Oncol. 2014;32:1031–1039. doi: 10.1200/JCO.2013.51.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamzic S, Kummer D, Froehlich TK, et al. Evaluating the role of ENOSF1 and TYMS variants as predictors in fluoropyrimidine-related toxicities: An IPD meta-analysis. Pharmacol Res. 2020;152:104594. doi: 10.1016/j.phrs.2019.104594. [DOI] [PubMed] [Google Scholar]