

Abstract
PURPOSE
The microsatellite instability (MSI) or deficient mismatch repair (dMMR) phenotype is usually regarded as a single biologic entity, given the absence of comparative analyses regarding prognosis and response to chemotherapy between sporadic and familial dMMR cancers.
PATIENTS AND METHODS
Patients with stage III colon cancers were randomly assigned to FOLFOX (leucovorin, fluorouracil, and oxaliplatin) with or without cetuximab in 2 large adjuvant phase III trials (N = 5,577). Among patients with MSI and KRAS exon 2 wild-type (WT) tumors, the prognostic and predictive impacts of sporadic versus familial dMMR cancers and BRAF V600E mutational status were determined. Multivariable Cox proportional hazards models were used to assess disease-free survival (DFS) by treatment arm, adjusting for age, sex, tumor grade, Eastern Cooperative Oncology Group performance status, pT/pN stage, and primary tumor location.
RESULTS
Among patients with MSI status with complete data for dMMR mechanism analysis (n = 354), 255 (72%) had sporadic (BRAF mutation and/or MLH1 methylation) and 99 (28%) had familial tumors (BRAF WT and unmethylated MLH1 or loss of MSH2/MSH6/PMS2 protein expression). A large proportion of dMMR sporadic tumors were mutated for BRAF (n = 200). In patients treated with FOLFOX, DFS did not differ statistically by dMMR mechanism, whereas in patients treated with FOLFOX plus cetuximab, those with sporadic tumors had worse DFS than those with familial cancers (multivariable hazard ratio, 2.69; 95% CI, 1.02 to 7.08; P = .04). Considering the predictive utility, the interaction between treatment and dMMR mechanism was significant (P = .03). Furthermore, a nonsignificant trend toward a deleterious effect of adding cetuximab to FOLFOX was observed in patients with BRAF-mutant but not BRAF WT tumors.
CONCLUSION
The addition of cetuximab to adjuvant FOLFOX was associated with shorter DFS in patients with sporadic dMMR colon cancer. Additional studies are needed to validate these results in metastatic disease.
INTRODUCTION
Colorectal cancer (CRC) is the second most common malignancy in developed countries. For patients with nonmetastatic colon cancer, surgical resection offers a potential cure. However, approximately one fourth of patients with stage III colon cancer develop recurrence and die as a result of metastatic disease despite adjuvant chemotherapy. Three phase III adjuvant trials demonstrated that adding oxaliplatin to fluoropyrimidine (fluorouracil [FU] or capecitabine) was associated with improvement of survival in patients with stage III colon cancer, thereby establishing FOLFOX (leucovorin, fluorouracil, and oxaliplatin) or XELOX (capecitabine plus oxaliplatin) as the standard of care.1 More recent adjuvant studies have not shown any benefit to adding targeted agents (bevacizumab or cetuximab) to FOLFOX or XELOX in patients with stage III colon cancer,2-4 despite improved outcomes with these combinations in the metastatic setting.5
Increasing evidence indicates that CRC is a biologically heterogeneous disease that develops via distinct pathways involving combinations of genetic and epigenetic changes. Defining tumor subtypes based on pathway-driven alterations has the potential to improve prognostication and guide therapy. Two well-described pathways of colorectal tumorigenesis include chromosomal instability and microsatellite instability (MSI), the latter being the consequence of deficient DNA mismatch repair (dMMR). MSI phenotype is observed in approximately 10% to 15% of patients with localized CRC and 3% to 5% of those with metastatic disease.6 dMMR can arise in a familial context from a germ line mutation in an MMR gene (MLH1, MSH2, MSH6, or PMS2), known as Lynch syndrome, or, more commonly, sporadically from epigenetic inactivation of MLH1 as a result of hypermethylation of promoter regions of cancer-specific genes, known as the CpG island methylator phenotype (CIMP).7,8 Sporadic dMMR CRCs frequently have an activating somatic V600E mutation in the BRAF oncogene (approximately 50%-70% of cases) that is absent in familial dMMR tumors.9,10
Molecular subtypes of CRC have been reported by our groups11,12 and others.13-15 These studies defined subtypes of CRC with statistically significant different clinical and pathologic characteristics as well as patient survival rates. In patients undergoing surgical resection of CRC, most studies found that the dMMR/MSI phenotype was associated with a better prognosis compared with cohorts of patients with proficient MMR (pMMR)/microsatellite stable (MSS) tumors.16,17 Furthermore, data regarding the predictive value of MMR status have suggested that patients with dMMR/MSI tumors do not benefit from FU,16,17 whereas the addition of oxaliplatin to FU may restore the efficacy of adjuvant chemotherapy in dMMR/MSI stage III colon cancer.18-20 However, few data exist regarding the prognostic and predictive impacts of dMMR/MSI tumors according to the origin of MMR inactivation (sporadic v familial) because of the relatively low incidence of these molecular characteristics.21 Moreover, analyses based on sporadic versus familial dMMR/MSI tumors should include BRAF V600E status because that mutation may have an independent prognostic value, as observed in pMMR/MSS tumors,11,12 and is significantly linked to the mechanism of sporadic dMMR.9,10
In this pooled analysis, we examined the prognostic as well as predictive utility of familial and sporadic dMMR tumors in patients with stage III colon cancer treated with FOLFOX with or without cetuximab in 2 phase III randomized trials: North Central Cancer Treatment Group (NCCTG, now part of the Alliance for Clinical Trials in Oncology) N01472 and PETACC8.3
PATIENTS AND METHODS
Study Population
This pooled analysis included all patients with available tumor blocks of resected stage III (any T, N1 or N2, M0) colon adenocarcinoma who participated in the NCCTG N01472 and PETACC83 adjuvant randomized phase III trials and provided institutional review board–approved biologic informed consent. The protocol and all amendments were approved by the institutional review board or ethics committee at each participating institution. Biospecimens were prospectively collected in both studies. Patients were randomly assigned after surgery to receive 6 months of adjuvant FOLFOX chemotherapy with or without cetuximab, as described previously.2,3 The NCCTG N0147 trial enrolled patients between February 2004 and November 2009; PETACC8 enrolled patients between December 2005 and November 2009. Amendments restricted enrollment to patients with KRAS exon 2 wild-type (WT) tumors in both studies. In both trials, the addition of cetuximab to FOLFOX failed to improve disease-free survival (DFS) overall as well as in patients with KRAS exon 2 WT tumors.2,3 Our study was restricted to patients with dMMR/MSI tumors without KRAS exon 2 mutations to evaluate the prognostic and predictive impacts of the dMMR mechanism (familial v sporadic origin) by treatment arm (FOLFOX alone or with cetuximab). The molecular analysis was centralized at the Mayo Clinic (Rochester, MN) for the NCCTG N0147 trial and at the Georges Pompidou European Hospital (Paris, France) for the PETACC8 trial.
MMR Status Determination
MMR tumor status was determined by immunohistochemistry (IHC) or by MSI testing when IHC was indeterminate, as previously described for each trial.12,22 dMMR was defined as loss of expression of ≥ 1 MMR proteins by IHC or exhibition of high-level MSI (MSI-H) on polymerase chain reaction (PCR) –based MSI testing. pMMR tumors were defined by showing intact MMR expression by IHC or MSS or low-level MSI (MSI-L) status on MSI testing.
DNA Extraction and Mutation Analysis
Mutation status was determined using genomic DNA extracted from formalin-fixed, paraffin-embedded (FFPE) tumor tissue that contained at least 50% tumor cells using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Testing for the c.1799T>A p.V600E BRAF mutation in exon 15 was performed as previously described.12,22
MLH1 Methylation
Methylation of MLH1 promoter was determined in tumors exhibiting a loss of MLH1 protein expression to distinguish patients with sporadic from familial dMMR status. For NCCTG N0147 and PETACC8 trials, tumor DNA was extracted from FFPE tissue and bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA). PCR primers were designed to detect differences between methylated and unmethylated DNA for the MLH1 promoter, as previously described.11
Determination of Sporadic Versus Familial dMMR/MSI Cases
As previously described and among patients with dMMR tumors, sporadic cases were defined as those exhibiting a loss of MLH1 protein expression with BRAF V600E mutation and/or showing methylation of MLH1 promoter.6 Familial cases were defined as patient tumors with loss of MSH2 or MSH6 (or both) or PMS2 (isolated) protein expression or loss of MLH1 protein expression with BRAF WT and unmethylated MLH1. For patient tumors tested with MSI assay, sporadic cases were defined as MSI-H tumors with BRAF V600E mutation or MLH1 methylation, and familial cases as MSI-H tumors without BRAF V600E mutation and unmethylated MLH1 promoter.6
Statistical Analysis
Biomarker status data were analyzed with investigators blinded to patient outcomes. DFS was defined as the time from random assignment to first documented local or metastatic recurrence or death linked to the disease, whichever occurred first. DFS curves were estimated with the Kaplan-Meier method. For comparisons of baseline characteristics, categorical outcomes were analyzed with χ2 tests. Continuous variables are presented as the median with interquartile range. The multivariable Cox model was used to estimate hazard ratios (HRs) with 95% CIs and to calculate P values for the impact of dMMR mechanism (sporadic v familial) and BRAF V600E mutational status (mutated v WT) on DFS. Adjusted factors included age, sex, tumor grade, Eastern Cooperative Oncology Group performance status, pT/pN stage, and primary tumor location. The prognostic analyses were initially performed separately in each treatment arm (FOLFOX alone and FOLFOX with cetuximab) to avoid any interaction between the potential effect of biomarkers and treatment. Then, to investigate the predictive ability of molecular subtype, a treatment–by–molecular subtype interaction term was fitted alongside the main effects of molecular subtype and treatment.
Analyses were carried out using a 2-sided statistical significance level of 5%. Analyses were performed using SAS software (version 9.4; SAS Institute, Cary, NC). The Alliance Statistics and Data Management Center conducted data integration and statistical analyses based on data frozen in October 2016. Data quality was ensured by review of data by the Alliance Statistics and Data Center and by the study chairperson following Alliance policies.
RESULTS
Patients Characteristics
From among the 5,577 patients included in the NCCTG N0147 and PETACC8 trials,2,3 we excluded patients who received irinotecan-based adjuvant chemotherapy per the initial design of the NCCTG N0147 trial (n = 379) and those who did not sign the biologic informed consent form or had unavailable material and/or technical failure for determination of MMR/MSI status (n = 524; Fig 1). Among the 4,674 patients tested for MMR status, we identified 499 patients with dMMR/MSI tumors for which 78 were excluded because their tumors had mutation of KRAS exon 2. Among the 421 patients with dMMR/MSI status included in this study population, 354 had available data to enable categorization as sporadic (n = 255; 72.0%) or familial tumors (n = 99; 28.0%), and 407 had available data for analysis of BRAF mutational status as WT (n=205; 50.4%) or mutated (V600E; n = 202; 49.6%; Fig 1). Demographic and clinical characteristics of the study population are listed in Table 1 and stratified by the dMMR mechanism (sporadic v familial) and BRAF V600E mutational status (Table 1). The median follow-up of the study population was 4.2 years.
FIG 1.

Flowchart of molecular study of NCCTG (North Central Cancer Treatment Group) N0147 and PETACC8 trials evaluating the impact of deficient DNA mismatch repair (dMMR) mechanism (sporadic v familial) and BRAF V600E mutational status in patients with dMMR/microsatellite instability (MSI) stage III colon cancer. MSS, microsatellite stable; pMMR, proficient DNA mismatch repair; WT, wild type.
TABLE 1.
Clinicopathologic Characteristics of Patients With KRAS Exon 2 WT dMMR/MSI Stage III Colon Cancer From PETACC8 and N0147 Trials

As expected, patients with sporadic dMMR/MSI tumors were significantly more likely to be older, be female, and have a proximal cancer compared with patients with dMMR/MSI familial disease6 (all P < .01; Table 1). BRAF V600E was detected in a majority of patients with sporadic dMMR/MSI disease (80%) and in 2 patients with familial dMMR/MSI tumors (1.4%10; P < .0001; Table 1). Patients with BRAF-mutated versus WT tumors were significantly more likely to be older, be female, and have a proximal tumor6 (all P < .0001; Table 1).
Prognostic and Predictive Impacts of dMMR Mechanism
In patients treated with FOLFOX alone, there was no significant difference in DFS between those with sporadic and those with familial dMMR disease (multivariable HR, 0.81; 95% CI, 0.29 to 2.22; P = .68; Fig 2A), whereas for those treated with FOLFOX plus cetuximab, patients with sporadic tumors had significantly worse DFS as compared with patients with familial tumors (multivariable HR, 2.69; 95% CI, 1.02 to 7.08; P = .04; Fig 2B).
FIG 2.
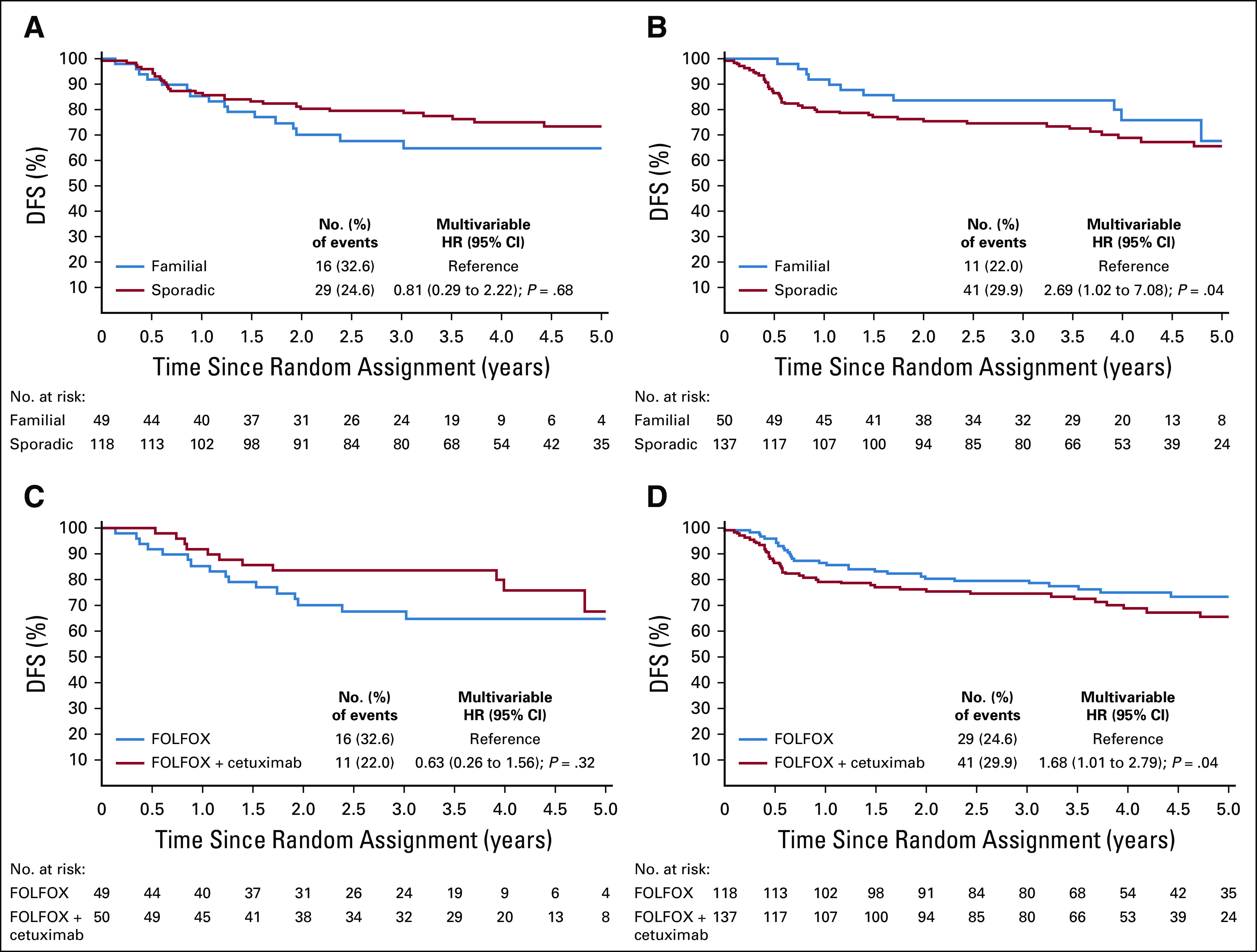
Disease-free survival (DFS) in patients with KRAS exon 2 wild-type deficient DNA mismatch repair (dMMR)/microsatellite instability (MSI) stage III colon cancer treated with (A) FOLFOX (leucovorin, fluorouracil, and oxaliplatin) or (B) FOLFOX plus cetuximab according to dMMR mechanism (familial v sporadic origin) and in (C) familial and (D) sporadic dMMR/MSI cases according to treatment arm (FOLFOX v FOLFOX + cetuximab). Multivariable Cox models adjusted for age, sex, tumor grade, Eastern Cooperative Oncology Group performance status, pT/pN stage, and primary tumor location. HR, hazard ratio.
Considering the predictive impact, we observed a significant interaction between the dMMR mechanism and treatment (P interaction = .03). In patients with dMMR tumors categorized as familial, there was a trend toward better outcome for those treated with FOLFOX plus cetuximab versus FOLFOX (multivariable HR, 0.63; 95% CI, 0.26 to 1.56; P = .32; Fig 2C). However, among patients with dMMR tumors categorized as sporadic, those treated with FOLFOX plus cetuximab had significantly worse DFS than those treated with FOLFOX alone (multivariable HR, 1.68; 95% CI, 1.01 to 2.79; P = .04; Fig 2D).
Furthermore, these results regarding the prognostic and predictive impacts of the dMMR mechanism according to treatment arm remained similar after the inclusion of patients with KRAS exon 2–mutated tumors (Appendix Table A1).
Prognostic and Predictive Impacts of BRAF V600E Status
There was no significant difference in DFS between WT and mutated BRAF tumors by treatment arm: FOLFOX (multivariable HR, 1.09; 95% CI, 0.52 to 2.25; P = .82; Fig 3A) versus FOLFOX plus cetuximab (multivariable HR, 1.28; 95% CI, 0.64 to 2.55; P = .48; Fig 3B).
FIG 3.
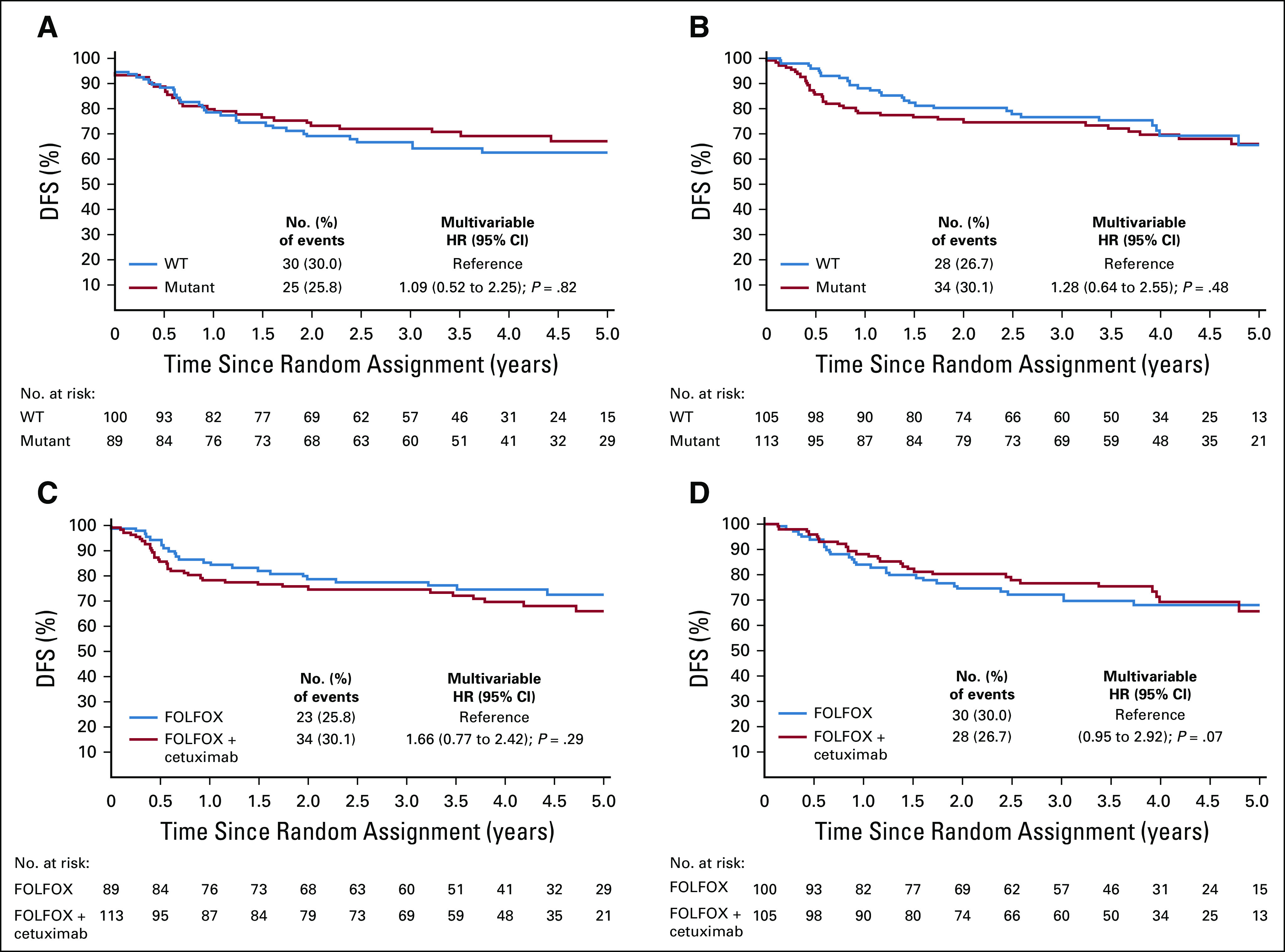
Disease-free survival (DFS) in patients with KRAS exon 2 wild-type (WT) deficient DNA mismatch repair (dMMR)/microsatellite instability (MSI) stage III colon cancer treated with (A) FOLFOX (leucovorin, fluorouracil, and oxaliplatin) or (B) FOLFOX plus cetuximab according to the BRAF V600E mutational status (WT v mutated) and in (C) BRAF mutated and (D) WT tumors according to treatment arm (FOLFOX v FOLFOX + cetuximab). Multivariate Cox models adjusted for age, sex, tumor grade, Eastern Cooperative Oncology Group performance status, pT/pN stage, and primary tumor location. HR, hazard ratio.
Furthermore, a nonsignificant trend toward a deleterious effect of adding cetuximab to FOLFOX was observed in patients with BRAF V600E–mutated tumors (multivariable HR, 1.66; 95% CI, 0.95 to 2.92; P = .07; Fig 3C), but not in those with WT BRAF (Fig 3D; P interaction = .40). The results of these analyses after inclusion of patients with KRAS exon 2–mutated tumors were not different (Appendix Table A2).
DISCUSSION
To our knowledge, our pooled analysis of data from 2 large randomized studies showed for the first time that addition of cetuximab to FOLFOX was associated with reduced DFS in patients with dMMR/MSI sporadic colon cancers, which was not observed for dMMR/MSI familial cases. The P value for interaction effect by dMMR mechanism (sporadic v familial) and treatment arm (FOLFOX v FOLFOX plus cetuximab) was statistically significant, highlighting the fact that the sporadic origin of dMMR/MSI tumors may be a predictive marker of a deleterious effect of cetuximab. Regarding the impact of BRAF mutational status, we observed a nonsignificant trend toward a deleterious effect of cetuximab for patients with BRAF V600E–mutated tumors. This observation is logically expected, because all dMMR/MSI tumors with BRAF mutation are of sporadic origin, but all dMMR/MSI sporadic cases are not mutated for BRAF (20% of BRAF WT in our study). Moreover, BRAF mutational status (mutated v WT) had no prognostic impact on DFS for the subgroup of patients with sporadic dMMR/MSI colon cancer, whatever the treatment arm (FOLFOX or FOLFOX plus cetuximab; Appendix Table A3).
The interaction between anti–epidermal growth factor receptor (EGFR) monoclonal antibodies and dMMR mechanism with impact on clinical outcome of patients with CRC has not been previously recognized. In the randomized COIN trial that compared oxaliplatin and fluoropyrimidine with or without cetuximab in metastatic CRC, addition of cetuximab was associated with shorter survival in patients with MSI as compared with MSS tumors.23 The association of cetuximab with adverse outcomes seemed to be limited to patients whose MSI tumors were mutated for BRAF V600E, which is consistent with a sporadic origin. These results, however, were not statistically significant, likely because of the small number of patients with MSI status analyzed in this series (N = 45, including 9 patients with BRAF-mutated tumors).23 More recently, the translational analysis of the randomized CALGB/SWOG 80405 trial showed that overall survival (OS) with first-line chemotherapy (FOLFOX or FOLFIRI) for patients with MSI metastatic CRC (n = 37) was shorter for those receiving cetuximab compared with bevacizumab (median OS, 11.9 v 30.0 months). In contrast, median OS was similar between cetuximab and bevacizumab treatment arms for those with MSS metastatic CRC (n = 586; median OS, 30.7 v 30.3 months).24 Despite the low number of patients with MSI status, it would be interesting to know the results according to dMMR mechanism, although sporadic cases are usually in the majority over familial cases. This negative effect of EGFR inhibitors in patients with (sporadic) MSI tumors is also supported by worse clinical outcomes in tumors originating in the right colon (enriched with MSI/BRAF mutation) compared with tumors originating in the left colon.25 Data suggest that the CIMP-positive phenotype could be associated with poorer survival in patients with metastatic CRC receiving anti-EGFR–based therapy.23 CIMP results from hypermethylation of multiple cancer-specific genes in addition to MLH1, which may underlie differential sensitivity to cetuximab.8 Of note, in CRCs, the CIMP-positive phenotype is not exclusively associated with dMMR/MSI tumors. Approximately 30% are observed in MSS tumors, a group in which mutation of BRAF V600E is infrequent (approximately 20%).26 Interestingly, others molecular alterations such as HER2 or MET amplification, NTRK or RET rearrangement, and PTEN or PIK3CA mutation were shown to be significantly associated with resistance to anti-EGFR agents in metastatic CRC. Moreover, dMMR/MSI status was significantly associated with these molecular determinants of primary resistance to anti-EGFR treatment.27
Another interesting finding in our study is that mutation of BRAF V600E in stage III colon cancer was not associated with poor prognosis in patients with dMMR/MSI status treated with standard FOLFOX (Fig 3A), in contrast to those with pMMR/MSS status.28 Evidence suggests that BRAF V600E mutation is associated with poorer survival after recurrence in both MSS and MSI tumors,29 as well as in metastatic CRCs with both MSS and MSI tumors.5,30 However, few data are available concerning the prognostic impact of BRAF V600E in patients with dMMR/MSI colon cancer receiving standard FOLFOX adjuvant chemotherapy. Previously, our groups evaluated the prognostic impact of BRAF V600E in patients with MSI stage III colon cancer included in the N0147 and PETACC8 trials.28 However, the data from the 2 treatment arms (FOLFOX and FOLFOX plus cetuximab) were pooled, limiting the robustness of the results, because our current study suggests that cetuximab may have a negative impact in the subgroup of patients with sporadic dMMR/MSI disease.
Our study has several strengths, which include prospective data and biomarker collection from patients enrolled in 2 large recent randomized studies. To our knowledge, our report includes the largest clinical series of patients with dMMR/MSI colon cancer evaluated for prognosis and prediction according to the dMMR mechanism. Study limitations include the lack of confirmation by germ line MMR mutation testing of categorization of dMMR/MSI tumors into suspected familial tumors. Furthermore, the algorithm used in our study was not able to identify Lynch-like syndrome (ie, dMMR/MSI status with BRAF WT and unmethylated MLH1 in tumor tissue, with no found germ line MMR gene mutation), because we did not perform germ line testing. Evidence indicates that such cases may result from double somatic mutations. We emphasize that definition of our categorization of sporadic and familial dMMR/MSI cases was inferred in this study cohort. In our series, we identified 2 patients with loss of MSH2 and PMS2, respectively (with intact MLH1 protein expression by IHC), and both were found to have BRAF V600E mutation. Parsons et al10 showed in a literature review that BRAF V600E mutation may occasionally be identified in patients with germ line mutation in an MMR gene, with a frequency of approximately 1% across all studies. Given the rarity of this association and the possibility of technical artifact, our study data were also analyzed excluding these 2 patients, and no significant differences were found in any of the results. Furthermore, the deleterious effect resulting from the addition of cetuximab to FOLFOX was observed in patients treated in the adjuvant setting, where anti-EGFR monoclonal antibodies are currently not recommended. Therefore, these results should be interpreted with caution, because the adjuvant and metastatic settings are not easily cross-comparable, and our findings need to be validated in the metastatic setting, where anti-EGFR agents are a therapeutic option for patients with RAS WT tumors.
In conclusion, our study showed that the addition of cetuximab to FOLFOX was associated with shorter DFS in patients with sporadic dMMR/MSI stage III colon cancers. Additional analysis based on molecular alterations or epigenetic changes involved in dMMR/MSI is needed to better understand the negative interaction between cetuximab and sporadic dMMR/MSI tumors. These data have important implications for use of anti-EGFR therapies in patients with dMMR/MSI status and warrant further validation, notably in cohorts of patients with RAS WT metastatic CRC, for whom anti-EGFR monoclonal antibodies are routinely used.
ACKNOWLEDGMENT
We thank the investigators of the NCCTG N0147 and PETACC8 trials for patient accrual contributions and the patients who participated in these clinical trials.
Appendix
TABLE A1.
Multivariable Analysis for DFS in Patients With dMMR/MSI Stage III Colon Cancer (including WT and mutated KRAS exon 2 tumor) According to Treatment Arm and dMMR Mechanism

TABLE A2.
Multivariable Analysis for DFS in Patients With dMMR/MSI Stage III Colon Cancer (including WT and mutated KRAS exon 2 tumor) According to Treatment Arm and BRAF V600E Status

TABLE A3.
Multivariable Analysis for DFS in Patients With Sporadic dMMR/MSI Stage III Colon Cancer (KRAS exon 2 WT) According to BRAF V600E Status

Footnotes
The PETACC8 study was sponsored by the Fédération Francophone de Cancérologie Digestive (FFCD) that was responsible for the study management. Merck KGaA and Sanofi-Aventis supported the study: Merck provided the study cetuximab and financial support for study management; Sanofi-Aventis provided financial support for the provision of oxaliplatin to Belgian sites when necessary. The N0147 trial was conducted as a collaborative trial of the North Central Cancer Treatment Group (NCCTG), Mayo Clinic, which is now part of the Alliance for Clinical Trials in Oncology. The study was supported in part by grants from the National Cancer Institute of the National Institutes of Health under Award Nos. R01 CA210509-01A1 (to FAS), U10CA025224 (to North Central Treatment Group), the NCCTG Biospecimen Resource (U24CA114740), and the Alliance for Clinical Trials in Oncology (U10CA1808821 and U10CA180882). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The study was also supported in part by funds from Bristol-Myers Squibb, ImClone Systems, Sanofi Aventis, and Pfizer. Bristol-Myers Squibb provided cetuximab to the NCCTG for conduct of the study. A.Z. is supported by the French National Society of Gastroenterology (SNFGE: Société Nationale Française de Gastro-Entérologie) (Bourse Robert Tournut).
EUDRACT 2005-003463-23 (PETACC8 trial); NCT00079274 (NCCTG N0147 trial).
AUTHOR CONTRIBUTIONS
Conception and design: Aziz Zaanan, Qian Shi, Julien Taieb, Steven R. Alberts, Richard M. Goldberg, Pierre Laurent-Puig, Frank A. Sinicrope
Financial support: Aziz Zaanan, Frank A. Sinicrope
Administrative support: Qian Shi, Frank A. Sinicrope
Provision of study material or patients: Aziz Zaanan, Steven R. Alberts, Ayman Zawadi, Enrico Mini, Gunnar Folprecht, Jean Luc Van Laethem, Pierre Laurent-Puig, Frank A. Sinicrope
Collection and assembly of data: Aziz Zaanan, Qian Shi, Julien Taieb, Steven R. Alberts, Thomas C. Smyrk, Catherine Julie, Ayman Zawadi, Josep Tabernero, Enrico Mini, Richard M. Goldberg, Gunnar Folprecht, Jean Luc Van Laethem, Daniel J. Sargent, Pierre Laurent-Puig, Frank A. Sinicrope
Data analysis and interpretation: Aziz Zaanan, Qian Shi, Julien Taieb, Steven R. Alberts, Jeffrey P. Meyers, Thomas C. Smyrk, Josep Tabernero, Enrico Mini, Richard M. Goldberg, Gunnar Folprecht, Karine Le Malicot, Daniel J. Sargent, Pierre Laurent-Puig, Frank A. Sinicrope
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Aziz Zaanan
Consulting or Advisory Role: Amgen, Eli Lilly, Merck Serono, Roche, Sanofi, Merck Sharp & Dohme, Servier, Baxter
Research Funding: Amgen
Julien Taieb
Consulting or Advisory Role: Roche, Merck, Amgen, Celgene, Eli Lilly, Servier, Sirtex Medical, Merck Sharp & Dohme, Pierre Fabre
Speakers’ Bureau: Servier, Amgen, Roche/Genentech, Sanofi, Merck, Eli Lilly, Merck Sharp & Dohme, Pierre Fabre
Catherine Julie
Honoraria: Novartis
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Josep Tabernero
Consulting or Advisory Role: Bayer, Boehringer Ingelheim, Eli Lilly, Merck Sharp & Dohme, Merck Serono, Novartis, Sanofi, Taiho Pharmaceutical, Merrimack, Peptomyc, Rafael Pharmaceuticals, Symphogen, Chugai Pharma, Ipsen, Merus, Pfizer, Seattle Genetics, Array BioPharma, AstraZeneca, BeiGene, Genentech, Genmab, Halozyme, Imugene, Inflection Biosciences, Kura, Menarini, Molecular Partners, Pharmacyclics, ProteoDesign, Roche, Servier, VCN Biosciences, Biocartis, Foundation Medicine, HalioDX SAS, Roche Diagnostics
Richard M. Goldberg
Honoraria: Amgen
Consulting or Advisory Role: Merck, Taiho Pharmaceutical, Merck, Novartis
Research Funding: Bristol-Myers Squibb
Travel, Accommodations, Expenses: Merck, Amgen
Gunnar Folprecht
Honoraria: Merck, Eli Lilly/ImClone, Roche/Genentech, Bristol-Myers Squibb, Sanofi/Aventis, Servier, Merck Sharp & Dohme
Consulting or Advisory Role: Roche/Genentech, Merck, Bristol-Myers Squibb, Sanofi/Aventis, Bayer, Mundipharma, Servier, Merck Sharp & Dohme, Amgen
Research Funding: Merck
Pierre Laurent-Puig
Honoraria: Amgen, AstraZeneca, Boehringer Ingelheim, Merck Serono, Merck, Roche, Sanofi, Biocartis, MSD Oncology, Bristol-Myers Squibb
Consulting or Advisory Role: Merck, Bristol-Myers Squibb, Boehringer Ingelheim
Patents, Royalties, Other Intellectual Property: Inventor of mir31-3p license to IntegraGen
Travel, Accommodations, Expenses: Roche, Merck
Frank A. Sinicrope
Stock and Other Ownership Interests: Illumina
Honoraria: Imedex, American Society of Clinical Oncology
Consulting or Advisory Role: Roche (Inst), Bristol-Myers Squibb (Inst), Ventana Medical Systems (Inst), HalioDx (Inst)
Research Funding: Ventana Medical Systems (Inst)
Travel, Accommodations, Expenses: Ventana Medical Systems
No other potential conflicts of interest were reported.
REFERENCES
- 1. Taieb J, André T, Auclin E. Refining adjuvant therapy for non-metastatic colon cancer, new standards and perspectives. Cancer Treat Rev. 2019;75:1–11. doi: 10.1016/j.ctrv.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 2. Alberts SR, Sargent DJ, Nair S, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–1393. doi: 10.1001/jama.2012.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taieb J, Tabernero J, Mini E, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): An open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:862–873. doi: 10.1016/S1470-2045(14)70227-X. [DOI] [PubMed] [Google Scholar]
- 4. Allegra CJ, Yothers G, O’Connell MJ, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: Results of NSABP protocol C-08. J Clin Oncol. 2011;29:11–16. doi: 10.1200/JCO.2010.30.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 6. Sinicrope FA. Lynch syndrome-associated colorectal cancer. N Engl J Med. 2018;379:764–773. doi: 10.1056/NEJMcp1714533. [DOI] [PubMed] [Google Scholar]
- 7. Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 9. Domingo E, Niessen RC, Oliveira C, et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24:3995–3998. doi: 10.1038/sj.onc.1208569. [DOI] [PubMed] [Google Scholar]
- 10. Parsons MT, Buchanan DD, Thompson B, et al. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: A literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49:151–157. doi: 10.1136/jmedgenet-2011-100714. [DOI] [PubMed] [Google Scholar]
- 11. Sinicrope FA, Shi Q, Smyrk TC, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taieb J, Zaanan A, Le Malicot K, et al. Prognostic effect of BRAF and KRAS mutations in patients with stage III colon cancer treated with leucovorin, fluorouracil, and oxaliplatin with or without cetuximab: A post hoc analysis of the PETACC-8 trial. JAMA Oncol. 2016;2:643–653. doi: 10.1001/jamaoncol.2015.5225. [DOI] [PubMed] [Google Scholar]
- 13. Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: Results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 15. Gavin PG, Colangelo LH, Fumagalli D, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: An assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18:6531–6541. doi: 10.1158/1078-0432.CCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zaanan A, Cuilliere-Dartigues P, Guilloux A, et al. Impact of p53 expression and microsatellite instability on stage III colon cancer disease-free survival in patients treated by 5-fluorouracil and leucovorin with or without oxaliplatin. Ann Oncol. 2010;21:772–780. doi: 10.1093/annonc/mdp383. [DOI] [PubMed] [Google Scholar]
- 19. André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: Updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J Clin Oncol. 2015;33:4176–4187. doi: 10.1200/JCO.2015.63.4238. [DOI] [PubMed] [Google Scholar]
- 20. Tougeron D, Mouillet G, Trouilloud I, et al. Efficacy of adjuvant chemotherapy in colon cancer with microsatellite instability: A large multicenter AGEO study. J Natl Cancer Inst. 2016;108:djv438. doi: 10.1093/jnci/djv438. [DOI] [PubMed] [Google Scholar]
- 21. doi: 10.1093/jnci/djr153. Sinicrope FA, Foster NR, Thibodeau SN, et al: DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst 103:863-875, 2011 [Erratum: J Natl Cancer Inst 103:1639, 2011] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinicrope FA, Mahoney MR, Smyrk TC, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31:3664–3672. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith CG, Fisher D, Claes B, et al. Somatic profiling of the epidermal growth factor receptor pathway in tumors from patients with advanced colorectal cancer treated with chemotherapy ± cetuximab. Clin Cancer Res. 2013;19:4104–4113. doi: 10.1158/1078-0432.CCR-12-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Innocenti F, Ou FS, Qu X, et al. Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol. 2019;37:1217–1227. doi: 10.1200/JCO.18.01798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28:1713–1729. doi: 10.1093/annonc/mdx175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barault L, Charon-Barra C, Jooste V, et al. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res. 2008;68:8541–8546. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 27. Cremolini C, Morano F, Moretto R, et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case-control study. Ann Oncol. 2017;28:3009–3014. doi: 10.1093/annonc/mdx546. [DOI] [PubMed] [Google Scholar]
- 28. Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst. 2016;109:djw272. doi: 10.1093/jnci/djw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sinicrope FA, Shi Q, Allegra CJ, et al. Association of DNA mismatch repair and mutations in BRAF and KRAS with survival after recurrence in stage III colon cancers: A secondary analysis of 2 randomized clinical trials. JAMA Oncol. 2017;3:472–480. doi: 10.1001/jamaoncol.2016.5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20:5322–5330. doi: 10.1158/1078-0432.CCR-14-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]