

SUMMARY
The identification of miRNA targets by Ago2 crosslinking-immunoprecipitation (CLIP) methods has provided major insights into the biology of this important class of non-coding RNAs. However, these methods are technically challenging and not easily applicable to an in vivo setting.
To overcome these limitations and facilitate the investigation of miRNA functions in vivo, we have developed a method based on a genetically engineered mouse harboring a conditional Halo-Ago2 allele expressed from the endogenous Ago2 locus. By using a resin conjugated to the HaloTag ligand, Ago2-miRNA-mRNA complexes can be purified from cells and tissues expressing the endogenous Halo-Ago2 allele. We demonstrate the reproducibility and sensitivity of this method in mouse embryonic stem cells, developing embryos, adult tissues, and autochthonous mouse models of human brain and lung cancers.
This method and the datasets we have generated will facilitate the characterization of miRNA-mRNA networks in vivo under physiological and pathological conditions.
eTOC
Li, Pritykin, Concepcion et al. report the development of Halo-enhanced Ago2 pulldown (HEAP), a method that streamlines the experimental identification of Ago2-miRNA-mRNA interaction sites in murine cells and tissues.
Graphical Abstract
INTRODUCTION
A key challenge in deciphering the biological functions of miRNAs remains the identification of their targets in vivo under physiological and pathological conditions. Although significant progress has been made in computational methods to predict miRNA binding sites (Agarwal et al., 2015; Bartel, 2009; Friedman et al., 2009; Grimson et al., 2007), these methods do not take into account several known and unknown variables that determine whether a ‘potential’ target site is in fact available and bound by a miRNA in a given cellular context. To complement computational approaches, biochemical methods to purify Ago2-miRNA-mRNA complexes have been developed (Chi et al., 2009; Grosswendt et al., 2014; Hafner et al., 2010; Helwak et al., 2013; Konig et al., 2010; Moore et al., 2015; Van Nostrand et al., 2016). Although the details vary, these methods rely on the use of antibodies to precipitate Argonaute-containing complexes, usually after UV crosslinking, followed by high-throughput sequencing of the associated mRNAs.
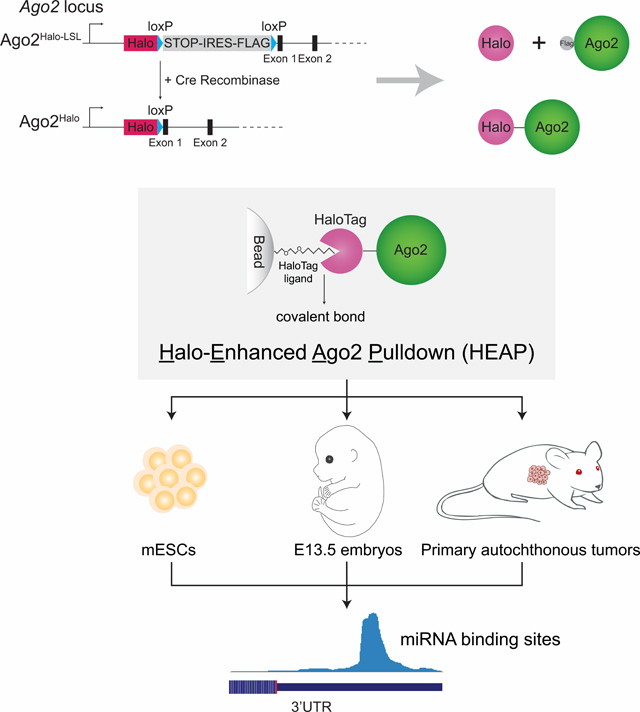
While these methods have been applied with substantial success to map miRNA-mRNA interactions in cell lines, they are used much less extensively in vivo due to their technical complexity and the lack of efficient ways to restrict the analysis to specific cell types within a tissue. To overcome these limitations, we have developed a method, Halo-enhanced Ago2 pulldown (HEAP), which utilizes a tagged version of the Ago2 protein and allows the direct purification of Ago2-containing complexes bypassing the need for radiolabeling, immunoprecipitation, and gel purification. To facilitate the application of this method in vivo, we have generated a mouse strain in which a conditional allele of Halo-tagged Ago2 is knocked into the endogenous Ago2 locus and is activated upon exposure to Cre recombinase.
To benchmark the HEAP method, we applied it to identify miRNA targets in diverse cellular contexts, including murine embryonic stem cells (mESCs), wild-type and miR-17~92-null mid-gestation mouse embryos, adult mouse lungs, adult mouse brains, and three distinct autochthonous mouse models of human lung and brain cancers. As a result, we have identified a large number of miRNA targets at high resolution and demonstrated the reproducibility and sensitivity of the HEAP method.
The datasets and the tools generated in this study reveal the complex landscape of miRNA targeting in vivo and will facilitate future studies aimed at characterizing the biological functions of this important class of small non-coding RNAs under physiological and pathological conditions.
RESULTS
A Halo-Ago2 fusion protein enables antibody-free purification of miRNA targets
The HaloTag is a 33 kDa haloalkane dehalogenase encoded by the DhaA gene from Rhodococcus rhodochrous that has been mutagenized to form an irreversible covalent bond to synthetic chloroalkane ligands (collectively known as HaloTag ligands) (Encell et al., 2012; Los et al., 2008). Linking the chloroalkane ligand to a solid substrate enables the efficient purification of fusion proteins containing the HaloTag (Figure 1A). Importantly, Gu and colleagues have recently used the HaloTag together with UV crosslinking to efficiently identify RNA targets of the RNA binding protein PTB (Gu et al., 2018).
Figure 1. Halo-enhanced Ago2 pulldown (HEAP).

A) Schematic of the Halo-Ago2 fusion protein covalently bound to a bead-conjugated HaloTag ligand. B) Ago2−/− immortalized MEFs transduced with MSCV-PIG, MSCV-PIG-Halo or MSCV-PIGHalo-Ago2 retroviruses were incubated with the HaloTag TMRDirect ligand and imaged. Notice the prevalently cytoplasmic localization of the Halo-Ago2 fusion protein. C) Ago2−/− MEFs transduced with retroviral vectors encoding HaloTag alone, full length Ago2 or the Halo-Ago2 fusion protein were transiently transfected with reporter plasmids expressing Firefly and Renilla luciferase and a plasmid expressing an shRNA against the Firefly luciferase. The ratio between Firefly and Renilla luciferase activity was measured 48 hours after transfection (upper panel). Whole-cell lysates from the same cells were probed with antibodies against Ago2 and β-actin (lower panel). Error bars: Mean ± SD. D) Schematic of the targeting strategy used to generate the Halo-Ago2 conditional knock-in allele. Halo: HaloTag; STOP: stop codon; IRES: internal ribosome entry site. E) Whole-cell lysates from mESCs with the indicated genotypes were probed with antibodies against Ago2, HaloTag and Tubulin. F) Outline of the strategy used to generate HEAP and input control libraries (upper panel) and a representative Halo-Ago2 binding site identified in mESCs (lower panel).
See also Figure S1A, S1B and Data S1.
To determine whether a similar strategy can be employed to purify complexes containing Ago2 proteins bound to miRNA and target mRNAs, we fused the HaloTag to the N-terminus of Ago2 (Halo-Ago2; Figure 1A). When expressed in Ago2−/− mouse embryonic fibroblasts (O’Carroll et al., 2007), the Halo-Ago2 fusion protein localized largely to the cytoplasm, while the HaloTag alone displayed uniform localization to both the cytoplasm and the nucleus (Figure 1B and Data S1). Importantly, the Halo-Ago2 construct was nearly as effective as wild-type Ago2 at rescuing RNAi in Ago2−/− MEFs, indicating that the Halo-Ago2 fusion protein retains slicing activity (Figure 1C).
To avoid artifacts due to ectopic expression of Halo-Ago2 and to enable the isolation of Ago2 complexes directly from murine tissues, we knocked-in the HaloTag cassette into the endogenous Ago2 locus in mESCs (Figure 1D). In this knock-in allele, the HaloTag is separated from the first coding exon of Ago2 by an in-frame loxP-STOP-IRES-FLAG-loxP (LSL) cassette (Ago2Halo-LSL). Cells harboring this allele express a bicistronic mRNA encoding for two proteins: the HaloTag and a FlagAgo2 fusion protein whose translation is initiated by an internal ribosomal entry site (IRES). Upon expression of the Cre recombinase, the LSL cassette is excised and the HaloTag is now brought in frame with the first coding exon of Ago2, thus resulting in expression of the Halo-Ago2 fusion protein (Figures 1D, 1E). The recombined allele expressing the Halo-Ago2 fusion will be hereafter referred to as Ago2Halo.
We first tested whether the Ago2Halo allele could be used to map miRNA-mRNA interactions in mESCs. For these experiments, we adapted the HITS-CLIP method originally developed by the Darnell group (Chi et al., 2009) with two significant streamlining modifications enabled by the covalent bond between Halo-Ago2 and the HaloTag ligand. First, instead of using anti-Ago2 antibodies to isolate Ago2-containing complexes, we used sepharose beads covalently linked to the HaloTag ligand. Second, the radiolabeling and SDS-PAGE purification step necessary in CLIP protocols to purify RNAs bound to Ago2 were omitted and replaced by extensive washes followed by direct RNA extraction from beads, library construction, and high-throughput sequencing of Halo-Ago2-bound miRNAs and mRNAs. We refer to this method as Halo-enhanced Ago2 pulldown (HEAP) (Figure 1F). By performing HEAP, two types of libraries are generated: a target library (mRNAs) and a miRNA library (Figures 1F and S1A). The former allows the identification of miRNA binding sites on their targets, while the latter provides an estimate of miRNA abundance.
When mapped to the mouse genome, HEAP mRNA libraries generated from Ago2Halo/+ mESCs—but not those generated from control Ago2Halo-LSL/+ cells—produced well-defined “clusters” of reads, hereafter referred to as ‘peaks’ (Figures S1A, S1B). To facilitate the identification of these peaks, we adapted the “SMinput” protocol used in eCLIP (Van Nostrand et al., 2016) and generated input control libraries from size-matched RNA fragments isolated after the limited RNase protection step (Figure 1F). We first identified putative peaks using the CLIPanalyze package (https://bitbucket.org/leslielab/clipanalyze), an improved peak-calling algorithm based on edge detection technique similar to methods from image processing (Hsin et al., 2018; Lianoglou et al., 2013; Loeb et al., 2012). CLIPanalyze uses the input control libraries as background to assign p-value to each peak, performing library size normalization based on reads aligned across the genome outside of putative peaks (see also STAR Methods for additional details).
To determine the sensitivity and reproducibility of the HEAP method, we generated HEAP libraries from three Ago2Halo/+ mESC clones (using 1.5 × 108 cells per library). By combining the three libraries, CLIPanalyze identified a total of 30,564 putative Ago2 binding sites at an adjusted p-value cutoff of 0.05. Previous studies have demonstrated that 3’-untranslated regions (3’UTRs) of mRNAs are the preferred, although not exclusive, sites of interaction between miRNAs and mRNAs (Bartel, 2018; Chi et al., 2009; Sarshad et al., 2018). Consistent with these findings, the majority of HEAP peaks we identified in mESCs mapped to 3’UTRs, followed by sites mapping to protein coding sequences (CDS) (Figures 2A and S1C). The fractions of 3’UTR and CDS peaks increased monotonically with their statistical significance, while intergenic and intronic peaks had the opposite behavior. For example, when examining the 1,000 most statistically significant peaks, greater than 50% of them mapped to 3’UTRs and less than 3% mapped to introns (Figure 2A). To measure reproducibility, we applied the CLIPanalyze algorithm independently to each library and performed pairwise Irreproducible Discovery Rate (IDR) (Li et al., 2011) analysis. On average, this analysis identified 80% of peaks as reproducible at IDR < 0.05, demonstrating the robustness of the HEAP method (Figure S1D). We also generated a series of HEAP libraries using decreasing numbers of mESCs (from 1.5 × 108 to 1 × 103). As expected, the total number of confidently identified peaks progressively decreased as the amount of starting material was reduced (Figure S1E). The most robust peaks could be identified in libraries generated from as few as 5 × 105 mESCs (Figure S1F), but for optimal results, we recommend starting from a minimum of 1 × 107 mESCs. Since mESCs have little cytoplasm, the detection limit is likely to be lower for cell types with more abundant cytoplasm.
Figure 2. Mapping Halo-Ago2 binding sites in mESCs.

A) Peaks identified in the HEAP libraries from mESCs were ranked by increasing adjusted p-value before calculating their distribution across genomic annotations. CDS: coding sequence; 5’UTR: 5’untranslated region; 3’UTR: 3’-untranslated region; LncRNA: long non-coding RNA. B) Enrichment for sequences complementary to murine miRNA seeds (7mers and 8mers) was calculated comparing 3’UTR sequences within and outside HEAP peaks. The bar plot shows enrichment for the top ten miRNA seed families ranked by 8mer enrichment scores. C) Scatter plot showing the correlation between number of 3’UTR peaks with 7mer or 8mer seed matches to individual miRNA families and abundance of their corresponding miRNAs as measured in HEAP miRNA libraries. Blue line: best fit linear regression, with 95% confidence interval in grey. D) Genome browser view of the Lefty2 3’UTR with tracks corresponding to libraries generated from wild-type mESC or cells harboring targeted mutations disrupting the predicted miR-291–3p binding site (Lefty2MUT1, Lefty2MUT2). E) Volcano plot of global changes in HEAP peak intensity between the Lefty2MUT and wild-type mESC libraries. Notice the selective loss of the Lefty2 3’UTR binding site (highlighted). F) Cumulative distribution function (CDF) plot for targets of miR-291–3p identified by iCLIP or HEAP. The log2 fold change was calculated in Ago1–4−/− mESCs upon ectopic FHAGO2 expression. HEAP only: targets identified uniquely by HEAP; iCLIP only: targets identified uniquely by iCLIP; shared: targets identified by both methods. P-value: two-sided Kolmogorov–Smirnov test.
See also Figure S1C–I, S2. See Data S2 for peaks identified in mESCs.
To gain additional insights into the nature of peaks identified by HEAP, we searched for enriched 7-mers in the sequences underlying peaks mapping to 3’UTRs (Figure S1G). Inspection of the resulting motifs revealed a marked enrichment for seed matches corresponding to miRNA families whose members are collectively highly expressed in mESCs (Figures 2B and S1H). We also observed a positive correlation between the relative abundance of individual miRNA families—estimated from the miRNA libraries—and the number of corresponding peaks identified by HEAP (Figure 2C).
To directly test whether the peaks identified by HEAP reflect true miRNA-mRNA interactions, we selected a robust peak identified in the 3’UTR of the Lefty2 mRNA (Figure 2D). The sequence underlying this peak includes a highly conserved 8-mer that is complementary to the miR-291–3p seed (Figure S1I). We used CRISPR-Cas9 and homologous recombination in mESCs to introduce point mutations designed to disrupt this seed match (Figure S1I). HEAP libraries generated from two independent Lefty2MUT clones showed complete and selective loss of the Lefty2 peak, further demonstrating the ability of the HEAP method to map bona fide miRNA-mRNA interactions in cells (Figures 2D, 2E).
To assess the ability of HEAP to identify functional miRNA binding sites, we analyzed an RNA-seq dataset generated by Bosson and colleagues from mESCs null for all four Argonaute proteins (Ago14−/−) in the presence or absence of exogenously expressed Flag- and HA-tagged AGO2 (FHAGO2) [(Bosson et al., 2014), GSE61348]. Introduction of FHAGO2 in Ago1–4−/− cells should restore miRNA function, causing repression of their targets. In agreement with this prediction, miRNA targets identified by HEAP were preferentially repressed upon FHAGO2 reintroduction (Figure S2A). The effect was particularly strong for targets assigned by HEAP to the most abundantly expressed miRNA families in mESCs. For example, we observed the strongest repression for targets of the miR-291–3p, miR-17–5p and miR-148–3p families, three miRNA families that account for greater than 12% of all miRNAs in mESCs (Figure S2A and data not shown). Peaks with lower adjusted p-values or higher log2 fold changes (HEAP vs. input control) were associated with stronger target repression (Figure S2B). As expected, peaks mapping to 3’UTRs were associated with strongest target repression compared to peaks mapping to other genomic annotations (Figure S2C).
This analysis also allowed us to compare miRNA targets identified by HEAP to those previously identified by Bosson et al. in Ago1–4−/−-FHAGO2 mESCs using iCLIP, a well-established variant of HITS-CLIP (Konig et al., 2010). By applying the CLIPanalyze peak calling algorithm, we identified 6,813 FHAGO2 binding sites in their iCLIP library, and nearly twice as many (on average 13,532) in each of the three HEAP mESC libraries. The iCLIP library also identified fewer peaks mapping to 3’UTR and more peaks mapping to intergenic regions compared to the HEAP libraries (Figure S2D). 3’UTR targets for miR-291–3p seed family identified by both methods were associated with strong repression of the corresponding genes upon FHAGO2 reintroduction (Figure 2F). The overlap between miR-291–3p binding sites identified by iCLIP and HEAP in 3’UTRs was partial, with the HEAP target pool being nearly twice as large (Figure S2E). Importantly, the targets identified only by HEAP also displayed strong repression upon FHAGO2 reintroduction, indicating that they are functional miRNA binding sites (Figure 2F). We further confirmed the ability of HEAP to identify functional miRNA targets by measuring mRNA and protein expression changes of HEAP targets upon inactivation of Dicer1, the key enzyme responsible for miRNA maturation, in mESCs (FigureS2F).
Collectively, these results show that HEAP provides an effective method to identify miRNA-mRNA interactions in cells.
A conditional Halo-Ago2 mouse enables identification of miRNA-mRNA interactions in vivo
The accurate identification of miRNA targets in vivo and in a cell-type specific context is essential to dissect the functions of miRNAs in development, homeostasis, and disease. To translate the HEAP method to an in vivo setting, we used mESCs harboring the Cre-inducible Halo-Ago2 allele to generate Ago2Halo-LSL/+ mice. We then crossed these animals to CAG-Cre mice (Sakai and Miyazaki, 1997) to delete the LSL cassette and induce ubiquitous expression of the endogenous Halo-Ago2 allele. PCR in mouse embryonic fibroblasts (MEFs) and immunoblot analysis in MEFs and tissues derived from these mice confirmed efficient deletion of the LSL cassette and expression of the Halo-Ago2 protein (Figures 3A and S3A, S3B). Although Ago2Halo/+ and Ago2Halo-LSL/+ mice were obtained at the expected Mendelian frequency and were phenotypically indistinguishable from wild-type mice, homozygous mice for the Ago2Halo or the Ago2Halo-LSL alleles were recovered at sub-Mendelian frequencies (9.9% and 11.9%, respectively, compared to the expected 25%, Figure 3B). The sub-Mendelian recovery of homozygous mice might reflect lower Ago2 expression levels compared to wild-type mice (Figures 3A and S3B) and/or an impaired miRISC formation or activity caused by the presence of the N-terminal tag. Size-exclusion chromatography in Ago2Halo/+ cells showed the HaloAgo2 fusion protein co-eluting with wild-type Ago2 in high molecular weight complexes (Figure S3C), and pull-down experiments confirmed the physical interaction between Halo-Ago2 and Tnrc6a, a core component of the miRISC (Figure S3D). Furthermore, reporter experiments using multiple luciferase reporter constructs harboring well-characterized miRNA binding sites, as well as a highly sensitive two-color fluorescent reporter system (Mukherji et al., 2011), showed no detectable differences in miRNA-mediated repression between wild-type and Ago2Halo/Halo MEFs (Figures S3E, S3F). A careful comparison of RNA-seq libraries generated from wild-type and Ago2Halo/Halo cells, however, revealed a slight preferential de-repression of targets of the most highly expressed miRNA families (Figure S3G). Due to the importance of miRNA-mediated gene regulation during embryonic development, it is possible that this modest perturbation of miRISC activity is responsible for the observed reduced viability of homozygous mice.
Figure 3. Identification of miR-17~92 targets in E13.5 embryos.

A) Expression of Ago2 fusion proteins in the lungs and kidneys of Ago2Halo-LSL and Ago2Halo mice. +/+: wild-type; LSL: Ago2Halo-LSL; Halo: Ago2Halo. B) Absolute numbers and frequencies of genotypes obtained from heterozygous intercrosses of Ago2Halo-LSL/+ or Ago2Halo/+ mice. P-value: Chi-Squared test. C) Outline of the HEAP experiments in E13.5 Ago2Halo/+ embryos wild-type, heterozygous or homozygous knockout for the miR-17~92 cluster. A schematic of the miR-17~92 cluster is shown at the bottom. miRNA members are color-coded based on their seed sequences. D) Heatmap and histogram of peak signal in an 800 bp region surrounding HEAP peaks obtained from miR-17~92WT, miR-17~92-HET and miR-17~92-KO E13.5 embryos. Peaks containing seed matches for the top 31 miRNA seed families ranked by abundance were chosen. Peaks with seed matches for miRNAs belonging to the miR-17~92 cluster are plotted in the upper panels, while the remaining peaks are plotted in the lower panels. E) Genome browser view of the miR-92a-1-dependent miRNA binding sites detected in the long non-coding RNA 1700020I14Rik (Cyrano). PhyloP: placental mammal basewise conservation by PhyloP. Notice the highly conserved 8mer seed match for miR92–3p under the second peak. F) RNA expression of Cyrano in the heart of E9.5 embryos harboring an allelic series of miR-17~92 mutant alleles. KO: embryos null for the entire miR-17~92 cluster; Δ17: embryos null for miR-17 and miR-20a; Δ18: embryos null for miR-18a; Δ19: embryos null for miR-19a and miR-19b-1, Δ92: embryos null for miR-92a-1; Δ17,18: embryos null for miR-17, miR18a and miR-20a; Δ17,18,92: embryos null for miR-17, miR-18a, miR-20a and miR-92a-1. Notice that Cyrano is only up-regulated in mutants in which miR-92a-1 is deleted. P-value: unpaired t-test. Data are represented as Mean ± SD.
See also Figure S3, S4. See Data S2 for peaks identified in E13.5 embryos.
To test whether endogenously expressed Halo-Ago2 can be used to identify miRNA targets in vivo, we crossed Ago2Halo/+ mice to mice harboring a targeted deletion of the miR-17~92 locus (Mirc1), a polycistronic miRNA cluster encoding six distinct miRNAs, which has been shown to be essential for mammalian development (Han et al., 2015; Ventura et al., 2008). We generated HEAP libraries from Ago2Halo/+; miR-17~92+/+ (miR-17~92-WT), Ago2Halo/+; miR-17~92+/− (miR-17~92-HET) and Ago2Halo/+; miR-17~92−/− (miR-17~92-KO) E13.5 embryos (Figure 3C). At an adjusted p-value cutoff of 0.01, HEAP identified a total of 8,661 peaks in these libraries, with a distribution across genomic annotations similar to that observed in mESCs (Figure S4A). Importantly, the intensity of peaks containing seed matches to members of the miR-17~92 cluster was markedly reduced—in a dose-dependent fashion—in the libraries generated from miR-17~92-HET and miR-17~92-KO embryos (Figure 3D). The murine genome contains two additional miRNA clusters that are paralogs to miR17~92 and encode similar miRNAs (Ventura et al., 2008), which may explain some residual HaloAgo2 binding to these sites even in the homozygous mutants. Using an RNA-seq dataset previously generated in the lab from E9.5 embryos harboring an allelic series of miR-17~92 mutant alleles [(Han et al., 2015), GSE63813], we demonstrated that HEAP targets containing seed matches for miR-17/20–5p, miR-19–3p and miR-92–3p mediated strong target repression (Figure S4B). The effect was particularly evident when considering genes harboring HEAP peaks for miR-17/20–5p and miR-92–3p, whose signal intensities were reduced in the miR-17~92-KO embryo, confirming the importance of combining biochemical and genetic approaches to study miRNA function.
Interestingly, we also identified a sizeable fraction of reproducible peaks (4%) mapping to noncoding RNAs. These included two previously uncharacterized miR-17~92-dependent sites matching the miR-92–3p seed in the long non-coding RNA Cyrano (Kleaveland et al., 2018; Ulitsky et al., 2011) (Figure 3E). Importantly, we observed significant upregulation of Cyrano in mouse E9.5 embryos lacking miR-92a-1, but not in mice harboring selective deletion of the other members of the cluster (Figure 3F) (Han et al., 2015), suggesting these binding sites are functional. These results demonstrate the usefulness of the Halo-Ago2 mouse strain in facilitating the identification of miRNA targets in vivo.
To directly compare the performance of HEAP to immunoprecipitation-based approaches in vivo, we next generated libraries from the cortex of P13 Ago2Halo/+ mice, a tissue from which high-quality miRNA-target libraries have been previously generated by HITS-CLIP and CLEAR-CLIP (Chi et al., 2009; Moore et al., 2015). Two HEAP libraries generated from the cortices of AgoHalo/+ mice produced 7,069 peaks at an adjusted p-value cutoff of 0.05. This number of miRNA-mRNA interaction sites is comparable to that identified by Moore and colleagues (CLEAR-CLIP, GSE73059, n = 7,927) using 12 biological replicates (Figures S4C, S4D). HEAP and CLEAR-CLIP identified similar numbers of targets for miR-124–3p, one of the most abundant miRNA families in the mouse cortex (Figure S4E). When benchmarked against a microarray gene expression dataset generated from neuroblastoma cells (CAD) ectopically expressing miR-124 [(Makeyev et al., 2007), GSE8498], HEAP and CLEAR-CLIP were equally effective at identifying miR-124 target sites that mediated target repression (Figure S4F). Collectively, these results demonstrate that the HEAP method provides a simple and cost-effective approach to identify miRNA-mRNA interactions during murine development and in primary tissues.
Identification of miRNA targets in normal adult tissues and in autochthonous tumors
We next tested whether the conditional Halo-Ago2 mouse could be used to identify miRNA-mRNA interactions in primary autochthonous tumors and in their tissues of origin. We first chose a mouse model of glioma driven by the Bcan-Ntrk1 gene fusion that we recently developed in our laboratory (Cook et al., 2017). In this model, Trp53fl/fl mice are injected intracranially with a mixture of two recombinant adenoviruses. The first expresses Cas9 and two sgRNAs (Ad-BN) designed to induce the Bcan-Ntrk1 rearrangement, an intra-chromosomal deletion resulting in the fusion between the N-terminal portion of Bcan and the kinase domain of Ntrk1. The second adenovirus expresses the Cre recombinase (Ad-Cre) to achieve concomitant deletion of Trp53 and allow glioma formation. By performing this procedure in 4~6-week-old Ago2Halo-LSL/+; Trp53fl/fl mice, we produced Bcan-Ntrk1 driven gliomas expressing the endogenous Halo-Ago2 allele.
We generated HEAP libraries from three independent Bcan-Ntrk1 gliomas and from the normal cortices of three age-matched Ago2Halo/+ mice. Quantification of miRNA abundance in HEAP miRNA libraries revealed drastic differences between the two tissues, with 77 miRNA seed families (26 broadly conserved) being significantly upregulated in gliomas, and 77 families (18 broadly conserved) downregulated (adjusted p-value < 0.05, absolute log2FC > 0.5, Figure 4A). Of note, the significantly downregulated families include miR-124–3p and miR-128–3p, two miRNA families that are highly expressed in the cortex of mice (Bak et al., 2008; Landgraf et al., 2007) and functionally important in the mouse central nervous system as suggested by genetic loss-of-function studies (Sanuki et al., 2011; Tan et al., 2013). Additionally, members of the oncogenic miRNA cluster miR-17~92 (He et al., 2005; Ota et al., 2004) were among the most strongly upregulated miRNAs in gliomas, suggesting the possibility that these miRNAs are functionally relevant in gliomagenesis.
Figure 4. Mapping miRNA-target interactions in normal brain and brain tumors.

A) Volcano plot of global changes in miRNA family expression between normal cortices and BcanNtrk1-driven gliomas, as determined by HEAP. Broadly conserved families are highlighted in circles and a few miRNA families of interest are colored and annotated. B) Total number and distribution across genomic annotations of peaks identified in the cortex and glioma HEAP libraries at adjusted p-value < 0.05. C) Top differentially enriched 8-mers in glioma and cortex HEAP peaks (peak selection cutoff: adjusted p-value < 0.05; absolute log2 (gliomas / cortices) > 0.5) by the HOMER de novo motif discovery algorithm. miRNA families whose seed sequences are complementary to these motifs are annotated. D) Changes in peak intensity correlate with changes in miRNA abundance. The area of each circle is proportional to the number of targets of each miRNA seed family as identified by HEAP. Only broadly conserved miRNA families with more than 40 HEAP targets are shown. E) Cumulative distribution function (CDF) plot for targets of miR-124–3p, miR-219a-5p and let-7–5p identified by HEAP. mRNA expression was estimated using read counts in input control libraries. The mRNA log2 fold change was calculated as gliomas vs. cortices. P-value: two-sided Kolmogorov–Smirnov test. F) Genome browser view of representative Halo-Ago2 binding sites detected exclusively in gliomas (top) or cortices (bottom).
See also Figure S5A. See Data S2 for peaks identified in gliomas and cortices.
Using an adjusted p-value cutoff 0.05, we identified 1,878 Halo-Ago2 binding sites in tumors and 2,688 sites in normal cortices, with an overlap of 1,335 sites. Peak distribution across genomic annotations was similar between the two tissues, with the majority of peaks mapping to 3’UTRs (Figure 4B). Analysis of seed matches under the peaks revealed marked differences between normal and neoplastic brains. Motifs complementary to the seeds of miR-219a–5p, miR-17–5p, miR-15/16–5p, miR-181–5p and miR-130–3p were preferentially enriched in peaks identified in gliomas, while motifs complementary to the seeds of miR-124–3p, miR-29–3p, miR-9–5p, miR-128–3p, miR137–3p, miR-138–5p and miR-7–5p were preferentially enriched in peaks from normal cortices (Figures 4C, 4F and S5A). Targets for the let-7–5p family of miRNAs were also abundant, but not differentially represented between the normal brain and tumors (Figures 4C and S5A). The enrichment for specific seed matches observed in the two conditions reflected in large part the differential expression of the corresponding miRNAs (Figure 4D) and resulted in differential gene regulation, as demonstrated by a statistically significant repression of miR-219a-5p targets in gliomas and of miR-124–3p targets in the normal cortices (Figure 4E).
Among all miRNA families, the miR-219a-5p family had the highest number of targets in gliomas (300 out of 1,878 peaks containing 6mer, 7mer or 8mer seed matches to miR-219a-5p). miR-219–5p has been reported to regulate oligodendrocyte (OL) differentiation and myelination in mice via targeting important regulators of oligodendrocyte progenitor cell (OPC) maintenance (Dugas et al., 2010; Emery, 2010; Fan et al., 2017; Wang et al., 2017; Zhao et al., 2010). Interestingly, we observed a strong interaction between miR-219a-5p and Pdgfra 3’UTR (Figure 4F), a characteristic marker of OPCs and a key player in gliomagenesis.
Finally, to extend the application of the HEAP method to other tumor types, we mapped miRNA-mRNA interactions in two murine models of non-small cell lung cancer (NSCLC): the Cre recombinase-mediated KRasLSL-G12D/+; Trp53fl/fl (KP) model (Jackson et al., 2001) and a CRISPR-Cas9 induced model driven by a chromosomal inversion resulting in the formation of the Eml4-Alk (EA) gene fusion (Maddalo et al., 2014). These two mouse models recapitulate two types of NSCLC observed in humans and differ not only in the initiating genetic lesions but also in the modality with which tumor formation is induced.
We generated HEAP libraries from Ago2Halo-LSL/+ mice bearing primary KP (N = 2) and EA (N = 3) tumors. Tumor-specific expression of the Halo-Ago2 allele was induced at the time of tumor initiation by intratracheal delivery of Ad-Cre, alone for the KP model or in combination with recombinant adenoviruses expressing Cas9 and the two gRNAs necessary to induce the Eml4-Alk rearrangement in the EA model (Ad-EA). In parallel, we also generated HEAP libraries from the lungs of two Ago2Halo/+ mice (Figure 5A).
Figure 5. Mapping miRNA-target interactions in lung adenocarcinomas.

A) Schematic of the experimental design. B) Total number and distribution across genomic annotations of peaks identified in normal lungs (two replicates) and in the KP and EA lung adenocarcinomas (two and three replicates, respectively) at adjusted p-value < 0.05. C) Scatter plots to compare peak intensity changes in EA vs. normal (y-axis) and KP vs. normal (x-axis). Peaks with seed matches for indicated miRNA families are colored. Left: highlighted are peaks containing seed matches for representative miRNA families whose targets were found in both normal lungs and lung adenocarcinomas. Right: highlighted are peaks with seed matches for miRNA families whose targets were preferentially found in lung adenocarcinomas. Selected motifs and their p-values as determined by the HOMER de novo motif discovery algorithm are shown on the side. D) Genome browser view of the miR-200bc-3p binding site in the 3’UTR of Zeb2. E) Targets identified by HEAP show preferential de-repression upon inactivation of the miRISC. Cumulative distribution function (CDF) plot of mRNA expression changes induced by T6B-YFP expression in murine KP cells (T6BWT-YFP / T6BMUT-YFP). Targets identified by HEAP for the indicated miRNA families were compared to background (“all genes”). P-value: two-sided Kolmogorov–Smirnov test.
See also Figure S5B–E. See Data S2 for peaks identified in normal lungs and lung adenocarcinomas.
The tumor libraries produced 1,899 peaks for the KP tumors and 2,127 peaks for the EA tumors. In contrast, only 417 peaks were identified in normal lungs (Figure 5B). This difference could not be attributed to differences in sequencing depth or Halo-Ago2 expression levels in normal lungs vs. tumors (Figure S5B). Rather, it may reflect reduced levels of fully assembled miRISC in the normal lung compared to lung tumors [(La Rocca et al., 2015) and La Rocca et al., manuscript in preparation].
Surprisingly, a direct comparison of the peaks identified in KP and EA tumors revealed strong similarity between the two tumor types (Figures 5C and S5C, S5D), suggesting that the miRNA targeting landscape is largely independent from the cancer initiation events in these two NSCLC models. Unbiased k-mer frequency analysis visualized as motif enrichment identified distinct miRNA seed-matches enriched in peaks in normal lungs and tumors. Binding sites for let-7–5p, miR-29–3p and miR-30–5p were strongly enriched in both tissues, while seed matches for several miRNAs implicated in tumorigenesis and metastasis, such as miR-200bc-3p (Davalos et al., 2012; Gibbons et al., 2009; Gregory et al., 2008; Sato et al., 2017; Si et al., 2017), miR-31–5p (Edmonds et al., 2016), miR-17–5p (He et al., 2005; Ota et al., 2004) and miR-25/92–3p (Ota et al., 2004) were dominant in the tumor libraries (Figures 5C and S5E). In human lung adenocarcinomas, miR-200 levels negatively correlate with tumor metastatic potential, at least in part because this miRNA can potently suppress epithelial-to-mesenchymal transition (EMT) (Davalos et al., 2012; Gibbons et al., 2009; Si et al., 2017). In agreement with this model, we observed a strong miR-200bc-3p binding site in the 3’UTR of Zeb2, a master regulator of EMT (Figure 5D).
To further validate the functional significance of these miRNA-mRNA interactions in lung cancer, we took advantage of a fusion protein (T6B-YFP) previously shown to bind Argonaute proteins and disrupt assembly of the miRISC complex, leading to a global de-repression of miRNA targets [(Hauptmann et al., 2015; Pfaff et al., 2013), LaRocca et al., manuscript in preparation]. We compared the transcriptome of mouse KP cancer cells expressing either T6B-YFP (T6BWT-YFP) or a mutant version (T6BMUT-YFP) that cannot bind Argonaute proteins and is therefore inactive. As shown in Figure 5E, genes harboring peaks identified by HEAP were preferentially de-repressed upon disruption of the miRISC, further confirming the ability of the HEAP method to identify functional miRNA-mRNA interactions in vivo.
DISCUSSION
We have demonstrated the ability of HEAP to identify miRNA-mRNA interaction sites in cells, developing embryos, normal adult tissues, and in primary autochthonous tumors. By mapping miRNA binding sites in mouse embryos lacking the miR-17~92 cluster, we identified direct targets of the miRNAs encoded by this cluster, including a long noncoding RNA that had not been previously reported to be regulated by this cluster. The HEAP method also allowed us to identify miRNA targets in primary autochthonous cancers in mice and in their tissues of origin, uncovering marked differences in the spectrum of miRNA targets between cancers and normal tissues.
When compared to standard immunoprecipitation-based approaches, HEAP offers several advantages. First, the covalent nature of the interaction between the HaloTag and the HaloTag ligands simplifies the isolation of Ago2-miRNA-mRNA complexes and removes the intrinsic variability of immunoprecipitation-based approaches. This feature is illustrated by the highly reproducible identification of miRNA-binding sites in murine embryonic stem cells, in developing embryos, in murine tissues and in tumors. Second, the conditional Cre-loxP-based nature of the Halo-Ago2 mouse strain enables the purification of Ago2-containing complexes and the identification of miRNA-mRNA interaction sites from a specific subset of cells, thus bypassing the need for microdissection and cell purification using cell surface markers. As proof of concept, we demonstrate this ability by mapping miRNA-mRNA interactions in three mouse models of human cancers driven by distinct combinations of oncogenes and tumor suppressor genes. We predict that the systematic application of HEAP will allow the construction of a detailed map of miRNA targets across tissues and cell types in mice.
We emphasize that the HEAP protocol can be easily modified to accommodate the many variations of the basic HITS-CLIP strategy, including those using ligation to generate chimeric reads between the mature miRNA and its target (CLASH, CLEAR-CLIP), and those designed to identify the crosslinking site at single base resolution (PAR-CLIP, iCLIP, eCLIP).
Although in this study we have focused exclusively on the identification of miRNA-mRNA interactions in cells and tissues, the conditional Halo-Ago2 mouse strain we have developed could prove useful for the biochemical characterization of Ago2-containing protein complexes in vivo and for imaging studies (Figure S3H and Data S1). Notably, fluorescent HaloTag ligands have been successfully used recently for super-resolution imaging of Halo-tagged proteins (Grimm et al., 2015). When applied to cells and tissues expressing the Halo-Ago2 knock-in allele, this strategy could provide insights into the subcellular localization and dynamics of this important RNA binding protein under different conditions and in response to external and internal cues.
Despite these advantages, some limitations of the HEAP method should be considered when planning experiments. First, as is true for any tagged protein, the presence of the HaloTag may have functional consequences. The reduced viability of the Halo-Ago2 homozygous animal we have observed does indicate that Halo-Ago2 is not entirely functionally identical to Ago2, perhaps due to reduced stability or to a subtle impairment of miRISC assembly and activity. Thus, it will be important to experimentally evaluate the functional relevance of individual miRNA-mRNA interactions identified using this approach. Second, although the conditional nature of the Halo-Ago2 allele is ideally suited for the direct identification of miRNA targets in rare cell populations within a tissue, the HEAP method requires a relatively large number of cells (ideally 1×107 cells or more) to produce robust results, and in some cases, it may be therefore necessary to pool tissues from multiple animals.
In conclusion, the HEAP method and the Cre-inducible Halo-Ago2 mouse strain described in this paper, combined with the growing array of strains expressing Cre in a temporally and spatially restricted fashion, will facilitate the generation of detailed maps of miRNA-mRNA interactions in vivo under physiological and pathological conditions.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrea Ventura (venturaa@mskcc.org).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The datasets generated during this study are available at GEO (GSE139349). CLIPanalyze is available for download at https://bitbucket.org/leslielab/clipanalyze. This published article includes algorithms and key parameters used during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
The Halo-Ago2 conditional knock-in mice were generated through gene targeting. The targeting construct was generated by modifying the pKO-II vector through three steps of cloning. First, a fragment comprising a 2 kb 5’ homology arm, the 5’UTR of Ago2, the HaloTag cDNA, the TEV protease recognition sequence, the coding sequence of Ago2 Exon1 and a portion of the first intron was inserted into the pKO-II vector immediately upstream of the frt-PGK-NEO-frt cassette. Second, a 5 kb 3’ homology arm was cloned into the HindIII site downstream of the frt-PGK-NEO-frt casette. Lastly, a loxP-STOP-IRES-FLAG-loxP cassette was inserted into the AsiSI site between the TEV cleavage sequence and Ago2 coding sequence.
V6.5 mESCs (obtained from the Rudolf Jaenisch laboratory at Whitehead Institute and Massachusetts Institute of Technology) were electroporated with the linearized targeting construct and selected in mESC medium containing G418 (Gibco) for 7 days. Recombinant clones were identified by Southern blot using probes designed against sequences outside the 5’ and 3’ homology regions. A validated clone was injected into C57BL/6 blastocyst to generate chimeric mice. Mice heterozygous for the targeted allele were crossed to the β-actin-Flpe mice (Rodriguez et al., 2000) to remove the frt-PGK-NEO-frt cassette, resulting in the generation of Ago2Halo-LSL/+ mice. The Ago2Halo/+ mice were obtained by crossing the Ago2Halo-LSL/+ mice to the CAG-Cre mice (Sakai and Miyazaki, 1997).
Mice carrying the knock-in alleles were genotyped using a three-primer PCR (p1, 5’-GCAACGCCACCATGTACTC-3’, final concentration 0.75 μM; p2, 5’-GAGGACGGAGACCCGTTG3’, final concentration 1.0 μM; p3, 5’-AGCCGTTCCTGAATCCTGTT-3’, final concentration 0.5 μM), which amplifies a 240-bp band from the wild-type allele (p1-p2), a 1281-bp band from the Ago2Halo-LSL allele and a 651-bp band from the Ago2Halo allele (p2-p3).
We also used miR-17~92−/− (Ventura et al., 2008), Trp53fl/fl (Marino et al., 2000) and KRasLSL-G12D/+ (Jackson et al., 2001) mice in this study. For the generation of E13.5 embryos, 6~10-week-old females were sacrificed at embryonic day 13.5. To generate P13 cortex HEAP libraries, cortices were harvested from 13-day-old Ago2Halo/+ mice. For the generation of gliomas, 4~6-week-old Ago2Halo-LSL/+; Trp53fl/fl mice were infected with recombinant adenoviruses and tumors were harvested approximately 80 days after injection. Normal cortices were harvested from age-matched Ago2Halo/+ mice. For the generation of lung adenocarcinomas, 10~12-week-old Ago2Halo-LSL/+ (EA model) and Ago2Halo-LSL/+; KRasLSL-G12D/+; Trp53fl/fl (KP model) mice were infected with recombinant adenoviruses and tumors were harvested 3 months after infection. Normal lungs were obtained from age-matched Ago2Halo/+ mice.
All studies and procedures were approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee.
Cells lines and cell culture conditions
Ago2Halo and Ago2Halo-LSL MEFs were generated by intercrossing Ago2Halo/+ and Ago2Halo-LSL/+ mice, respectively and derived using standard protocols. MEFs were immortalized with retrovirus expressing the SV40 large T antigen (Addgene:13970) (Zhao et al., 2003). Ago2−/− MEFs were a kind gift from Alexander Tarakhovsky (Rockefeller University). Murine KP cells were derived from murine KRasG12D; Trp53−/− lung adenocarcinomas.
Cells were maintained in a humidified incubator at 37 °C, 5% CO2. mESCs were grown on irradiated MEFs in KnockOut DMEM (Gibco) supplied with 15% FBS (Gibco), leukemia inhibitory factor (Millipore, 10 U / mL), penicillin/streptomycin (Gibco, 50 U/ mL), GlutaMax (Gibco), non-essential amino acids (Sigma-Aldrich), nucleosides (Millipore) and 2-Mercaptoethanol (Bio-Rad, 100 μM). MEFs were cultured in DMEM (Gibco) containing 10% FBS, penicillin/streptomycin (100 U/mL) and L-glutamine. KP cells were cultured in Advanced DMEM/F12 (Gibco, 1:1) containing 5% FBS, HEPES (Gibco, 10 mM), GlutaMax and penicillin/streptomycin (100 U/mL).
Method Details
Luciferase Assay
Ago2−/− MEFs were transduced with the MSCV-PIG (Mayr and Bartel, 2009) (Addgene: 21654), MSCV-PIG-Halo, MSCV-PIG-Halo-Ago2 or MSCV-PIG-Ago2 retroviruses to generate cell lines stably expressing HaloTag, the Halo-Ago2 fusion or Ago2. The dual-luciferase reporter assay system (Promega) was used to measure the cleavage activity of Halo-Ago2 and Ago2. Luciferase reporter plasmids pIS0 (luc+, Firefly luciferase, Addgene: 12178) (Yekta et al., 2004) and pIS1 (Rluc, Renilla luciferase, Addgene: 12179) were co-transfected into MEFs, along with a pSico vector expressing an shRNA against the Firefly luciferase or a control shRNA against CD8 (Ventura et al., 2004). The ratio between Firefly and Renilla luciferase activity was measured following manufacturer’s instructions at 48 hrs after transfection.
mESC mutagenesis
The Dicer1 knockout cells were generated from Ago2Halo/+ mESCs using CRISPR-Cas9. A pX333 vector (Addgene: 64073) (Maddalo et al., 2014) expressing Cas9 and a pair of guide RNAs designed to delete a portion of the RNase III 1 domain of Dicer1, was transiently transfected into Ago2Halo/+ mESCs. Single clones were isolated and genotyped by PCR.
Lefty2 mutant clones were generated from Ago2Halo/+ mESCs using CRISPR-Cas9-mediated homologous recombination. PX330 vectors (Addgene: 42230) (Cong et al., 2013; Ran et al., 2013) expressing Cas9 and guide RNAs targeting the predicted miR-291–3p binding site in the 3’UTR of Lefty2 were transiently transfected, together with single-stranded template DNAs, into Ago2Halo/+ mESCs. Clones undergoing homologous recombination were enriched using the method developed by Flemr and Buhler with plasmid pMB1610_pRR-Puro (Addgene: 65853) containing a fragment of guide RNA target sequence (Flemr and Buhler, 2015). Clones homozygous for the desired mutations were identified by PCR and Sanger sequencing. See also Table S1 for oligo information.
Protein analysis by mass spectrometry
Tandem Mass Tag mass spectrometry
Five independent Dicer1 knockout and five wild-type mESC clones were used in the proteomic analysis. Frozen cell pellets were lysed in 8 M urea and 200 mM EPPS, pH 8.5 with protease inhibitor (Roche) and lysates were additionally passed 10 times through a 21-gauge needle. Disulfide bonds were reduced using 5 mM tris(2-carboxyethyl)phosphine (30 min, RT) and alkylated with 10 mM iodoacetamide (30 min, RT in the dark). Alkylation reaction was quenched with 10 mM dithiotreitol for 15 min at RT. Per sample 100 μg protein (protein concentration determined prior to reduction/alkylation by BCA assay) were precipitated using methanol-chloroform precipitation and digested at RT with Lys-C protease (Wako Chemicals) in 200 mM EPPS, pH 8.5 at a 50:1 protein:enzyme ratio overnight. More complete protein digestion was achieved through addition of trypsin (100:1 protein:enzyme ratio, Promega) for an additional 6 hrs at 37 °C. Acetonitrile was added to sample to a concentration of approximately 30%, and peptides were labelled with 0.2 mg TMT isobaric label reagent (Thermo Fisher Scientific) per sample for 1 hr at RT. Labelling reactions were quenched with the addition of hydroxylamine to 0.3% (v/v). Samples were combined at a 1:1:1:1:1:1:1:1:1:1 ratio and dried down by vacuum centrifugation. Excess TMT label was removed by C18 solid-phase extraction (Waters). The pooled sample was fractioned by off-line basic pH reversed-phase HPLC over a 50 min 5–35% acetonitrile gradient in 10 mM ammonium bicarbonate pH 8.0 into 96 fractions using an Agilent 300Extend C18 column (Wang et al., 2011). Collected fractions were combined into 48 fractions, of which 24 non-adjacent fractions were desalted using StageTips, dried by vacuum centrifugation and peptides were solubilized in 5% acetonitrile and 5% formic acid for subsequent LC-MS/MS analysis (Paulo et al., 2016). Approximately 2 μg of each sample was analyzed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) coupled to a Proxeon EASY-nLC 1200 liquid chromatography pump (Thermo Fisher Scientific) and a 100 μm × 35 cm microcapillary column packed with Accucore C18 resin (2.6 μm, 150 Å, Thermo Fisher). Peptides were fractionated over a 150 min gradient of 3 – 25% acetronitrile in 0.125% formic acid. An MS3-based TMT method was used, as described previously (McAlister et al., 2014; Paulo et al., 2016; Ting et al., 2011). MS1 spectra were acquired with a resolution of 120,000, 350–1400 Th, an automatic gain control (AGC) target of 5e5, and a maximum injection time of 100 ms in the Orbitrap mass analyzer. The ten most intense ions were fragmented by collision-induced dissociation (CID) and analyzed in a quadrupole ion trap with AGC 2e4, normalized collision energy (NCE) 35, q-value 0.25, maximum injection time 120 ms, and an isolation window of 0.7 Th. MS3 spectra were acquired in the Orbitrap mass analyzer (AGC 2.5e5, NCE 65, maximum injection time 150 ms, 50,000 resolution at 400 Th) after fragmentation of MS2 ions by HCD. Isolation windows were chosen depended on charge state z (z=2 1.3 Th, z=3 1 Th, z=4 0.8 Th, z=5 0.7 Th).
Mass spectrometry data processing
Spectra were searched using Sequest (Eng et al., 1994) with a 50 ppm precursor mass tolerance, 0.9 fragment ion tolerance and a maximum of two internal cleavage sites. Methionine oxidation was included as a variable modification, with a maximum of three modifications per peptide. Cysteine alkylation and TMT addition on lysines and peptide N-termini were set as fixed modifications. Spectra were searched against the Uniprot mouse proteome sequence database (downloaded on February 7th, 2014) containing both SwissProt and TrEMBL entries. Common contaminants were added to the database. The database was sorted in the following order: contaminant, SwissProt entries, TrEMBL entries and protein length within each category. All peptide sequences in the database were reversed and appended. FDR was estimated by linear discriminant analysis (Elias and Gygi, 2007; Peng et al., 2003), a 1% FDR filtering was applied at the peptide and protein level. Peptides were collapsed into a minimal number of protein identification as described by Huttlin and colleagues (Huttlin et al., 2010). This resulted in a filtered matrix of protein abundance values for 8,056 proteins. Then log2FC of abundance was calculated for each protein by summing values within five replicates of each condition, adding 1 to each sum, and then taking log2 of the ratio of the sums.
Halo-Ago2 imaging
Ago2−/− MEFs transduced with retroviruses MSCV-PIG, MSCV-PIG-Halo or MSCV-PIG-Halo-Ago2 were treated with 100 nM HaloTag TMRDirect ligand (Promega) overnight and imaged on a ZEISS AXIO A1 microscope with AXIOCam MRC (ZEISS). A LD Plan-NEOFLUAR 20X/0.4 Ph2 korr objective was used.
Ago2+/+, Ago2Halo-LSL/Halo-LSL and Ago2Halo/Halo MEFs were treated with 200 nM Janelia Fluor 646 HaloTag ligands or Janelia Fluor 549 HaloTag ligands (Promega) at least 1hr prior to experiment. Before imaging, media containing HaloTag ligands was replaced with warm media without phenol red (Gibco). Cells were kept at 37°C with 5% CO2 and 100% humidity while imaging. Confocal imaging was performed on a ZEISS LSM880 microscopy (Carl Zeiss) using the Airyscan module. A 63X 1.4 NA oil objective was used. Time lapse images were acquired with a Zeiss alpha Plan-Apochromat 100X/1.46NA objective on an Axio Observer.Z1 in widefield using a Hamamatsu ORCA Flash4.0 v2 camera. Interval between frames was 500 ms with 250 ms exposures. Images were processed using ZEN (Zeiss) and Fiji (NIH).
Size exclusion chromatography
Cells were lysed with Sup6–150 buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.5, 2.5 mM MgCl2, 0.01% Triton X-100, protease inhibitor (Roche) and phosphatase inhibitor (Roche)). Lysates were fractionated using the Superose 6 10/300 GL prepacked column (GE Healthcare) coupled with the AKTA FPLC system as described in (La Rocca et al., 2015; Olejniczak et al., 2013). Eluted proteins were concentrated by trichloroacetic acid (TCA)/acetone precipitation, analyzed by immunoblot and imaged using the Odyssey CLx imaging system (Li-Cor).
Isolation of Halo-Ago2/Tnrc6 complexes
Ago2Halo-LSL/Halo-LSL and Ago2Halo/Halo MEFs were lysed with HaloTag protein purification buffer (150 mM NaCl, 50 mM HEPES, pH 7.5, 0.005% IGEPAL CA-630) and lysates were incubated with HaloTag magnetic beads (Promega) for 90 min at room temperature on a rotator. After three washes with the HaloTag protein purification buffer, proteins were released by TEV protease (Invitrogen) digestion at 30 °C for 1 hr. Eluted proteins were analyzed by immunoblot and visualized using ECL (GE Healthcare).
Dual luciferase reporter assay
Fragments of the 3’UTRs of Pten and Adrb2 containing miRNA binding sites for miR-29–3p and let-75p, respectively, were amplified from cDNA and cloned into the multiple cloning site of the psiCHECK2 vector (Promega) by HiFi assembly (New England Biolabs). Control vectors were created by mutagenizing the predicted miRNA seed match in each of these vectors by PCR using 5’ phosphorylated primers followed by ligation. For Taf7, 3’UTR fragments containing wild-type or mutant binding site for miR-21–5p was synthesized and cloned into the psiCHECK2 vector by HiFi assembly. The luciferase reporters were transfected into MEFs in triplicates. Luciferase activity was measured using the dual-luciferase reporter assay system (Promega) according to manufacturer’s instructions 48 hrs post transfection. See also Table S1 for oligo information.
Two-color fluorescent reporter assay
MEFs were engineered to stably express the reverse tetracycline-controlled transactivator (rtTA) using a lentiviral vector rtTA-N144 (Addgene: 66810) (Richner et al., 2015). The two-color fluorescent reporter pTRETightBI-RY-0 (Addgene: 31463), pTRETightBI-RY-1pf (Addgene: 31467) and pTRETightBI-RY-4 (Addgene: 31465) were transfected into the rtTA-expressing MEFs. 48 hrs after transfection, fluorescent signals were measured using flow cytometry. Signals were processed as described by Mukherji and colleagues (Mukherji et al., 2011). Mean and standard deviation of autofluorescence in eYFP and mCherry channels were obtained from untransfected cells. The mean autofluorescence plus twice the standard deviation was subtracted from each cell’s eYFP and mCherry signals. Cells with eYFP signals lower than 0 were removed. The fluorescent signals were binned along the eYFP axis and mean mCherry signals were calculated in each bin.
Recombinant adenovirus delivery
Recombinant adenoviruses used for inducing chromosomal rearrangements (Ad-BN, Ad-EA) (Cook et al., 2017; Maddalo et al., 2014) and Ad-Cre were purchased from ViraQuest.
For the generation of Bcan-Ntrk1-driven gliomas, a 1:1 mixture of Ad-BN and Ad-Cre, in total ~3 × 109 infectious particles, was administrated to Ago2Halo-LSL/+; Trp53fl/fl mice (4~6 weeks old), via stereotactic intracranial injection as described in Cook et al., 2017. Gliomas were harvested approximately 80 days after injection, when mice became symptomatic.
For the generation of Eml4-Alk-driven lung adenocarcinomas, 10~12-week-old Ago2Halo-LSL/+ mice were intratracheally infected with a 1:1 mixture of Ad-EA and Ad-Cre (in total ~6 × 1010 infectious particles). To generate KRasG12D; Trp53−/− lung tumors, 10~12-week-old Ago2Halo-LSL/+; KRasLSL-G12D/+; Trp53fl/fl mice were intratracheally infected with Ad-Cre (~2.5 × 107 PFU). Lung tumors were harvested approximately 3 months after infection.
T6B peptide
Mouse KP cells were transduced with retroviruses expressing the T6BWT-YFP or the T6BMUT-YFP fusion protein, in which five tryptophan residues were mutated to alanines [(Hauptmann et al., 2015; Pfaff et al., 2013) and LaRocca et al., manuscript in preparation].
RNA sequencing
Total RNAs from mESCs, MEFs, lung adenocarcinomas and normal lung tissues were extracted using TRIzol Reagent (Invitrogen) and subjected to DNase (QIAGEN) treatment followed by RNeasy column clean-up (QIAGEN). After quantification and quality control, 500ng of total RNA underwent poly(A) selection and TruSeq library preparation using the TruSeq Stranded mRNA LT Kit (Illumina) according to the manufacturer’s instructions. Samples were barcoded and run on a HiSeq 2500 or a Hiseq 4000 in a 50bp/50bp paired end run.
Total RNAs of T6B-YFP-expressing KP cells were isolated using TRIzol Reagent and subjected to DNase treatment and isopropanol re-precipitation. After quantification and quality control, 1 ug of total RNA underwent ribosomal depletion and library preparation using the TruSeq Stranded Total RNA LT Kit (Illumina). Samples were run on a HiSeq 4000 in a 50bp/50bp paired end run.
Reads were aligned to the standard mouse genome (mm10) using Hisat2 (v0.1.6-beta) (Kim et al., 2019) or STAR v2.5.3a (Dobin et al., 2013). RNA reads aligned were counted at each gene locus. Expressed genes were subjected to differential gene expression analysis by DESeq2 v1.20.0 (Love et al., 2014).
Analysis of public datasets
RNA-seq data generated from E9.5 miR-17~92 mutant embryos were obtained from the authors and are available in GEO (GSE63813) (Han et al., 2015). In this study, gene expression was profiled in triplicates in heart, mesoderm and all remaining tissues of wild-type (WT) embryos and embryos null for miR-17 and miR-20a (Δ17), null for miR-18a (Δ18), null for miR-19a and miR-19b-1 (Δ19), and null miR-92a-1 (Δ92), null for miR-17, miR-18a and miR-20a (Δ17,18), null for miR-17, miR-18a, miR-20a and miR-92a-1 (Δ17,18,92), and null for the entire cluster (KO). Embryos were of different genders. The data was aligned using HISAT v0.1.6-beta. In each tissue, differential gene expression analysis was performed using DESeq2 v1.6.3 using multi-factorial model “~ d17 + d18 + d19 + d92 + gender”, where factor “d17” encoded for conditions that were Δ17, factor “d18” encoded for conditions that were Δ18, etc., and factor “gender” encoded for the genders of the embryos. This allowed us to estimate the log2FC of expression associated with each individual miRNA family in each tissue when accounting for contribution from other miRNA families and the gender.
The microarray dataset from CAD cell expressing miR-124 was obtained from (GSE8498) (Makeyev et al., 2007) using function getGEO() from GEOquery v2.50.5 (Davis and Meltzer, 2007). Differential expression analysis was run using functions lmFit() and eBayes() from limma v3.38.3 (Ritchie et al., 2015).
The TT-FHAGO2 RNA-seq, iCLIP (GSE61348) (Bosson et al., 2014) and CLEAR-CLIP (GSE73059) (Moore et al., 2015) datasets were processed and aligned to the UCSC mm10 mouse genome using STAR v2.5.3a. Reads mapping to multiple loci or with more than 5 mismatches were discarded.
miRNA targets z-score calculation
For conserved miRNA families, the mean log2 fold change of predicted targets compared to the rest of the transcriptome (background) was calculated. The means were converted to z-scores using an approach developed by Kim and Volsky (Kim and Volsky, 2005). Z-score = (Sm - μ) × m1/2 / SD, where Sm is the mean of log2 fold changes of genes for a given gene set, m is the size of the gene set, and μ and SD are the mean and the standard deviation of background log2 fold change values.
HEAP and input control library preparation
mESCs were harvested and irradiated with UV at dose 400 mJ/cm2 in cold PBS on ice. Fresh tissues were harvested, homogenized and irradiated with UV for three times at dose 400 mJ/cm2. Cell or tissue pellets were snap frozen on dry-ice and stored at −80 °C.
Frozen pellets were thawed, lysed with mammalian lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100 and 0.1% Na deoxycholate) containing protease inhibitor cocktail (Promega) and treated with RQ1 DNase (Promega) for 5 min at 37 °C. In order to get the “footprint” Halo-Ago2, lysates were treated with RNase A (Affymetrix, 1:50,000 diluted in TBS) for 5 min at 37 °C. ~2% of the lysates were saved for input control library preparation. The remaining lysates were diluted with buffer TBS (700 μL TBS per 300 μL lysates). For each sample, 300 μL Halolink resin (Promega) was used. The Halolink resin was equilibrated with TBS buffer containing 0.05% IGEPAL CA-630 and incubated with the TBS-diluted lysates at room temperature for 1.5 hr. After incubation, the resin was washed extensively with a series of buffers: SDS elution buffer (50 mM Tris-HCl, pH 7.5 and 0.1% SDS, one wash for 30 min at room temperature on a rotator), LiCl wash buffer (100 mM Tris-HCl, pH 8.0, 500 mM LiCl, 1% IGEPAL CA-630 and 1% Na deoxycholate, three times), 1× PXL buffer (1× PBS with 0.1% SDS, 0.5% Na deoxycholate and 0.5% IGEPAL CA-630, two times), 5× PXL buffer (5× PBS with 0.1% SDS, 0.5% Na deoxycholate and 0.5% IGEPAL CA-630, two times) and PNK buffer (50 mM Tris-HCl, pH 7.4, 10 mM MgCl2 and 0.5% IGEPAL CA-630, two times).
After dephosphorylation with calf intestinal alkaline phosphatase (Promega) at 37 °C for 20 min and washes with buffer PNK-EGTA (50 mM Tris-HCl, pH 7.4, 20 mM EGTA and 0.5% IGEPAL CA-630, two times) and PNK (two times), a 3’ RNA adaptor with a phosphate on its 5’ end (RL3) was ligated to the 3’ end of RNAs using T4 RNA ligase 1 (NEB) at 16 °C overnight. Next day, the resin was sequentially washed with buffer 1× PXL (once), 5× PXL (once) and PNK (three times). RNAs on the resin were treated with T4 PNK (NEB) at 37 °C for 20 min and washed with buffer PNK (three times), Wash/Eq (once) and PK (100 mM Tris-HCl, pH 7.5, 50 mM NaCl and 10 mM EDTA, once). To release RNAs from the resin, proteins were digested with 4 mg/mL proteinase K (Roche) in PK buffer at 37 °C for 20 min and further inactivated by 7 M urea dissolved in PK buffer at 37 °C for 20 min. Free RNAs were extracted using phenol/chloroform and precipitated with ethanol/isopropanol at −20 °C overnight. Next day, RNAs were pelleted, washed with 70% cold ethanol and resuspended in DEPC-treated H2O. A 5’ RNA adaptor (RL5) with six degenerate nucleotides and a common ‘G’ on its 3’ end (RL5-NNNNNNG, RL5D-6N) was ligated to the purified RNAs using T4 RNA ligase 1 at 16 °C for 5 hrs. Then, the RNAs were treated with RQ1 DNase at 37 °C for 20 min to remove residual DNAs and purified by phenol/chloroform extraction and ethanol/isopropanol precipitation.
Purified RNAs were reverse transcribed using the DP3 primer (final concentration: 0.5 μM) and Superscript III reverse transcriptase (Invitrogen). The resulting cDNAs were amplified with primers DP3 and DP5 (final concentrations: 0.5 μM) and Accuprime Pfx DNA polymerase (Invitrogen) to the optimal amplification point. The optimal amplification cycle (defined as the cycle before the PCR reaction reaching a plateau) was preliminarily determined by a diagnostic PCR visualized on gel or a real-time PCR with SYBR green (Invitrogen). PCR products of miRNAs (HEAP miRNA library, expected size: 65 bp) and targets (HEAP mRNA library, expected size range: 75~200 bp) were resolved on a 15% TBE-Urea polyacrylamide gel (Invitrogen) and extracted separately (Figure S1A). To construct library for high-throughput sequencing, DNA primers DP3-barcodeX (“X” stands for barcode index) and DSFP5 (final concentrations: 0.33 μM) containing Illumina adaptors, sequencing primer binding sites and Illumina TruSeq indexes for multiplexing were introduced to the HEAP miRNA and mRNA libraries by PCR. Sequencing libraries were run on a 6% TBE polyacrylamide gel (Invitrogen) and purified.
To prepare input control library, RNAs in the lysates saved before the Halolink resin pulldown were dephosphorylated with calf intestinal alkaline phosphatase and phosphorylated using with T4 PNK. RNAs were then cleaned up using the MyONE Silane beads (ThermoFisher Scientific) as described in (Van Nostrand et al., 2016). Then, the 3’ RNA adaptor (RL3) was ligated to the purified RNAs at 16 °C overnight. Next day, the ligated RNAs were purified using the MyONE Silane beads. Similar to the preparation of HEAP libraries, the RNAs were ligated to the 5’ RNA adaptor (RL5D-6N) at 16 °C for 5 hrs, treated with RQ1 DNase, purified, reverse transcribed to cDNAs and amplified by PCR using primers DP3 and DP5. PCR products ranging from 75 to 200 bp were resolved on a 15% TBE-Urea polyacrylamide gel and used for input library preparation (see HEAP library preparation).
See also Table S1 for oligos and adaptors used in library construction and sequencing.
HEAP mRNA and miRNA libraries, along with the matched input control libraries, were submitted to the Integrated Genomics Operation Core at Memorial Sloan Kettering Cancer Center for high-throughput sequencing. After quantification and quality control, libraries were pooled and run on a HiSeq 2500 in Rapid mode in a 100 bp or 125 bp single end run.
HEAP library preprocessing
Barcode removal
The 6 nt degenerate barcodes and the last nucleotide ‘G’ coming from the 5’ adaptor RL5D-6N (in total 7 nt) were removed from the beginning of reads and appended to the original read names, which later were used to distinguish duplicated reads produced at PCR amplification steps.
Adaptor removal and read quality control
The 3’ adaptor (5’-GTGTCAGTCACTTCCAGCGGGATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3’) and bases with Phred quality score lower than 20 were trimmed from reads using cutadapt v1.15 or v1.17 (Martin, 2011). After trimming, reads shorter than 18 nt were discarded.
Alignment
Processed reads were aligned to the UCSC mm10 mouse genome using STAR v2.5.3a. Reads mapping to multiple loci or with more than 5 mismatches were discarded.
PCR duplicate removal
Reads mapped to the same locus with identical barcodes were considered PCR duplicates and therefore collapsed. This was achieved by storing aligned reads using chromosome names, strand information, positions of the first bases and the 7 nt barcodes as keywords. Representative reads of these unique events were written into a new BAM file, which was used for peak calling.
Peak calling
Peak calling was done using the unpublished package CLIPanalyze (https://bitbucket.org/leslielab/clipanalyze). The function findPeaks() was used to run multiple steps of analysis. First, the combined signal from uniquely aligned and PCR-duplicate-corrected reads from multiple replicates was convolved with the second derivative of a Gaussian filter. Zero-crossings of the convolved signal corresponded to edges of putative peaks. Second, read counting was run in putative peaks and in GENCODE-annotated gene exons with putative peaks subtracted, for both HEAP replicates and input control replicates. Library sizes for both HEAP and input control replicates were estimated using the read counts in exons outside of putative peaks. Third, using these library size estimates, differential read count analysis was performed between HEAP and input control read counts in putative peaks using DESeq2, and FDR-corrected p-values (adjusted p-values) were assigned to each peak. Peaks of size > 20 nt and read count log2FC > 0 in HEAP vs. input control were selected for downstream analysis. Peaks were annotated as overlapping with 3’UTR, 5’UTR, exons, introns, intergenic regions, lncRNA, in that order, using GENCODE (vM17) annotation. Peaks overlapping with genes of types “lincRNA”, “antisense”, “processed_transcript”, according to GENCODE, were annotated as lncRNA peaks.
For mESCs, peak calling was run with the following parameters in findPeaks(): count.threshold = 10, extend.slice = 10, bandwidth = 80, extend.peaks.in.genes = 150. The full set of peaks was generated by comparing three independent HEAP libraries against two input control libraries. To identify peaks in each individual replicate to assess reproducibility and in the cell number titration experiment, a single library was compared to the two input control libraries. For iCLIP, peak calling was run using a single iCLIP library (TT-FHAGO2) against a single control library (TT-AGO2) with the following parameters: count.threshold = 5, extend.slice = 50, bandwidth = 60, extend.peaks.in.genes = 150. For comparison with iCLIP, peak calling with the same parameters was run for each single HEAP library of comparable size against a single input control library (Figures 2F and S2D, S2E).
For embryos, peak calling was run using HEAP in one wildtype (miR-17~92-WT), two heterozygous (miR-17~92-HET) and one homozygous knockout (miR-17~92-KO) embryo against the four matching input control libraries using the following parameters: count.threshold = 5, extend.slice = 10, bandwidth = 80, extend.peaks.in.genes = 150. Then differential HEAP read count analysis was performed using DESeq2 v1.22.1 in miR-17~92-KO against miR-17~92-WT and miR-17~92-HET libraries to determine miR-17~92-dependent peaks.
For cortices of P13 mice, peak calling was run using the two HEAP libraries against the two matching input control libraries using the same parameters as for embryos. The same parameters were used for peak calling using CLEAR-CLIP in 12 replicates vs. the input control libraries generated for HEAP. Differential HEAP read count analysis in HEAP vs. input control was performed using DESeq2 v1.20.0.
For gliomas and cortices in adult mice, three HEAP libraries from each context were generated. Before peak calling, size factors Y of the six HEAP libraries were estimated using the byte sizes of corresponding BAM files. Then, BAM files for two glioma replicates and three cortex replicates were downsampled to similar sizes to the smallest glioma replicate using samtools v1.3.1 (Li et al., 2009) with scaling factors X = 1/Y. Peak calling was run using the six scaled HEAP libraries against the six matching input control libraries, using the same parameters as for embryos, to identify the set of putative peaks. As usual, only peaks of size > 20 nt and with log2FC > 0 in HEAP vs. input control were used in downstream analysis. Furthermore, only peaks with average normalized read count > 10 in the three glioma replicates or in the three cortex replicates were selected. To identify significant peaks in gliomas, DESeq2 v1.20.0 for read counts in these selected peaks was run using the three glioma replicates against the three matching input control replicates. To identify significant peaks in cortices, DESeq2 for read counts in these selected peaks was run using the three cortex replicates against the three matching input control replicates. Differential HEAP read counts analysis between gliomas and cortices was run in peaks with adjusted p-value < 0.05 (in HEAP vs. input control).
For lung tumors, peak calling was run using two HEAP libraries generated from normal lungs, two HEAP libraries from KP tumors and three HEAP libraries from EA tumors against seven matching input control libraries, using the same parameters as for embryos. Peaks of size > 20 nt and with log2FC > 0 in HEAP vs. input control were used in downstream analysis. Furthermore, only peaks with average normalized read count > 10 in the two normal lung replicates, in the two KP tumor replicates or in the three EA tumor replicates were selected. To identify significant peaks in each tumor type, DESeq2 v1.20.0 for read counts in these selected peaks was run using the tumor replicates against their matching input control replicates. To identify significant peaks in normal lungs, DESeq2 for read counts in the selected peaks was run using the two normal lung replicates against the two matching input control replicates. To compare peak intensities between KP and EA tumors, DESeq2 for read counts in peaks with adjusted p-value < 0.05 (in HEAP vs. input control) was run using the three EA tumor replicates against the two KP tumor replicates. Since peak intensities in EA and KP highly correlate with each other, the five tumor replicates were grouped and used for downstream analysis. To compare peak signals between tumors and normal lungs, differential HEAP read count analysis was perform in peaks with adjusted p-value < 0.05 (in HEAP vs. input control) between the five tumor replicates and the two normal lung replicates.
miRNA abundance estimates
Reads in the HEAP miRNA libraries were processed and filtered following the “Barcode removal” and the “Adaptor removal and read quality control” steps described in the “HEAP library preprocessing” section. Processed small RNA reads were aligned to a miRNA genome index built from 1,915 murine pre-miRNA sequences from miRbase version 21 (Kozomara et al., 2018) (ftp://mirbase.org/pub/mirbase/21/) using Bowtie v2.3.4 (Langmead and Salzberg, 2012), and these reads were considered true miRNA counts if they fell within ± 4 bps at each of the 5’ and 3’ end of the annotated mature miRNAs. PCR duplicates were removed as described in the “PCR duplicate removal” step in the “HEAP library preprocessing” section.
miRNA seed family data were downloaded from the TargetScan website at http://www.targetscan.org/mmu_71/mmu_71_data_download/miR_Family_Info.txt.zip. For miRNA family level analysis, read counts mapping to members of the same miRNA family were summed up.
mRNA abundance estimates
Input control libraries generated from gliomas and cortices were used to estimate mRNA abundance. Reads were counted at each gene locus using featureCounts v1.6.3 (Liao et al., 2014) with GENCODE (vM22) primary annotation. Differential gene expression analysis was performed using DEseq2 v1.20.0.
Motif discovery
Unbiased motif enrichment analysis
Frequencies of all k-nucleotide-long sequences (k-mers, k = 7) were calculated for sequences in selected peaks (Freqselected) and background sequences (Freqbg). The enrichment score for these 7-mers was calculated as log2FC = log2 ((Freqselected + c) / (Freqbg + c)), where c was a small corrective value that depended on k, the number and size of peaks. K-mers with the highest log2FCs were then reported. This analysis was performed using functions calculateKmerBackground() and findKmerEnrich() in CLIPanalyze. For mESCs, peaks mapping to 3’UTR were selected and background sequences were defined as sequences of 3’UTRs outside of peaks. For brain and lung cancers, peaks differentially present in tumors and their tissues of origin (adjusted p-value < 0.1, absolute log2FC (tumor vs. normal) > 0.5) were selected and compared against background sequences, defined as exon sequences of genes, in which peaks were identified.
Enrichment score calculation for miRNA seed matches
log2 enrichment score of miRNA seed matches in Figure 2B was calculated as log2 (Freq3’UTR / Freqbg). Freq3’UTR was frequencies of 7mer or 8mer seed matches for miRNA seed families in 3’UTR peaks, while Freqbg was frequencies of these seed matches calculated in background sequences. Background sequences were defined as 3’UTR sequences outside of peaks.
HOMER de novo motif discovery
In mESC libraries, for the top 50 7-mers found by unbiased motif enrichment analysis, positions of their exact occurrences in 3’UTR peaks were found. Sequences of a 15-bp region around these occurrences were extracted and subjected to HOMER de novo motif discovery (Heinz et al., 2010), using 15-bp windows shifted 100 bp and 200 bp on both sides of the 7-mers (and excluding those overlapping with any of the 3’UTR peaks) as background sequences. Similarly, for glioma and cortex libraries, the top 50 7-mers found in each context were mapped to corresponding peak set and subjected to HOMER de novo motif discovery. For normal lung and lung tumor libraries, the top 70 7-mers from each context were used.
IDR analysis
IDR analysis was run using the python package at https://github.com/nboley/idr (Li et al., 2011). All putative peaks (size > 20 nt, log2FC > 0 for HEAP vs. input control) were provided via parameter “-- peak-list”. Peaks called for individual replicates were scored using log2FC in HEAP vs. input control and provided via parameter “--samples”, separately for each pair of replicates. Peaks at IDR < 0.05 were considered reproducible.
HEAP coverage analysis
bigWig files for visualization of HEAP and input control libraries at 1 bp resolution were produced in the following way. First, deep Tools bam Coverage v3.1.3 (Ramirez et al., 2016) with parameter “-bs 1—scale Factor X” was used to produce bed Graph files. Here, size factors Y were estimated using DESeq2 applied to read counts in exons outside of peaks in all HEAP and input control libraries in a particular experimental model (mESCs, embryos, etc.) and then reciprocals X = 1 / Y were used as BAM coverage scaling factors. Only bed Graph signal in the standard chromosomes was selected. Then “bed tools sort” (bed tools v2.23.0 (Quinlan and Hall, 2010)) and bedGraphToBigWig v4 (Kent et al., 2010) were used to produce big Wig files. HEAP libraries were visualized using UCSC genome browser or IGV (Robinson et al., 2011).
To measure HEAP coverage of various peak sets in embryo libraries, peaks were first assigned to miRNA seed families by searching for the corresponding 7mer and 8mer seed matches in peak sequences. All miRNA seed families were ranked by abundance measured in miR-17~92-WT embryo. Peaks containing seed matches for the top 31 miRNA families were chosen. Score matrices of 800 bp windows surrounding these peaks were generated from size-factor-corrected bigWigs using the ScoreMatrixList() function from the genomation package v1.14.0 (Akalin et al., 2015). Histograms of average score were produced using the function plotMeta(). Heatmaps were generated using the multiHeatMatrix() function and extreme values were removed before plotting using the winsorize parameter with values c(0,98).
QUANTIFICATION AND STATISTICAL ANALYSIS
Luciferase assays in Figure 1C and S3E were performed in triplicates and data were represented as mean ± SD. For Figure 2C and S5D, the fit with confidence intervals was produced using function ‘geom_smoonth()’ in R package ggplot using parameter ‘method = “lm”‘ and all other parameters being default. For cumulative distribution function plots, p-values were determined using two-sided Kolmogorov-Smirnov tests between indicated gene sets. For Figure 3B, p-values were calculated using Chi-squared tests. For Figure 3F, normalized counts determined by RNA-seq were plotted as mean ± SD and p-values were determined using unpaired t-tests. For Figure S5B, normalized mRNA counts were plotted as mean ± SD. All other statistical and quantitative analysis was described in detail in the previous sections.
Supplementary Material
Time-lapse imaging of Halo-Ago2 with Janelia Fluor 549 HaloTag ligands in Ago2Halo/Halo MEFs. Scale bar: 10 μm.
Table S1. List of oligonucleotides used in this study, Related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Ago2 | Cell Signaling Technology | Cat#2897; Clone: C34C6; RRID: AB_2096291 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat#A2228; Clone: AC-74; RRID: AB_476697 |
| Mouse monoclonal anti-Tubulin | Sigma-Aldrich | Cat#T9026; Clone: DM1A; RRID: AB_477593 |
| Mouse monoclonal anti-HaloTag | Promega | Cat#G9211; RRID: AB_2688011 |
| Rabbit polyclonal anti-FLAG | Sigma-Aldrich | Cat#F7425; RRID: AB_439687 |
| Rabbit polyclonal anti-GW182 (TNRC6A) | Bethyl | Cat#A302–329A; RRID: AB_1850240 |
| Donkey anti-Rabbit IgG polyclonal antibody (IRDye 800CW) | LI-COR Biosciences | Cat#926–32213; RRID: AB_621848 |
| Donkey anti-Mouse IgG polyclonal antibody (IRDye 680RD) | LI-COR Biosciences | Cat#926–68072; RRID: AB_10953628 |
| Donkey anti-Rabbit IgG, HRP-conjugated | GE Healthcare | Cat#NA934; RRID: AB_772206 |
| Sheep anti-Mouse IgG, HRP-conjugated | GE Healthcare | Cat#NA931; RRID: AB_772210 |
| Bacterial and Virus Strains | ||
| pAd-Cas9-EA (Ad-EA) | ViraQuest Inc.; Maddalo et al., 2014 | N/A |
| pAd-Cas9-BN (Ad-BN) | ViraQuest Inc.; Cook et al., 2017 | N/A |
| Ad-Cre | ViraQuest Inc. | N/A |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1960 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| TruSeq Stranded mRNA LT Kit | Illumina | Cat#RS-122–2102 |
| TruSeq Stranded Total RNA LT Kit | Illumina | Cat#RS-122–1202 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HaloTag Ligands TMRDirect | Promega | Cat#G2991 |
| Janelia Fluor 646 HaloTag ligand | Promega | Cat#GA1120 |
| Janelia Fluor 549 HaloTag ligand | Promega | Cat#GA1110 |
| Amersham ECL Western Blotting Detection Reagent | GE Healthcare | Cat#RPN2106 |
| Odyssey Blocking Buffer (TBS) | LI-COR Biosciences | Cat#927–50003 |
| KnockOut DMEM | Gibco | Cat#10829018 |
| Fetal Bovine Serum, embryonic stem cell-qualified, US origin | Gibco | Cat#16141079 |
| ESGRO Leukemia Inhibitory Factor (LIF) | Millipore | Cat#ESG1107 |
| Penicillin-Streptomycin (5,000 U/mL) | Gibco | Cat#15070063 |
| GlutaMax Supplement | Gibco | Cat#35050061 |
| MEM non-essential amino acid solution (100X) | Sigma-Aldrich | Cat#M7145 |
| EmbryoMax Nucleosides (100X) | Millipore | Cat#ES-008-D |
| 2-Mercaptoethanol | BIO-RAD | Cat#1610710, CAS: 60–24-2 |
| DMEM | Gibco | Cat#11965118 |
| DMEM, no phenol red | Gibco | Cat#21063029 |
| Advanced DMEM/F12 | Gibco | Cat#12634028 |
| HEPES (1M) | Gibco | Cat#15630080 |
| Geneticin Selective Antibiotic (G418 Sulfate) | Gibco | Cat#10131035 |
| Halolink Resin | Promega | Cat#G1914 |
| Magne HaloTag Beads | Promega | Cat#G7282 |
| PhosStop, Phosphatase inhibitor | Roche | Cat#4906837001 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#11836170001 |
| AcTEV protease | Invitrogen | Cat#12575015 |
| Protease Inhibitor Cocktail, 50x | Promega | Cat#G6521 |
| RQ1 RNase-Free DNase | Promega | Cat#M6101 |
| RNase A | Affymetrix | Cat#70194Z |
| Alkaline Phosphatase, Calf Intestinal (CIAP) | Promega | Cat#M1821 |
| T4 RNA Ligase 1 | New England Biolabs | Cat#M0204 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat#M0201 |
| SuperScript III Reverse Transcriptase | Invitrogen | Cat#18080044 |
| Accuprime Pfx Supermix | Invitrogen | Cat#12344040 |
| Proteinase K, recombinant, PCR Grade | Roche | Cat#3115836001 |
| Phenol solution | Sigma-Aldrich | Cat#P4682 |
| Chloroform – isoamyl alcohol mixture | Sigma-Aldrich | Cat#25668 |
| Novex TBE-Urea Gels, 15%, 10 well | Invitrogen | Cat#EC6885BOX |
| Novex TBE Gels, 6%, 10 well | Invitrogen | Cat#EC6265BOX |
| SYBR Green I Nucleic Acid Gel Stain | Invitrogen | Cat#S7563 |
| Dynabeads MyOne Silane | ThermoFisher Scientific | Cat#37002D |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| RNase-Free DNase Set | QIAGEN | Cat#79254 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat#E2621L |
| cOmplete Protease Inhibitor Cocktail (Mass Spectrometry) | Roche | Cat#11836153001 |
| Trypsin Gold, Mass Spectrometry Grade | Promega | Cat#V5280 |
| Lysyl Endopeptidase (Lys-C) | Wako Chemicals | Cat#129–02541 |
| TMT10plex Isobaric Label Reagent Set plus TMT11–131C Label Reagent | Thermo Fisher Scientific | Cat#A34808 |
| Oligonucleotides | ||
| See Table S1. | N/A | N/A |
| Deposited Data | ||
| HEAP libraries and RNA-seq datasets | This paper | GSE139349 |
| TT-FHAgo2 mESC iCLIP | Bosson et al., 2014 | GSE61348 |
| TT-FHAgo2 mESC RNAseq | Bosson et al., 2014 | GSE61348 |
| CLEAR-CLIP | Moore et al., 2015 | GSE73059 |
| miR-17~92 E9.5 embryo RNA-seq | Han et al., 2015 | GSE63813 |
| miR-124 overexpression CAD | Makeyev et al., 2007 | GSE8498 |
| miRbase version 21 | Kozomara et al., 2018 | ftp://mirbase.org/pub/mirbase/21/ |
| TargetScan | Agarwal et al., 2015 | http://www.targetscan.org/mmu_71/mmu_71_data_download/miR_Family_Info.txt.zip |
| Experimental Models: Cell Lines | ||
| Ago2-/- MEFs | Alexander Tarakhovsky Laboratory; O’Carroll et al., 2007 | N/A |
| Ago2Halo-LSL/+ mESCs | This paper | N/A |
| Ago2Halo/+ mESCs | This paper | N/A |
| Ago2Halo-LSL MEFs | This paper | N/A |
| Ago2Halo MEFs | This paper | N/A |
| KRasG12D;Trp53-/- (KP) NSCLC cell lines | This paper | N/A |
| V6.5 mESCs | Rudolf Jaenisch Laboratory | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: KRasLSL-G12D/+: B6.129S4-Krastm4Tyj/J | Jackson et al., 2001 | JAX Stock No. 008179 |
| Mouse: Trp53fl/fl: B6.129P2-Trp53tm1Brn/J | Marino et al., 2000 | JAX Stock No. 008462 |
| Mouse: β-actin-Flpe: B6;SJL-Tg(ACTFLPe)9205Dym/J | Memorial Sloan Kettering Cancer Center Mouse Genetics Core Facility; Rodriguez et al., 2000 | JAX Stock No. 003800 |
| Mouse: CAG-Cre: Tg(CAG-cre)13Miya | Memorial Sloan Kettering Cancer Center Mouse Genetics Core Facility; Sakai and Miyazaki, 1997 | N/A |
| Mouse: Ago2Halo-LSL | This paper | N/A |
| Mouse: Ago2Halo | This paper | N/A |
| Mouse: miR-17~92-/- | Ventura et al., 2008 | N/A |
| Recombinant DNA | ||
| MSCV-PIG-Empty | Mayr and Bartel, 2009 | Addgene: 21654 |
| MSCV-PIG-Halo | This paper | N/A |
| MSCV-PIG-Ago2 | This paper | N/A |
| MSCV-PIG-Halo-Ago2 | This paper | N/A |
| pKOII-Halo-lox-IRES-lox-Ago2 | This paper | N/A |
| pMB1610_pRR-puro | Flemr et al., 2015 | Addgene: 65853 |
| PX330 | Cong et al., 2013, Ran et al., 2013 | Addgene: 42230 |
| PX333 | Maddalo et al., 2014 | Addgene: 64073 |
| piS0 | Yekta et al., 2004 | Addgene: 12178 |
| piS1 | Addgene | Addgene: 12179 |
| psico luc + Cre | Ventura et al., 2004 | N/A |
| psico CD8 + Cre | Ventura et al., 2004 | N/A |
| pBABE-SV40-puro | Zhao et al., 2003 | Addgene:13970 |
| pTRETightBI-RY-0 | Mukherji et al., 2011 | Addgene: 31463 |
| pTRETightBI-RY-1pf | Mukherji et al., 2011 | Addgene: 31467 |
| pTRETightBI-RY-4 | Mukherji et al., 2011 | Addgene: 31465 |
| rtTA-N144 | Richner et al., 2015 | Addgene: 66810 |
| psiCheck2 | Promega | C8021 |
| psiCheck2-Pten-miR-29-WT | This paper | N/A |
| psiCheck2-Pten-miR-29-MUT | This paper | N/A |
| psiCheck2-Adrb2-let-7-WT | This paper | N/A |
| psiCheck2-Adrb2-let-7-MUT | This paper | N/A |
| psiCheck2-Taf7-miR-21-WT | This paper | N/A |
| psiCheck2-Taf7-miR-21-MUT | This paper | N/A |
| Turn-T6BWT-EYFP | This paper | N/A |
| Turn-T6BMUT-EYFP | This paper | N/A |
| Software and Algorithms | ||
| STAR (v2.5.3a) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Cutadapt (v1.15 and v1.17) | Martin, 2011 | https://cutadapt.readthedocs.io |
| Bowtie2 (v2.3.4) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2 |
| DESeq2 (v1.20.0, v1.6.3 and 1.22.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| IDR | Li et al., 2011 | https://github.com/nboley/idr |
| Deeptools (v3.1.3) | Ramirez et al., 2016 | https://deeptools.readthedocs.io |
| Bedtools (v2.23.0) | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io |
| kentUtils | Kent et al., 2010 | https://github.com/ENCODE-DCC/kentUtils |
| Limma (v3.38.3) | Ritchie et al., 2015 | http://bioconductor.org/packages/release/bioc/html/limma.html |
| featureCounts (v1.6.3) | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts |
| Genomation (v1.14.0) | Akalin et al., 2015 | https://bioconductor.org/packages/release/bioc/html/genomation.html |
| HISAT2 (v0.1.6-beta) | Kim et al., 2019 | https://ccb.jhu.edu/software/hisat2 |
| GEOquery (v2.50.5) | Davis and Meltzer, 2007 | https://bioconductor.org/packages/release/bioc/html/GEOquery.html |
| Samtools (v1.3.1) | Li et al., 2009 | http://www.htslib.org |
| Parametric Analysis of Gene Set Enrichment | Kim and Volsky, 2005 | N/A |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| CLIPanalyze (v0.0.8) | unpublished | https://bitbucket.org/leslielab/clipanalyze |
| Sequest | Eng et al., 1994 | http://proteomicsresource.washington.edu/protocols06/sequest.php |
| Odyssey | LI-COR Biosciences | https://www.licor.com/bio/ |
| IGV | Robinson et al., 2011 | http://software.broadinstitute.org/software/igv/ |
| ZEN | ZEISS | https://www.zeiss.com/microscopy/int/downloads.html |
| Fiji | NIH | https://imagej.net/Fiji |
| Other | ||
| Superose 6 column | GE Healthcare | Cat#10/300 GL |
| Sep-Pak C18 3 cc Vac Cartridge, 200 mg | Waters | Cat#WAT054945 |
HIGHLIGHT.
The authors describe a mouse strain harboring a Cre-regulated Halo-Ago2 knock-in allele
The model streamlines the experimental identification of miRNA-mRNA interactions
The authors identify miRNA targets in mESCs, embryos, normal tissues, and tumors
ACKNOWLEDGMENTS
We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This work was funded by grants from the NIH (NCI R01CA149707, AV), The Pershing Square Sohn Cancer Research Alliance (AV), The Starr Cancer Consortium (AV and DB), and the Geoffrey Beene Cancer Research Foundation (AV). YP was supported by the AACR-Bristol-Myers Squibb Immuno-oncology Research Fellowship, Grant Number 19–40-15-PRIT. CPC was supported by the NCI F31 training grant, Grant Number 1F31CA168356–01A1.
We thank Gregory Hannon for suggesting the use of the HaloTag to pull down Argonaute-containing complexes, Joana de Campos Vidigal for assistance with the design of the Halo-Ago2 targeting construct and for the gene targeting experiments, and members of the Ventura, Leslie, and Benezra laboratories for discussion and suggestions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, Franke V, Vlahovicek K, Mason CE, and Schubeler D (2015). Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 31, 1127–1129. [DOI] [PubMed] [Google Scholar]
- Bak M, Silahtaroglu A, Møller M, Christensen M, Rath MF, Skryabin B, Tommerup N, and Kauppinen S (2008). MicroRNA expression in the adult mouse central nervous system. Rna 14, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018). Metazoan MicroRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, and Sharp PA (2014). Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, and Darnell RB (2009). Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PJ, Thomas R, Kannan R, de Leon ES, Drilon A, Rosenblum MK, Scaltriti M, Benezra R, and Ventura A (2017). Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat Commun 8, 15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos V., Moutinho C., Villanueva A., Boque R., Silva P., Carneiro F., and Esteller M. (2012). Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31, 2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, and Meltzer PS (2007). GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, and Barres BA (2010). Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron 65, 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds MD, Boyd KL, Moyo T, Mitra R, Duszynski R, Arrate MP, Chen X, Zhao Z, Blackwell TS, Andl T, et al. (2016). MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J Clin Invest 126, 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Emery B (2010). Regulation of oligodendrocyte differentiation and myelination. Science 330, 779–782. [DOI] [PubMed] [Google Scholar]
- Encell LP, Friedman Ohana R, Zimmerman K, Otto P, Vidugiris G, Wood MG, Los GV, McDougall MG, Zimprich C, Karassina N, et al. (2012). Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr Chem Genomics 6, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fan HB, Chen LX, Qu XB, Ren CL, Wu XX, Dong FX, Zhang BL, Gao DS, and Yao RQ (2017). Transplanted miR-219-overexpressing oligodendrocyte precursor cells promoted remyelination and improved functional recovery in a chronic demyelinated model. Sci Rep 7, 41407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemr M, and Buhler M (2015). Single-Step Generation of Conditional Knockout Mouse Embryonic Stem Cells. Cell Rep 12, 709–716. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, and Bartel DP (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, et al. (2009). Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev 23, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA., Bert AG., Paterson EL., Barry SC., Tsykin A., Farshid G., Vadas MA., Khew-Goodall Y., and Goodall GJ. (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10, 593–601. [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12, 244–250, 243 p following 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, and Bartel DP (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, and Rajewsky N (2014). Unambiguous Identification of miRNA:Target Site Interactions by Different Types of Ligation Reactions. Molecular Cell 54, 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wang M, Yang Y, Qiu D, Zhang Y, Ma J, Zhou Y, Hannon GJ, and Yu Y (2018). GoldCLIP: Gel-omitted Ligation-dependent CLIP. Genomics, Proteomics & Bioinformatics 16, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr., Jungkamp AC, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YC, Vidigal JA, Mu P, Yao E, Singh I, Gonzalez AJ, Concepcion CP, Bonetti C, Ogrodowski P, Carver B, et al. (2015). An allelic series of miR-17 approximately 92-mutant mice uncovers functional specialization and cooperation among members of a microRNA polycistron. Nat Genet 47, 766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann J, Schraivogel D, Bruckmann A, Manickavel S, Jakob L, Eichner N, Pfaff J, Urban M, Sprunck S, Hafner M, et al. (2015). Biochemical isolation of Argonaute protein complexes by AgoAPP. Proc Natl Acad Sci U S A 112, 11841–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. (2005). A microRNA polycistron as a potential human oncogene. Nature 435, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, and Tollervey D (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin JP, Lu Y, Loeb GB, Leslie CS, and Rudensky AY (2018). The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat Immunol 19, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL., Jedrychowski MP., Elias JE., Goswami T., Rad R., Beausoleil SA., Villén J., Haas W., Sowa ME., and Gygi SP. (2010). A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-Y, and Volsky DJ (2005). PAGE: parametric analysis of gene set enrichment. In BMC Bioinformatics, pp. 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleaveland B, Shi CY, Stefano J, and Bartel DP (2018). A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 174, 350–362.e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, and Ule J (2010). iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Birgaoanu M, and Griffiths-Jones S (2018). miRBase: from microRNA sequences to function. Nucleic Acids Research 47, D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rocca G, Olejniczak SH, Gonzalez AJ, Briskin D, Vidigal JA, Spraggon L, DeMatteo RG, Radler MR, Lindsten T, Ventura A, et al. (2015). In vivo, Argonaute-bound microRNAs exist predominantly in a reservoir of low molecular weight complexes not associated with mRNA. Proc Natl Acad Sci U S A 112, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann Appl Stat 5, 1752–1779. [Google Scholar]
- Lianoglou S, Garg V, Yang JL, Leslie CS, and Mayr C (2013). Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev 27, 2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). feature Counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, and Rudensky AY (2012). Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell 48, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV., Encell LP., McDougall MG., Hartzell DD., Karassina N., Zimprich C., Wood MG., Learish R., Ohana RF., Urh M., et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, et al. (2014). In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, and Maniatis T (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal; Vol 17, No 1: Next Generation Sequencing Data AnalysisDO - 1014806/ej171200. [Google Scholar]
- Mayr C, and Bartel DP (2009). Widespread Shortening of 3′UTRs by Alternative Cleavage and Polyadenylation Activates Oncogenes in Cancer Cells. Cell 138, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Analytical Chemistry 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Scheel TK, Luna JM, Park CY, Fak JJ, Nishiuchi E, Rice CM, and Darnell RB (2015). miRNA-target chimeras reveal miRNA 3’-end pairing as a major determinant of Argonaute target specificity. Nat Commun 6, 8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GXY, Tsang JS, Sharp PA, and van Oudenaarden A (2011). MicroRNAs can generate thresholds in target gene expression. Nature Genetics 43, 854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll D, Mecklenbrauker I, Das PP, Santana A, Koenig U, Enright AJ, Miska EA, and Tarakhovsky A (2007). A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev 21, 1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejniczak SH, La Rocca G, Gruber JJ, and Thompson CB (2013). Long-lived microRNA-Argonaute complexes in quiescent cells can be activated to regulate mitogenic responses. Proc Natl Acad Sci U S A 110, 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, and Seto M (2004). Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 64, 3087–3095. [DOI] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, and Gygi SP (2016). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, and Gygi SP (2003). A proteomics approach to understanding protein ubiquitination. Nature Biotechnology 21, 921–926. [DOI] [PubMed] [Google Scholar]
- Pfaff J, Hennig J, Herzog F, Aebersold R, Sattler M, Niessing D, and Meister G (2013). Structural features of Argonaute-GW182 protein interactions. Proc Natl Acad Sci U S A 110, E3770–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F., Ryan DP., Gruning B., Bhardwaj V., Kilpert F., Richter AS., Heyne S., Dundar F., and Manke T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner M, Victor MB, Liu Y, Abernathy D, and Yoo AS (2015). MicroRNA-based conversion of human fibroblasts into striatal medium spiny neurons. Nature Protocols 10, 1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nature Biotechnology 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, and Dymecki SM (2000). High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25, 139–140. [DOI] [PubMed] [Google Scholar]
- Sakai K, and Miyazaki J. i. (1997). A Transgenic Mouse Line That Retains Cre Recombinase Activity in Mature Oocytes Irrespective of thecreTransgene Transmission. Biochemical and Biophysical Research Communications 237, 318–324. [DOI] [PubMed] [Google Scholar]
- Sanuki R, Onishi A, Koike C, Muramatsu R, Watanabe S, Muranishi Y, Irie S, Uneo S, Koyasu T, Matsui R, et al. (2011). miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat Neurosci 14, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Sarshad AA, Juan AH, Muler AIC, Anastasakis DG, Wang X, Genzor P, Feng X, Tsai PF, Sun HW, Haase AD, et al. (2018). Argonaute-miRNA Complexes Silence Target mRNAs in the Nucleus of Mammalian Stem Cells. Mol Cell 71, 1040–1050.e1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Shien K, Tomida S, Okayasu K, Suzawa K, Hashida S, Torigoe H, Watanabe M, Yamamoto H, Soh J, et al. (2017). Targeting the miR-200c/LIN28B axis in acquired EGFR-TKI resistance non-small cell lung cancer cells harboring EMT features. Sci Rep 7, 40847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si L, Tian H, Yue W, Li L, Li S, Gao C, and Qi L (2017). Potential use of microRNA-200c as a prognostic marker in non-small cell lung cancer. Oncol Lett 14, 4325–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CL, Plotkin JL, Veno MT, von Schimmelmann M, Feinberg P, Mann S, Handler A, Kjems J, Surmeier DJ, O’Carroll D, et al. (2013). MicroRNA-128 governs neuronal excitability and motor behavior in mice. Science 342, 1254–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, and Bartel DP (2011). Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, and Jacks T (2004). Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A 101, 10380–10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A., Young AG., Winslow MM., Lintault L., Meissner A., Erkeland SJ., Newman J., Bronson RT., Crowley D., Stone JR., et al. (2008). Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Moyano AL, Ma Z, Deng Y, Lin Y, Zhao C, Zhang L, Jiang M, He X, Ma Z, et al. (2017). miR-219 Cooperates with miR-338 in Myelination and Promotes Myelin Repair in the CNS. Dev Cell 40, 566–582.e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, et al. (2011). Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. PROTEOMICS 11, 2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta S, Shih IH, and Bartel DP (2004). MicroRNA-directed cleavage of HOXB8 mRNA. Science 304, 594–596. [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, and Hahn WC (2003). Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3kinase. Cancer Cell 3, 483–495. [DOI] [PubMed] [Google Scholar]
- Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, Hoang T, Xu X, Mi QS, Xin M, et al. (2010). MicroRNA-mediated control of oligodendrocyte differentiation. Neuron 65, 612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse imaging of Halo-Ago2 with Janelia Fluor 549 HaloTag ligands in Ago2Halo/Halo MEFs. Scale bar: 10 μm.
Table S1. List of oligonucleotides used in this study, Related to STAR Methods.
Data Availability Statement
The datasets generated during this study are available at GEO (GSE139349). CLIPanalyze is available for download at https://bitbucket.org/leslielab/clipanalyze. This published article includes algorithms and key parameters used during this study.