

Abstract
PURPOSE
Homologous DNA repair–deficient (HRD) ovarian cancers (OCs), including those with BRCA1/2 mutations, have higher levels of genetic instability, potentially resulting in higher immunogenicity, and have been suggested to respond better to immune checkpoint inhibitors (ICIs) than homologous DNA repair–proficient OCs. However, clinical evidence is lacking. The study aimed to evaluate the associations between BRCA1/2 mutations, HRD, and other genomic parameters and response to ICIs and survival in OC.
METHODS
This is a single-institution retrospective analysis of women with recurrent OC treated with ICIs. BRCA1/2 mutation status and clinicopathologic variables were abstracted from the medical records. Targeted and whole-exome sequencing data available for a subset of patients were used to assess tumor mutational burden (TMB), HRD, and fraction of genome altered (FGA). ICI response was defined as lack of disease progression for ≥ 24 weeks. Associations of BRCA1/2 status and genomic alterations with progression-free survival (PFS) and overall survival (OS) were determined using Cox proportional hazards models.
RESULTS
Of the 143 women treated with ICIs, 134 had known BRCA1/2 mutation status. Deleterious germline or somatic BRCA1/2 mutations were present in 31 women (24%). There was no association between presence of BRCA1/2 mutations and response (P = .796) or survival. Genomic analysis in 73 women found no association between TMB (P = .344) or HRD (P = .222) and response, PFS, or OS. There were also no significant differences in somatic genetic alterations between responders and nonresponders. High FGA was associated with an improvement in PFS (P = .014) and OS (P = .01).
CONCLUSION
TMB, BRCA1/2 mutations, and HRD are not associated with response or survival, cautioning against their use as selection criteria for ICI in recurrent OC. FGA should be investigated further as a biomarker of response to immunotherapy in OC.
INTRODUCTION
Alterations in the DNA mismatch repair pathway increase susceptibility to immune checkpoint inhibition (ICI),1 potentially through increased tumor mutational burden (TMB) and generation of neoantigens.2 High TMB has been shown to improve outcomes after ICI in some cancers3; however, the role of alterations in other DNA repair pathways as predictive biomarkers of response to immunotherapies remains unknown.
CONTEXT
Key Objective
Although homologous repair deficiency (HRD), as seen in BRCA1/2-mutated ovarian cancer (OC), has been hypothesized to confer increased immunogenicity, clinical data showing improved responses to immune checkpoint inhibition (ICI) are lacking. We sought to evaluate whether the presence of BRCA mutations and other genomic markers of HRD predict response to ICI and improved survival in OC.
Knowledge Generated
We found that tumor mutational burden and traditional markers of HRD, including BRCA1/2 mutations, did not predict response to ICI in OC. However, fraction of genome altered (FGA), a marker of global copy number alterations, was associated with improved survival in those receiving ICI.
Relevance
These findings caution against using BRCA1/2 status and HRD as a rationale for ICI in OC and highlight the need for other biomarkers. In this study, we identified FGA as a potential marker of response to ICI in OC that warrants further investigation.
High-grade serous ovarian cancer (HGSOC) is characterized by homologous recombination DNA repair deficiencies (HRDs), and 20%-25% of ovarian cancers (OCs) harbor germline or somatic deleterious mutations in BRCA1/2.4 Alterations in other genes may also give rise to HRD, and a plethora of genomic biomarkers for HRD assessment, including large-scale state transitions (LSTs)5 and mutational signatures,6 have been developed to assess HRD in human cancers.5
Although controversial, HRD tumors have been proposed to have increased immunogenicity,7 potentially resulting in increased response to ICI. Case reports have demonstrated efficacy with ICI in BRCA1/2-mutated OCs, and this has been hypothesized to increase response to ICI.8 Supporting these findings, genomic instability associated with BRCA1/2-mutated tumors may drive generation of cytosolic DNA and activation of the cGAS-STING signaling axis,9 which has been shown to be an important mediator of tumor immune recognition and response to ICI. Conversely, chronic cytosolic DNA signaling associated with chromosomally unstable cancers has been demonstrated to drive activation of immunosuppressive pathways and metastases through noncanonical nuclear factor-κB (NF-κB) signaling,10 which suggests that BRCA1/2-mutated tumors may actually be less immunogenic. Supporting this hypothesis, an extensive analysis of BRCA1/2-mutated breast tumors has demonstrated that HRD may be associated with decreased immunogenicity.11
Responses to ICI in OC have been modest,12-14 and it is unclear whether response to ICI varies according to BRCA1/2 mutation status, other markers of HRD, or TMB. Emerging data suggest that the combination of ICI and poly (ADP-ribose) polymerase (PARP) inhibitors may act in an additive or synergistic manner,15-19 spawning a number of large trials evaluating ICIs in combination with PARP inhibitors. Although these studies highlight that PARP inhibition could engender immunogenicity in OCs, possibly through activation of the cGAS-STING signaling axis,9 they fail to answer whether BRCA1/2-mutated OCs are inherently more sensitive to ICI because it is difficult to separate the contribution of each therapy within such a setting.
To address these questions, we characterized a cohort of patients with OC who received ICI alone, without additional cytotoxic or targeted therapy. Specifically, we sought to investigate whether pathogenic BRCA1/2 mutations, HRD status, TMB, or other genomic alterations are associated with ICI response in OC.
METHODS
Data Collection
Data collection and analysis were performed using a retrospective research protocol approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board. ICI was defined as therapy with antibodies targeting PD-1, PD-L1, CTLA-4, LAG-3, or a combination. Retrospective review identified 143 women with recurrent OC treated with ICIs from January 2013 to April 2019, in whom ICI was administered without combination with chemotherapy, PARP inhibitors, or other targeted agents. Patient characteristics and BRCA1/2 status were abstracted and double-verified. BRCA1/2 mutation was defined as presence of any pathogenic somatic or germline BRCA1/2 mutations, excluding variants of unknown significance. Patients were considered to have missing data if results of both somatic and germline testing were not documented in the medical record. Age, stage, and histology were defined at pathologic diagnosis. Initial treatment and surgical outcomes were abstracted from the clinical record. CA-125, albumin, body mass index, and platinum-free interval were defined at the initiation of ICI. Platinum resistance was defined as disease progression per imaging within 6 months of last platinum treatment.
Genomics Cohort Selection
Somatic genomic analyses were available for 73 patients, using Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT; n = 64)20 and/or whole-exome sequencing (WES; n = 20; not overlapping with MSK-IMPACT, n = 9) to assess the repertoire of somatic mutations, copy number alterations (CNAs), genomics features of HRD based on LST scores,21 and fraction of genome altered (FGA; Fig 1). For LST and FGA analyses, WES (n = 20) was used when available, and MSK-IMPACT (n = 53) was used for the remaining patients. For TMB analysis, only MSK-IMPACT (n = 64) data were used because of differences in depth between MSK-IMPACT and WES sequencing. Results from the molecular diagnostic reports were used and further curated by a board-certified molecular pathologist (D.M.).
FIG 1.

Patient selection, BRCA mutations, and genomic analysis. FGA, fraction of genome altered; g, germline; ICI, immune checkpoint inhibition; IMPACT, Integrated Mutation Profiling of Actionable Cancer Targets targeted sequencing; LST, large-scale transition; s, somatic; TMB, tumor mutation burden; WES, whole-exome sequencing.
DNA Sequencing and Bioinformatics Analyses
In all patients, DNA was obtained from archival tissue collected during the initial debulking surgery. DNA samples extracted from formalin-fixed, paraffin-embedded OCs (all histologies) and matched blood or normal tissue were subjected to MSK-IMPACT targeted sequencing (n = 64) and/or WES (n = 20), as previously described.22,23 Briefly, raw sequence reads were aligned to the reference human genome GRCh37 using the Burrows‐Wheeler Aligner (BWA 0.7.15).24 Local realignment, duplicate read removal, and base quality score recalibration were performed using the Genome Analysis Toolkit (GATK 3.7).25 Somatic single nucleotide variants (SNVs) were called using MUTECT (1.1.7),26 and small insertions and deletions (indels) were identified using Strelka (1.0.15),27 VarScan2 (2.3.7),28 Lancet (1.0.0),29 and Scalpel (0.5.3)30 and further curated by manual inspection. SNVs and indels outside of target regions were filtered out, as were SNVs and indels for which the variant allele fraction (VAF) in the tumor sample was < 5 times that of the paired normal VAF, as previously described.22,23 Finally, SNVs and indels found at > 5% global minor allele frequency in the Single Nucleotide Polymorphism Database (dbSNP; build 137) and > 5% global allele frequency in EXAC (0.3.1) were discarded. Somatic CNAs and loss of heterozygosity were obtained using FACETS.22,23,31 Mutation hotspots were assigned according to Chang et al.32
TMB was calculated by dividing the number of nonsynonymous mutations by the total size of the capture panel in mega base pairs. To define the mutational signatures, we assessed the mutational context of synonymous and nonsynonymous SNVs in samples subjected to MSK-IMPACT targeted sequencing using SigMA,33 using the OC tumor type setting and targeted sequencing setting. For samples subjected to WES, deconstructSigs34 was used at default settings with COSMIC signature metrics, as previously described.35 Mutational signatures were defined only for the samples with at least 5 SNVs using SigMA or at least 20 SNVs for deconstructSigs, as previously described.35,36
On the basis of the CNAs obtained by FACETS, FGA was defined as the length of segments with a log2 or linear CNA value > 0.2 divided by the length of all segments measured. LST scores, defined as a chromosomal breakpoint resulting in allelic imbalance between adjacent regions of at least 10 Mb, were determined, and a cutoff score ≥ 15 was used for LST-high cases, as previously described.21,22,36,37
Statistical Analysis
Duration of ICI therapy was defined as time from first dose to date of progression or death as a result of disease (progression-free survival [PFS]). Progression of disease was defined as clinical progression or radiographic progression using RECIST 1.1 criteria. Patients who discontinued therapy for reasons other than progression (n = 9) were observed until progression or start of new therapy. Response to ICI was defined as lack of disease progression for ≥ 24 weeks.38 Overall survival (OS) was defined from initiation of ICI to death or last follow-up in those still living.
Patient characteristics and clinical variables were reported using summary statistics. BRCA1/2 status and genomic markers were compared between responders and nonresponders. All comparisons between BRCA1/2 mutant versus wild-type (WT) and responders versus nonresponders were calculated using Fisher’s exact test for categorical variables and Wilcoxon rank sum test for continuous variables. Kaplan-Meier survival analysis was used to estimate median PFS and OS and 1-year PFS and OS rates by BRCA1/2 status and other genomic markers (TMB, LST, and FGA). Because of low overall TMB, an exploratory analysis of its impact on outcome was performed with cutoffs set at the median and upper quartile. Additional exploratory analyses of FGA with outcomes were performed with cutoffs defined as the median and using the maximally selected standardized log-rank statistic method.39-41 Log-rank and Wilcoxon rank test P values were calculated to examine results using different weights (higher weight on earlier time points for Wilcoxon test); however, only the log-rank P values are reported. A Cox proportional hazards model was used to assess continuous variables in survival univariately. A multivariable Cox proportional hazards model was built to examine the relationship between PFS or OS and BRCA1/2 status, adjusting for covariates. For somatic genomic markers, analyses were only performed for the subset of patients with complete genomic marker information.
RESULTS
Of the 143 women with recurrent OC treated with ICI, 76 (53%) received an anti–PD-1/PD-L1 antibody, 46 (32%) received an anti–PD-1/PD-L1 antibody in combination with an anti–CTLA-4 antibody, and the remaining women (n = 21) received other ICI regimens. The majority of patients (n = 116) were treated on protocols, and 27 women were treated under compassionate use programs. Median age at diagnosis was 57 years (range, 23-77 years). The majority of patients (n = 116) had high-grade serous histology, and 27 patients had other histologies (clear cell, n = 14; endometrioid, n = 6; low-grade serous, n = 4; carcinosarcoma, n = 1; mixed serous/endometrioid, n = 1; and adenocarcinoma, n = 1). Most women (83%) were platinum resistant at ICI and had received a median of 4 lines (range, 1-13 lines) of treatment before ICI. Responses to ICI were observed in 25 women overall (18%; Table 1). At a median follow-up time of 28.6 months, median PFS was 2.5 months (95% CI, 2.0 to 2.8 months), and median OS was 16.5 months (95% CI, 11.1 to 20.5 months) from initiation of ICI (Tables 2 and 3) for the entire cohort.
TABLE 1.
Patient Characteristics, Overall and Stratified by BRCA1/2 Status

TABLE 2.
Univariable PFS Analysis

TABLE 3.
Univariable OS Analysis

Of the 143 women included in this study, 134 had known BRCA1/2 status (germline testing, n = 119; somatic testing, n = 83), of whom 31 (24%) had pathogenic germline or somatic BRCA1/2 mutations (Fig 1, Data Supplement). Patients with BRCA1/2 mutations, compared with patients with WT BRCA1/2, had more prior treatment lines (median, 5 v 4 lines, respectively; P = .018), longer time from diagnosis to ICI (53.6 v 37.8 months, respectively; P = .013), higher rates of prior PARP inhibitor treatment (26% v 5%, respectively; P = .002), and a trend toward more HGSOC (94% v 78%, respectively; P = .064; Table 1). Response to ICI did not vary according to BRCA1/2 status (20% in BRCA1/2 WT v 16% in BRCA1/2 mutated; P = .796; Table 1 and Fig 2B). Of the 64 patients with available MSK-IMPACT targeted sequencing data, 11 had germline or somatic BRCA1/2 mutations, of which 8 were found to be biallelic. Similarly, there was no enrichment for responders in patients with either BRCA1- or BRCA2-mutated tumors or in patients with biallelic BRCA1/2 loss, although the numbers of patients in these analyses were small (Data Supplement). BRCA1/2 status was also not associated with PFS or OS in univariable or multivariable models, even after adjusting for clinical variables such as CA-125, albumin, platinum-resistant status, or prior treatment lines (Fig 2C, Tables 2 and 3, Data Supplement).
FIG 2.
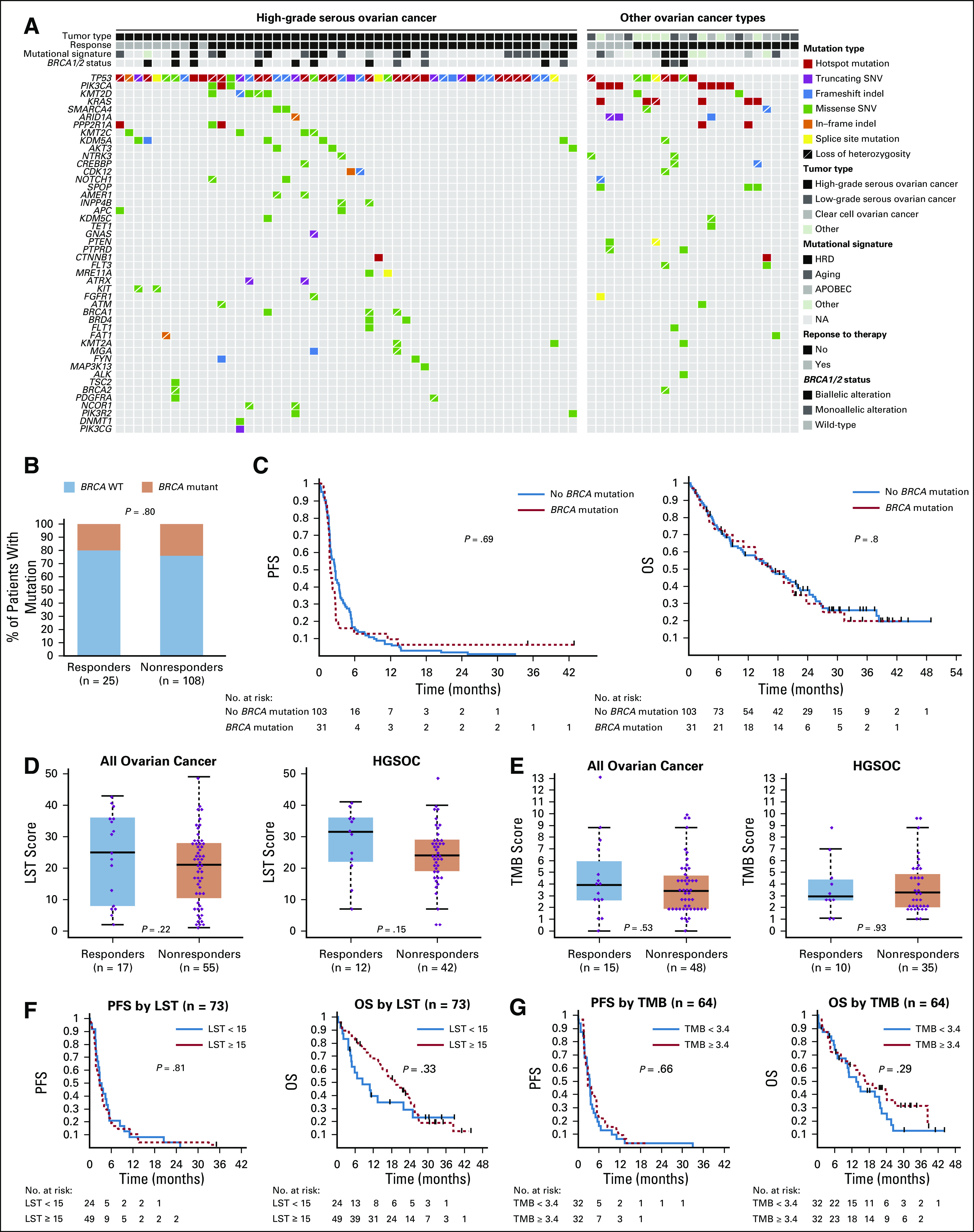
Association of immune checkpoint inhibitor (ICI) response with molecular alterations in ovarian cancer. (A) Somatic mutations in high-grade serous ovarian cancer (HGSOC; left) and non-HGSOC (right). Mutation types, mutational signatures, response to ICI, and BRCA1/2 status are indicated by the labels. (B) Relative percentages of patients with deleterious germline and somatic BRCA1 or BRCA2 mutations in each response group. (C) Differences in progression-free survival (PFS; left) and overall survival (OS; right) from the start of ICI as a function of presence of deleterious germline or somatic BRCA1/2 mutations. (D) Association of homologous DNA repair deficiency (HRD) status (defined by large-scale transition [LST] scores) with response in all patients (left) and the HGSOC-only cohort (right). (E) Association of tumor mutational burden (TMB) with response in all patients (left) and the HGSOC-only cohort (right). (F) Differences in PFS (left) and OS (right) from the start of ICI as a function of HRD status. (G) Differences in PFS (left) and OS (right) from the start of ICI as a function of TMB at a median cutoff. (C, F, and G) Log-rank P value is displayed. NA, not applicable; SNV, single nucleotide variant; WT, wild-type.
In the subset of women with somatic genomic data, there were no differences in the repertoire of somatic mutations, gene CNAs, or mutational signatures by response to ICI in the overall population (n = 64) or in the HGSOC cohort (n = 41; Fig 2A). All tumors were microsatellite stable. There were no differences in TMB or LST scores between responders and nonresponders, overall or in HGSOC cohort (Figs 2D and 2E). Although TMB and LST were significantly higher in those with BRCA1/2 mutations (Data Supplement), neither was associated with PFS or OS (Figs 2F and 2G), including when the upper quartile of the TMB cutoff was used (Data Supplement). Only 1 patient (non-HGSOC) displayed a TMB > 10 (Fig 2E). There were no obvious differences in CNAs between responders and nonresponders, overall or in the HGSOC cohort (Data Supplement).
FGA represents the percentage of the genome affected by copy number gains or losses. Although FGA was not associated with BRCA1/2 mutation status (Data Supplement), there was a trend toward a higher FGA in responders, both overall and in the HGSOC cohort (Fig 3A), although this did not reach statistical significance (P = .097 and P = .099, respectively). We performed an exploratory, hypothesis-generating analysis to ascertain the impact of FGA on PFS and OS of patients with OC treated with ICI, both overall and in the HGSOC cohort only. This analysis revealed that increasing FGA (continuous) was associated with improved PFS (hazard ratio [HR], 0.31; 95% CI, 0.11 to 0.92; P = .035) and OS (HR, 0.28; 95% CI, 0.08 to 0.96; P = .043; Tables 2 and 3) overall. Using a cutoff of 34%, derived from the maximally selected standardized log-rank method, high FGA was associated with improved OS (HR, 0.49; 95% CI, 0.28 to 0.86; log-rank P = .01) and improved PFS (HR, 0.54; 95% CI, 0.32 to 0.89; log-rank P = .014) in the overall cohort (Figs 3A and 3B, Tables 2 and 3), although this cutoff needs to be interpreted with caution and requires validation in larger cohorts once data are available. In the HGSOC-only cohort, FGA as a continuous variable was associated with PFS (HR, 0.23; 95% CI, 0.06 to 0.92; log-rank P = .037), and FGA using our cutoff of 34% had a borderline association with PFS (HR, 0.53; 95% CI, 0.27 to 1.03; log-rank P = .057; Fig 3C, Data Supplement). In the HGSOC-only cohort, FGA was not significantly associated with OS. Interestingly, although BRCA status, TMB, and LST were not associated with improved PFS, there was high overlap between tumors with BRCA mutations and high TMB, LST, and FGA (Data Supplement), suggesting that, in addition to LST and point mutations, additional genome rearrangements in FGA-high tumors may be contributing to the apparent immunogenicity of these tumors. To ensure that FGA was not a prognostic indicator in HGSOC overall regardless of therapy, we examined the association of high FGA with OS in a cohort of 242 women with HGSOC who underwent MSK-IMPACT testing and had available survival data from a similar time period (2015-2018) but who did not receive ICI and found that high FGA was not associated with OS (Data Supplement), suggesting that the predictive ability of FGA may be specific for response to ICI.
FIG 3.

Association of fraction of genome altered (FGA) with response to immune checkpoint inhibitor (ICI) and survival. (A) FGA by response to ICI (left: overall cohort, right: high-grade serous ovarian cancer [HGSOC] only). (B) Association of FGA with progression-free survival (PFS; left) and overall survival (OS; right) in all overall cancer. Log-rank P values are shown. (C) Association of FGA with PFS (left) and OS (right) in the HGSOC subset only. Log-rank P values are shown. Cutoff of 34%, derived using maximally selected standardized log-rank statistic, was used for survival analysis.
DISCUSSION
In this cohort of heavily pretreated, mostly platinum-resistant patients with OC, pathogenic mutations affecting BRCA1/2 were not found to be predictors of ICI response, which is consistent with prior reports from smaller cohorts with known BRCA1/2 status.13,14 These findings indicate that, despite the suggested greater immunogenicity of BRCA1/2 mutation–associated OCs,7 these cancers do not exhibit improved responses to ICI. Furthermore, our results highlight that TMB, a biomarker of response to ICI in several other cancers, carries limited predictive value in OC, likely because of the overall low TMB in these cancers.
The exact mechanism for the lack of superior responses to ICI in BRCA-mutated and HRD OC is unclear. Although presence of BRCA mutations was associated with higher TMB and LST scores, this did not correlate with response to ICI or survival, suggesting that higher levels of potential neoantigens may not correlate with immune response in OC or that the threshold of immunogenicity has not been met by the higher mutational load in BRCA-mutated cancers. A recent analysis of genomic markers in breast cancer corroborated our findings.11 The analysis found that despite increased TMB and neoantigen load, HRD-high tumors had lower immunogenicity as measured by cytolytic activity and lymphocyte infiltration. Furthermore, HRD-high and BRCA-mutated tumors exhibited lower levels of type I interferon signaling and decreased activation of transforming growth factor-β and NF-κB pathways, key drivers of immune response, when compared with HRD-low tumors.11 In addition, others have suggested that tumor aneuploidy is associated with decreased cytotoxic immune cell infiltration and immune evasion, potentially through weakening of the major histocompatibility complex and antigen presentation.42 Both studies reveal heterogeneity between tumor types and BRCA mutations and monoallelic versus biallelic loss that should be further investigated in larger studies.
Our exploratory analyses provide an indication that, in OC, high FGA may be associated with improved PFS and OS after ICI. These findings need to be interpreted with caution because an optimal cutoff for FGA as a predictor of response to ICI will require validation in a larger study. We also found that FGA was not a prognostic indicator in patients with HGSOC who received other therapies, suggesting its predictive value may be specific to ICI. Nevertheless, these findings generate a rationale for investigation of the biologic basis underlying the immunogenicity of FGA-high OCs, including the potential propensity of FGA-high genomes to generate immunogenic neoantigens and/or association of genomic instability of these tumors with cytosolic DNA signaling and type I interferon response.9,16 Analyses of biomarkers of response in melanoma and non–small-cell lung cancer to ICI have highlighted the importance of clonality of TMB and neoantigen burden as a key determinant of T-cell immunoreactivity.43 Future studies aiming to establish FGA and other metrics of genomic instability as biomarkers of ICI response will need to resolve the role of neoantigens and intratumor heterogeneity, ideally leveraging whole-genome and single-cell technologies.44
To our knowledge, to date, this study represents the largest cohort of women with recurrent OC treated with ICI and known BRCA1/2 mutation status. An additional strength of our study is the focus on the genomic predictors of response exclusively in patients who received ICI without combined chemotherapy or PARP inhibition, which otherwise could confound the results. However, this study is limited by its retrospective nature, heterogeneity of the treatment population, and limited rate of somatic BRCA1/2 testing. There are also inherent limitations of targeted sequencing, including the use of archival rather than fresh samples. Overall, the clinical benefit rate of 17.6% observed in this study (defined as lack of progression at 24 weeks) was consistent with prior trials. Different treatment types were well balanced between the arms (Table 1); thus, it is unlikely that heterogeneity in treatments had a significant impact on the outcomes of this study. Furthermore, because BRCA and HRD status are not expected to change much from baseline, especially in the absence of prior PARP inhibitor therapy, we feel that genetic analyses of baseline samples provide an accurate representation of the major mutational signatures present in these patients’ tumors at the time of ICI.
In conclusion, our findings indicate that BRCA1/2 mutations, HRD, and increasing TMB are not associated with clinical benefit from ICI in heavily pretreated patients with OC and caution against using these biomarkers as selection tools for ICI in patients with OC. However, we found that increasing FGA may be associated with improved survival after ICI in OC, and additional studies are warranted to test FGA as a biomarker of response to immunotherapy in patients with OC.
PRIOR PRESENTATION
Presented in part at the 55th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 31-June 4, 2019.
SUPPORT
Supported by the Young Investigator Award from the Conquer Cancer Foundation (Y.L.L.), the Ovarian Cancer Research Foundation Liz Tilberis Award (D.Z.), Department of Defense Ovarian Cancer Research Academy Grant No. OC150111 (D.Z.), Memorial Sloan Kettering Cancer Center Support Grant No. P30 CA008748, the Breast Cancer Research Foundation (J.S.R.-F.), the Sarah Jenkins Fund (J.S.R.-F.), Congressionally Directed Medical Research Program W81XWH-15-1-0547 (J.S.R.-F.), and grants from the Breast Cancer Research Foundation (B.W.), Cycle for Survival (B.W.), and Stand Up to Cancer (B.W.).
AUTHOR CONTRIBUTIONS
Conception and design: Ying L. Liu, Alexia Iasonos, Julia Boland, Daniel J. Powell Jr, Carol Aghajanian, Britta Weigelt, Dmitriy Zamarin
Financial support: Dmitriy Zamarin
Administrative support: Ying L. Liu, Dmitriy Zamarin
Provision of study materials or patients: Ying L. Liu, Claire Friedman, Rachel Grisham, Jason A. Konner, Dmitriy Zamarin
Collection and assembly of data: Ying L. Liu, Margaret Callahan, Noah Z. Feit, Julia Boland, Ahmet Zehir, Rachel Grisham, Roisin E. O’Cearbhaill, Dmitriy Zamarin
Data analysis and interpretation: Ying L. Liu, Pier Selenica, Qin Zhou, Alexia Iasonos, Ignacio Vazquez-Garcia, Diana Mandelker, Robert A. Burger, Daniel J. Powell Jr, Claire Friedman, Karen Cadoo, Jason A. Konner, Roisin E. O’Cearbhaill, Carol Aghajanian, Jorge S. Reis-Filho, Britta Weigelt, Dmitriy Zamarin
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Alexia Iasonos
Consulting or Advisory Role: Mylan, Brightpath, Intelligencia
Margaret Callahan
Employment: Bristol Myers Squibb (I), Celgene (I), Kleo Pharmaceuticals (I)
Consulting or Advisory Role: AstraZeneca, Moderna Therapeutics, Merck, Immunocore
Research Funding: Bristol Myers Squibb (Inst)
Other Relationship: Clinical Care Options, Potomac Center for Medical Education
Ignacio Vazquez-Garcia
Stock and Other Ownership Interests: CRISPR Therapeutics, Editas Medicine
Ahmet Zehir
Honoraria: Illumina
Robert A. Burger
Employment: Genentech
Stock and Other Ownership Interests: Genentech
Consulting or Advisory Role: AstraZeneca, Tesaro, Merck, Genentech, Morphotek, Myriad Genetics
Research Funding: Incyte (Inst), Astra Zeneca (Inst), and Genzyme (Inst)
Travel, Accommodations, Expenses: Tesaro, Genentech
Daniel J. Powell Jr
Stock and Other Ownership Interests: Atara Biotherapeutics, InsTIL Bio
Consulting or Advisory Role: Neon Therapeutics, Iovance Biotherapeutics, Tmunity Therapeutics, InsTIL Bio, Bellicum Pharmaceuticals
Research Funding: Eli Lilly, Tmunity Therapeutics, Incyte, Monojul, AstraZeneca/MedImmune
Patents, Royalties, Other Intellectual Property: I hold patents in the field of chimeric antigen receptor (CAR) T-cell therapy in oncology and have received royalties related to their licensing to Novartis; I hold patents in the field of CAR T cell therapy in Oncology and have received royalties related to their licensing to Tmunity
Travel, Accommodations, Expenses: Iovance Biotherapeutics
Claire Friedman
Consulting or Advisory Role: AstraZeneca/MedImmune
Research Funding: Bristol Myers Squibb (Inst), Arcus Biosciences (Inst), Genentech (Inst), Merck (Inst)
Other Relationship: Guidepoint Global
Uncompensated Relationships: Genentech, Merck
Open Payments Link: https://openpaymentsdata.cms.gov/physician/477023/summary
Karen Cadoo
Honoraria: OncLive
Consulting or Advisory Role: GSK Tesaro
Research Funding: AstraZeneca (Inst), Syndax (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Rachel Grisham
Consulting or Advisory Role: Mateon Therapeutics, Clovis Oncology, Regeneron
Research Funding: Context Therapeutics (Inst)
Travel, Accommodations, Expenses: EMD Serono
Other Relationship: Prime Oncology, MCM Education, OncLive
Jason A. Konner
Consulting or Advisory Role: Immunogen, AstraZeneca, Tesaro, AbbVie
Research Funding: Genentech
Travel, Accommodations, Expenses: AstraZeneca
Roisin E. O'Cearbhaill
Consulting or Advisory Role: Tesaro, GlaxoSmithKline
Research Funding: Juno Therapeutics (Inst), Sellas Life Sciences (Inst), Ludwig Institute for Cancer Research (Inst), Stem CentRx (Inst), TapImmune (Inst), TCR2 Therapeutics (Inst), Regeneron (Inst), Genmab (Inst)
Carol Aghajanian
Consulting or Advisory Role: Tesaro, Mersana, Eisai, Roche
Research Funding: Genentech (Inst), AbbVie (Inst), Clovis Oncology (Inst), AstraZeneca (Inst), Clovis Oncology (Inst), AstraZeneca (Inst)
Jorge S. Reis-Filho
Consulting or Advisory Role: Genentech, Invicro, Ventana Medical Systems, Volition RX, Paige AI, Goldman Sachs, Novartis, Repare Therapeutics
Britta Weigelt
Consulting or Advisory Role: Genentech (I), Invicro (I), Ventana Medical Systems (I), Volition RX (I), Page AI (I), Goldman Sachs (I), Repare Therapeutics (I)
Dmitriy Zamarin
Consulting or Advisory Role: Merck, Synlogic, Western Oncolytics, Tesaro, Agenus, Trieza Therapeutics, ACM Biolabs
Research Funding: Genentech (Inst)
Patents, Royalties, Other Intellectual Property: I hold a patent regarding the use of recombinant Newcastle disease virus (NDV) for cancer therapy (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Germano G, Lamba S, Rospo G, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 3.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marquard AM, Eklund AC, Joshi T, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 2015;3:9. doi: 10.1186/s40364-015-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai Y, Sun C, Feng Y, et al. Potent immunogenicity in BRCA1-mutated patients with high-grade serous ovarian carcinoma. J Cell Mol Med. 2018;22:3979–3986. doi: 10.1111/jcmm.13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuo K, Spragg SE, Ciccone MA, et al. Nivolumab use for BRCA gene mutation carriers with recurrent epithelial ovarian cancer: A case series. Gynecol Oncol Rep. 2018;25:98–101. doi: 10.1016/j.gore.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mouw KW, Goldberg MS, Konstantinopoulos PA, et al. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7:675–693. doi: 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–472. doi: 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. doi: 10.1158/1078-0432.CCR-18-0468. Kraya AA, Maxwell KN, Wubbenhorst B, et al: Genomic signatures predict the immunogenicity of BRCA-deficient breast cancer. Clin Cancer Res 25:4363-4374, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu YL, Zamarin D. Combination immune checkpoint blockade strategies to maximize immune response in gynecological cancers. Curr Oncol Rep. 2018;20:94. doi: 10.1007/s11912-018-0740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Disis ML, Taylor MH, Kelly K, et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: Phase 1b results from the JAVELIN solid tumor trial. JAMA Oncol. 2019;5:393–401. doi: 10.1001/jamaoncol.2018.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matulonis UA, Shapira-Frommer R, Santin AD, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann Oncol. 2019;30:1080–1087. doi: 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 15.Konstantinopoulos PA, Waggoner S, Vidal GA, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5:1141. doi: 10.1001/jamaoncol.2019.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. doi: 10.1016/j.celrep.2018.11.054. Ding L, Kim HJ, Wang Q, et al: PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 25:2972-2980.e5, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen J, Zhao W, Ju Z, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–319. doi: 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JM, Botesteanu DA, Tomita Y, et al. Patients with BRCA mutated ovarian cancer may have fewer circulating MDSC and more peripheral CD8+ T cells compared with women with BRCA wild-type disease during the early disease course. Oncol Lett. 2019;18:3914–3924. doi: 10.3892/ol.2019.10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JM, Cimino-Mathews A, Peer CJ, et al. Safety and clinical activity of the programmed death-ligand 1 inhibitor durvalumab in combination with poly (ADP-ribose) polymerase inhibitor olaparib or vascular endothelial growth factor receptor 1-3 inhibitor cediranib in women’s cancers: A dose-escalation, phase I study. J Clin Oncol. 2017;35:2193–2202. doi: 10.1200/JCO.2016.72.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riaz N, Blecua P, Lim RS, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8:857. doi: 10.1038/s41467-017-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weigelt B, Bi R, Kumar R, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst. 2018;110:1030–1034. doi: 10.1093/jnci/djy028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pareja F, Lee JY, Brown DN, et al. The genomic landscape of mucinous breast cancer. J Natl Cancer Inst. 2019;111:737–741. doi: 10.1093/jnci/djy216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saunders CT, Wong WS, Swamy S, et al. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 28.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narzisi G, Corvelo A, Arora K, et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol. 2018;1:20. doi: 10.1038/s42003-018-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narzisi G, O’Rawe JA, Iossifov I, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods. 2014;11:1033–1036. doi: 10.1038/nmeth.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8:174–183. doi: 10.1158/2159-8290.CD-17-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gulhan DC, Lee JJ, Melloni GEM, et al. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51:912–919. doi: 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 34.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashley CW, Da Cruz Paula A, Kumar R, et al. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol Oncol. 2019;152:11–19. doi: 10.1016/j.ygyno.2018.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. doi: 10.1200/PO.19.00103. Smith ES, Paula ADC, Cadoo KA, et al: Endometrial cancers in BRCA1 or BRCA2 germline mutation carriers: Assessment of homologous recombination DNA repair defects. JCO Precis Oncol 10.1200/PO.19.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popova T, Manié E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 38. doi: 10.1016/j.jtho.2016.07.017. Shukuya T, Mori K, Amann JM, et al: Relationship between overall survival and response or progression-free survival in advanced non-small cell lung cancer patients treated with anti-PD-1/PD-L1 antibodies. J Thorac Oncol 11:1927-1939, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lausen B, Hothorn T, Bretz F, et al. Assessment of optimally selected prognostic factors. Biom J. 2004;46:364–374. [Google Scholar]
- 40. Lausen B, Sauerbrei W, Schumacher M: Classification and regression trees (CART) used for the exploration of prognostic factors measured on different scales, in Dirschedl P, Ostermann R (eds): Computational Statistics 25th Conference on Statistical Computing. Heidelberg, Germany, Physica-Verlag, 1994, pp 483-496. [Google Scholar]
- 41.Lausen B, Schumacher M. Maximally selected ranked statistics. Biometrics. 1992;48:73–75. [Google Scholar]
- 42.Davoli T, Uno H, Wooten EC, et al. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355:eaaf8399. doi: 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laks E, McPherson A, Zahn H, et al. Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell. 2019;179:1207–1221.e22. doi: 10.1016/j.cell.2019.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]