

Abstract
PURPOSE
Circulating tumor DNA (ctDNA) has been used for disease monitoring in several types of cancer. The aim of our study was to investigate whether ctDNA can be used for response monitoring in neuroblastoma.
METHODS
One hundred forty-nine plasma samples from 56 patients were analyzed by quantitative polymerase chain reaction (qPCR) for total cell free DNA (cfDNA; albumin and β-actin) and ctDNA (hypermethylated RASSF1A). ctDNA results were compared with mRNA-based minimal residual disease (qPCR) in bone marrow (BM) and blood and clinical patient characteristics.
RESULTS
ctDNA was detected at diagnosis in all patients with high-risk and stage M neuroblastoma and in 3 of 7 patients with localized disease. The levels of ctDNA were highest at diagnosis, decreased during induction therapy, and not detected before or after autologous stem-cell transplantation. At relapse, the amount of ctDNA was comparable to levels at diagnosis. There was an association between ctDNA and blood or BM mRNA, with concordant results when tumor burden was high or no tumor was detected. The discrepancies indicated either low-level BM infiltration (ctDNA negative/mRNA positive) or primary tumor/soft tissue lesions with no BM involvement (ctDNA positive/mRNA negative).
CONCLUSION
ctDNA can be used for monitoring disease in patients with neuroblastoma. In high-risk patients and all patients with stage M at diagnosis, ctDNA is present. Our data indicate that at low tumor load, testing of both ctDNA and mRNA increases the sensitivity of molecular disease monitoring. It is likely that ctDNA can originate from both primary tumor and metastases and may be of special interest for disease monitoring in patients who experience relapse in other organs than BM.
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor of childhood. In approximately 50%, patients present with high-risk (HR) disease and are treated with intensive multimodality treatment protocols that encompass induction therapy, primary tumor surgery, myeloablative chemotherapy with autologous stem-cell rescue, local irradiation, and anti-GD2–based immunotherapy1 (Appendix Fig A1). Despite this intensive therapy, in approximately one half of HR patients, the tumor will relapse and result in a fatal outcome.2 Assessment of treatment response is based on the International Neuroblastoma Response Criteria. Meta-iodobenzylguanidine (MIBG) scintigraphy, imaging (magnetic resonance imaging/positron emission tomography scans), and bone marrow (BM) examinations by histology or (immuno)cytology are combined to assess the extent of disease.3 Because the median age at diagnosis is 18.8 months,4 response evaluation that is based on imaging and BM testing often must be performed under general anesthesia. Therefore, alternative methods for monitoring response would potentially result in fewer risks to these patients. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) is a more sensitive technique for detection and monitoring of minimal residual disease (MRD) in neuroblastoma. Several prospective studies investigating the clinical significance of this technique for mRNA-based MRD detection in HR neuroblastoma are ongoing or have been published.5-8
CONTEXT
Key Objective
Can hypermethylated RASSF1A be used as a circulating tumor DNA (ctDNA) marker for minimal residual disease detection in neuroblastoma?
Knowledge Generated
When testing cell free DNA, we were able to detect tumor-derived hypermethylated RASSF1A in all patients with stage M disease at diagnosis. ctDNA levels decreased during treatment and were high again at relapse. Comparison between ctDNA and blood- or bone marrow (BM)–derived mRNA revealed that discrepancies were found when BM infiltration was low or when there were primary tumor lesions without BM involvement.
Relevance
ctDNA is an interesting source for monitoring disease in patients with neuroblastoma. Our data indicate that testing of both ctDNA and mRNA increases the sensitivity of molecular disease monitoring.
However, even with mRNA-based RT-qPCR, many patients with low or negative MRD results during treatment will experience recurrent disease.5,9 As an alternative to mRNA, circulating tumor DNA (ctDNA) might be a valuable source of tumor-derived material. ctDNA comprises circulating DNA fragments (cell free DNA [cfDNA]) that carry tumor-specific alterations, which can be found in the plasma of patients with cancer.10-12 Because of the invasiveness of tumor biopsy and the lack of repeated biopsies during follow-up, the use of liquid biopsies is being investigated. Several studies have shown the feasibility of detecting mutations or tumor-specific translocations in the ctDNA by high-depth targeted sequencing or mutation-specific PCR to monitor disease in various types of adult cancer.13-19 However, neuroblastoma tumors, like many pediatric tumors, lack recurrent mutations and translocations. Broader analysis, such as whole-exome sequencing (WES) and shallow whole-genome sequencing (sWGS), to detect tumor-specific mutations or copy number alterations have been performed successfully using cfDNA of patients with neuroblastoma at diagnosis and relapse.20-26 Nevertheless, these techniques are only informative when the ctDNA content is approximately ≥ 10%27 and are, therefore, less suited for the detection of MRD, when the tumor burden is low.
In contrast to the copy number alterations and tumor-specific mutations, a methylation-specific qPCR assay could potentially be a more general and sensitive ctDNA marker. Previously, our group demonstrated that the RASSF1A gene is inactivated by hypermethylation in all stage M and MS and in 86% of localized neuroblastoma tumors. Hypermethylated RASSF1A (RASSF1Am) can be detected in BM with a similar sensitivity as mRNA and has shown added value in mRNA-negative BM.28 In addition, RASSF1Am already has been described as a prognostic ctDNA marker at diagnosis.29 The aim of this study was to investigate the feasibility of using ctDNA (RASSF1Am in plasma) to monitor treatment response in patients with HR/stage M neuroblastoma. We retrospectively performed qPCR for RASSF1Am on cfDNA from stored remains of previously collected plasma samples of patients with localized or metastatic neuroblastoma at diagnosis and for patients with HR/stage M neuroblastoma during treatment and at relapse. To test the additional value of ctDNA monitoring, we compared it with other techniques for disease monitoring: MIBG scans, urinary catecholamines, immunocytology, and RT-qPCR RNA-based MRD detection in BM and peripheral blood (PB).
METHODS
Between 2013 and 2016, from all consecutively diagnosed patients who were included in this study (N = 56), 149 PB samples for mRNA and cfDNA and 105 BM samples for mRNA were tested. Because stored remains were used, not all patients had samples for all time points. In this feasibility study, both HR and non-HR patients were included. Patients were treated at the Amsterdam University Medical Center (UMC), Erasmus Medical Center, or the Princess Máxima Center for Pediatric Oncology. Written informed consent from parents or guardians was obtained for all patients. The study was approved by the medical research ethics committee of Amsterdam UMC (MEC07/219#08.17.0836). Clinical data (including urinary catecholamines (homovanillic acid and vanillylmandelic acid) and imaging data (primary tumor [longest diameter], MIBG Curie score30) were collected from electronic patient files. Seventy-three PB samples from healthy adult male volunteers were collected as controls from Sanquin Blood Supply (Amsterdam, the Netherlands). Because RASSF1Am is also present in plasma of pregnant women, women were excluded as control donors. Pediatric PB control samples were collected from the Amsterdam UMC (Appendix Table A1).
Sample Collection, DNA Isolation, Bisulfite Conversion, and Real-Time qPCR
Methods for sample collection, DNA isolation, bisulfite conversion, and real-time qPCR for RASSF1Am28 and mRNA markers31 can be found in the Appendix and Appendix Table A2.
Data Analysis
Total cfDNA was quantified by qPCR for albumin (ALB) or β-actin (ACTB). A maximum of a 3.3-Ct difference between preconverted ALB and postconverted ACTB was accepted to ensure decent conversion. Samples with a ΔCT between ALB and ACTB > 3.3 are not included in the analysis for RASSF1Am. RASSF1Am was scored positive not quantifiable (PNQ) if not all wells of the triplicate were positive or one of the replicates had a Ct value > 1.5 than the mean Ct of the replicates. Quantification of RASSF1Am was performed relative to the neuroblastoma cell line IMR-32. For quantification with mRNA markers, relative values were calculated using the equation 2ΔΔCT (ΔCT sample – ΔCT IMR-32) × 100%. The median relative expression of 5 markers was used for the analysis. cfDNA and ctDNA levels were not normally distributed and are presented as median (interquartile range). Kruskal-Wallis tests were used for comparison of cfDNA or ctDNA levels. McNemar’s test was used for concordance between ctDNA and PB and BM mRNA MRD levels. All statistical analyses were performed with SPSS version 23 (IBM Corporation, Chicago, IL) or GraphPad Prism 8 (GraphPad Software, La Jolla, CA) software.
RESULTS
Patients and Samples
From 48 patients with HR and/or stage M and 8 patients with non-HR neuroblastoma, 149 samples were tested in this study (Fig 1). From the 8 patients with non-HR neuroblastoma, only diagnostic samples were tested. Patient characteristics are listed in Table 1. Six of the 149 patient samples and 12 of 73 healthy control samples were not included for RASSF1Am qPCR because too much DNA had been lost during bisulfite conversion (Fig 1). In 2 of 61 adult control samples, RASSF1Am amplification was observed (Ct value, 40.1 and 37.1), but this occurred in only 1 of 3 replicates. In the 20 pediatric control samples, no amplification of RASSF1Am was found.
FIG 1.
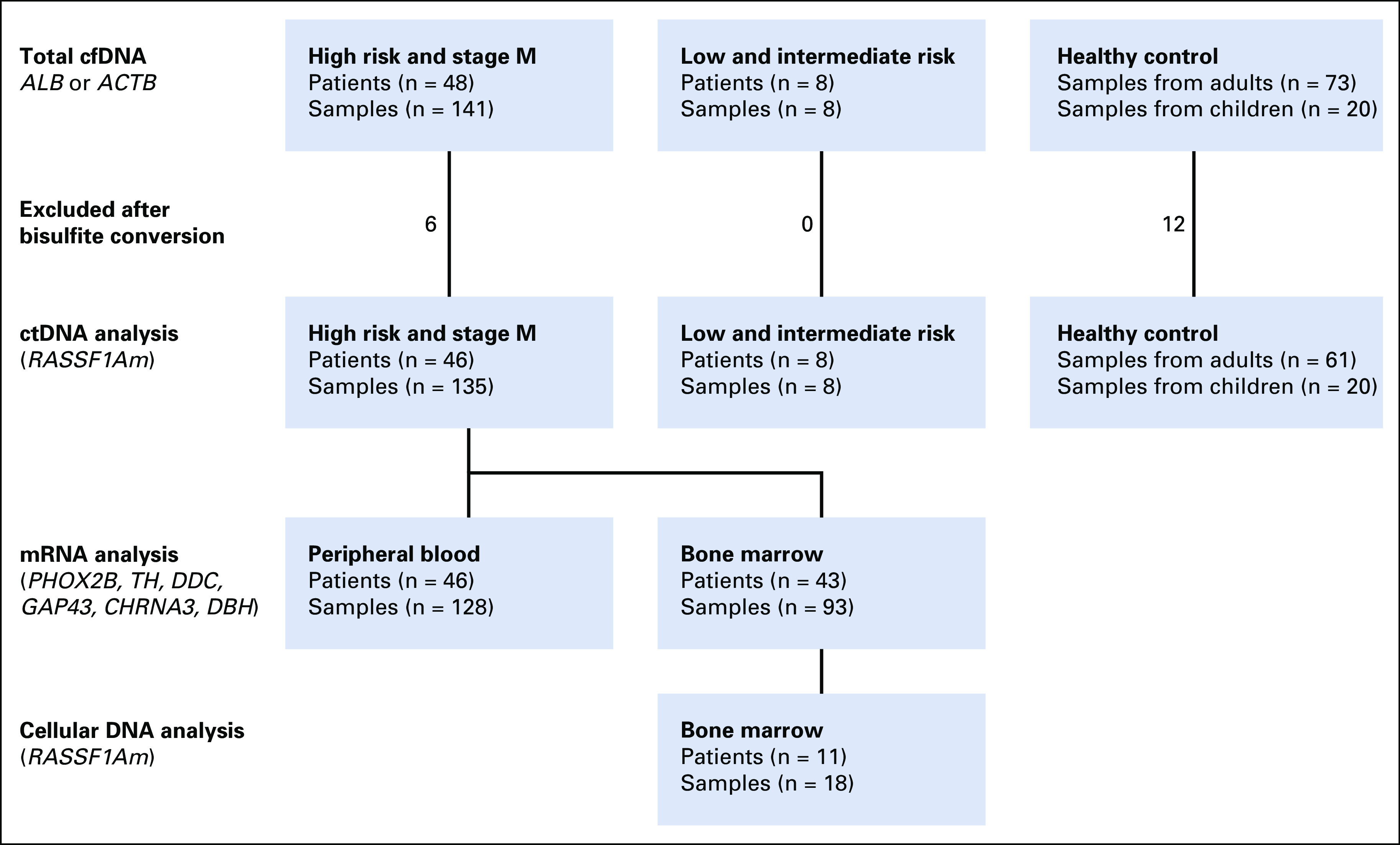
Flowchart of samples tested for total cell free DNA (cfDNA), number of samples excluded after too much DNA had been lost after bisulfite conversion, and number of samples tested for circulating tumor DNA (ctDNA) by hypermethylated RASSF1A (RASSF1Am). ACTB, β-actin; ALB, albumin.
TABLE 1.
Patient Characteristics
Amount of cfDNA
The amount of cfDNA per milliliter of plasma was determined by ALB or ACTB qPCR in 73 adult and 20 pediatric control samples and compared with 31 samples from patients with neuroblastoma (diagnosis or relapse). Compared with adult or pediatric control samples, samples from patients with neuroblastoma at diagnosis or relapse (all stages) had significantly more cfDNA (median, 1.5 ng/mL [interquartile range, 0.4-4.2 ng/mL], 3.1 ng/mL [interquartile range, 1.4-6.7 ng/mL], and 22.07 ng/mL [interquartile range, 5.7-98.90 ng/mL]; P < .0001 and P = .0045, respectively). Patients with stage M disease at diagnosis had the highest cfDNA levels (median, 73.1 ng/mL; interquartile range, 5.2-285.5 ng/mL; Fig 2A). There was no significant difference in total cfDNA levels during treatment and follow-up (Fig 2B). In the 28 samples where ctDNA was detected and quantified, the cfDNA levels were higher compared with the 86 patient samples where no ctDNA was detected (median, 34.2 ng/mL [range, 9.2-98.7 ng/mL] v 7.9 ng/mL [range, 3.5-25.8 ng/mL]; P = .044). Twenty-nine samples with detectable but not quantifiable ctDNA had significantly higher cfDNA levels (median, 12.9 ng/mL; range, 4.4-39.7 ng/mL) compared with adult control samples (median, 1.5 ng/mL; range, 0.4-4.2 ng/mL; P < .001). When no ctDNA was detected, cfDNA levels were still higher compared with adult control samples (median, 7.9 ng/mL; range, 3.5-25.8 ng/mL; P < .001; Fig 2C). Compared with pediatric control donors, only patients with quantifiable ctDNA levels had significantly higher levels of cfDNA (P = .0007).
FIG 2.
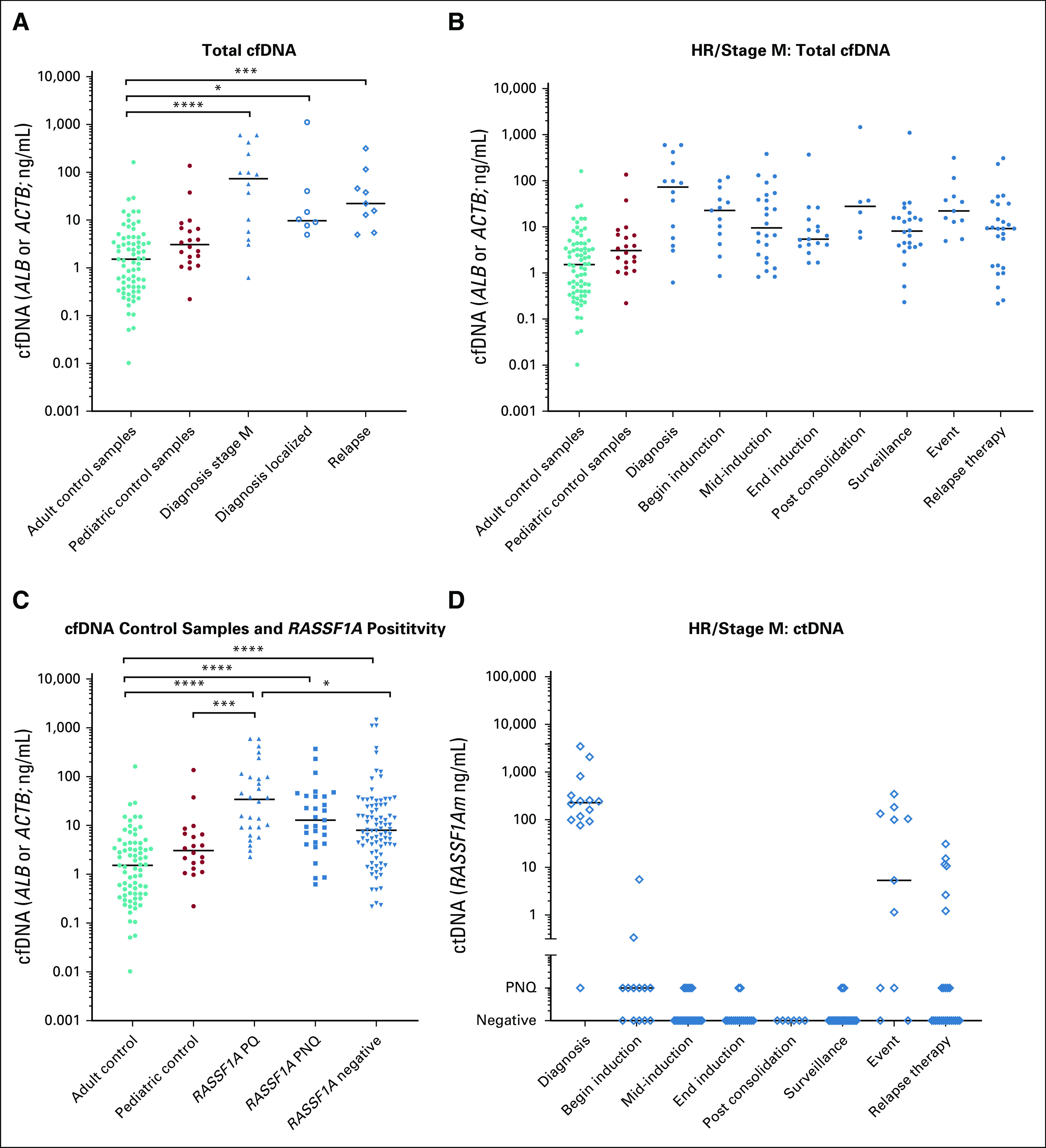
Amount of cell free DNA (cfDNA) and circulating tumor DNA (ctDNA). (A) Comparison of amount cfDNA (measured by albumin [ALB] or β-actin [ACTB]) between patients with neuroblastoma and healthy control donors. (B) Amount of cfDNA (measured by ALB or ACTB) at different time points during treatment. (C) Amount of cfDNA in samples with ctDNA compared with samples from healthy control donors and samples without ctDNA detected. (D) Amount of ctDNA (measured by hypermethylated RASSF1A [RASSF1Am]) at different time points during treatment. Begin induction indicates until 2 courses of induction therapy; mid-induction indicates after 3-5 courses of induction chemotherapy, unless additional courses were given after 6 courses, and samples before last course were also included at this time point; end induction indicates at the end of induction therapy; surveillance indicates during follow-up or at relapse suspicion; and event indicates relapse or progression. HR, high risk; PNQ, positive not quantifiable; PQ, positive and quantified. (*) P < .05, (**) P < .01, (***) P < .001, (****) P < .0001.
Level of ctDNA
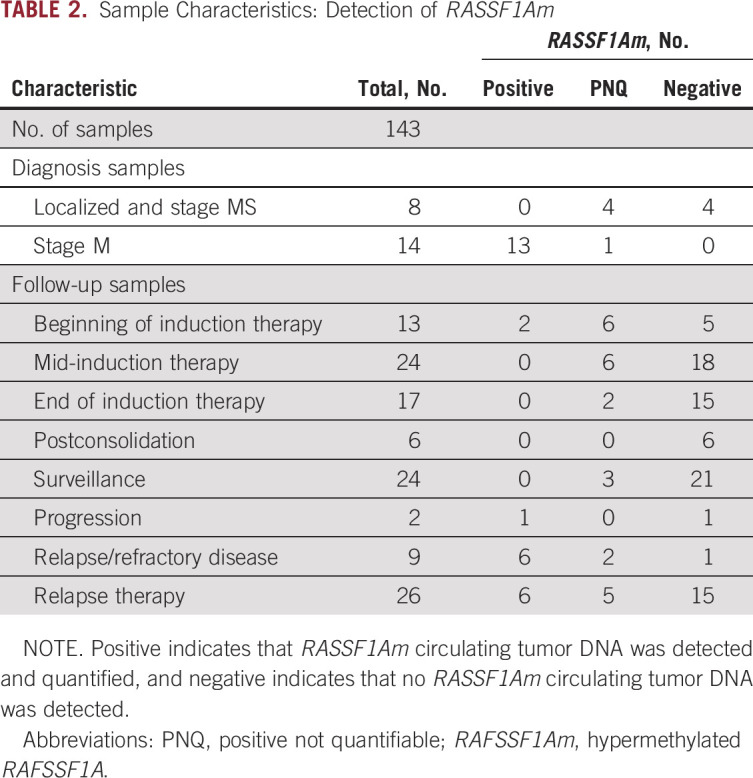
In all diagnostic samples from patients with stage M neuroblastoma, RASSF1Am was detected. In 3 of 7 diagnostic samples from patients with localized disease and in 1 sample from a patient with stage MS disease, ctDNA was detected, although not in the quantitative range. During induction chemotherapy (patients with HR/stage M disease only), in 14 (38%) of 37 patients, ctDNA was detected (median Ct value, 30.6; min-max range, 24.7-33.8). At surveillance, 3 samples were positive, and these patients eventually experienced recurrent disease. In 8 of 9 samples from patients with relapse at the time of sampling, ctDNA was detected. Results are listed in Table 2 and in more detail in Appendix Table A3. The levels of ctDNA were highest at diagnosis, decreased during induction therapy, and undetectable at the end of induction chemotherapy. At relapse, ctDNA levels were comparable to levels at diagnosis (Fig 2D). The percentage of ctDNA of total cfDNA, calculated with the equation [RASSF1Am / (RASSF1Am + unmethylated RASSF1A) × 100], was 94% (range, 82%-98%) in the 14 diagnostic samples from patients with stage M disease. In the 28 samples where RASSF1Am could be quantified, the median percentage of ctDNA was 87% (range, 0.7%-99.9%); 29 additional samples were positive for RASSF1Am but could not be quantified.
TABLE 2.
Sample Characteristics: Detection of RASSF1Am
Comparison of ctDNA and the Detection of Neuroblastoma mRNA in PB and BM
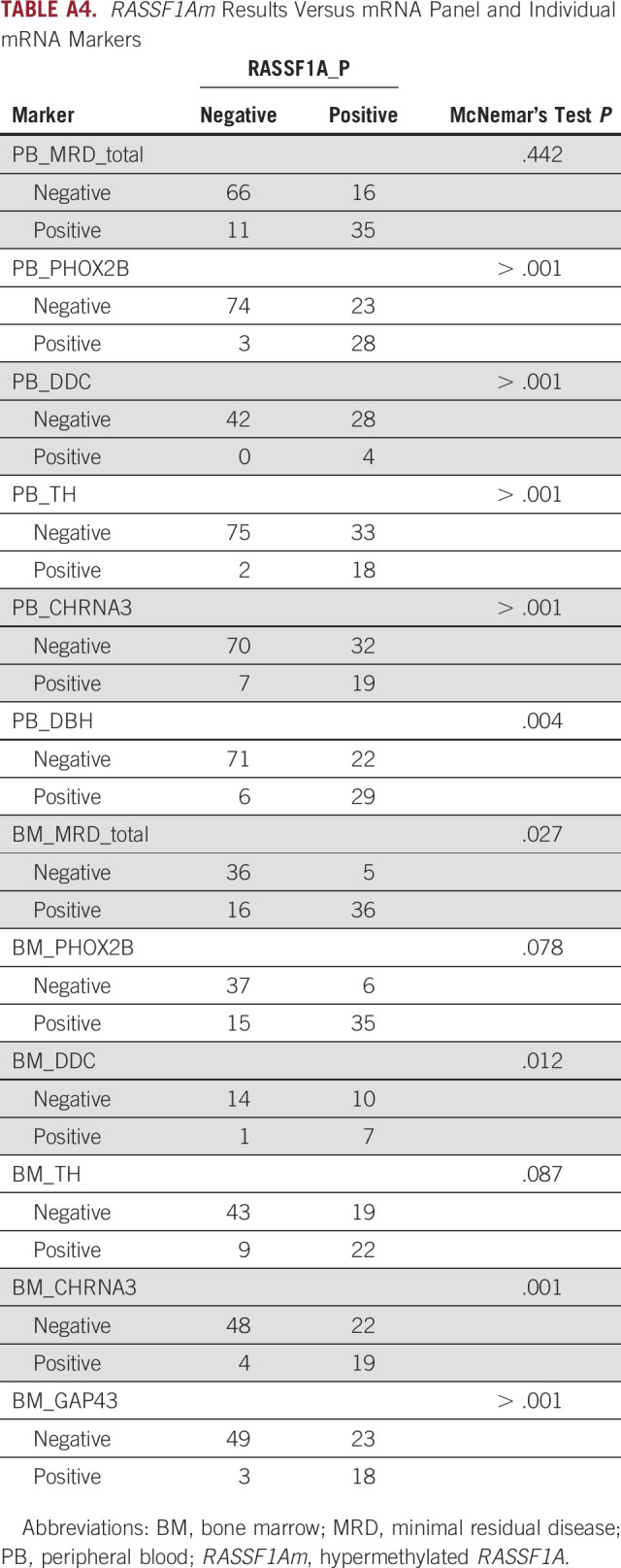
To study whether ctDNA, measured as RASSF1Am, can be used as an MRD marker in patients with HR/stage M disease, we compared it with our panel of mRNA markers.31 In 128 matched PB samples, ctDNA could be compared with neuroblastoma mRNA, which demonstrated 79% concordant results (Fig 3A). Compared with the individual mRNA markers, RASSF1Am was more often positive, but the combined mRNA markers identified the same positive samples as RASSF1Am (Appendix Table A4).
FIG 3.
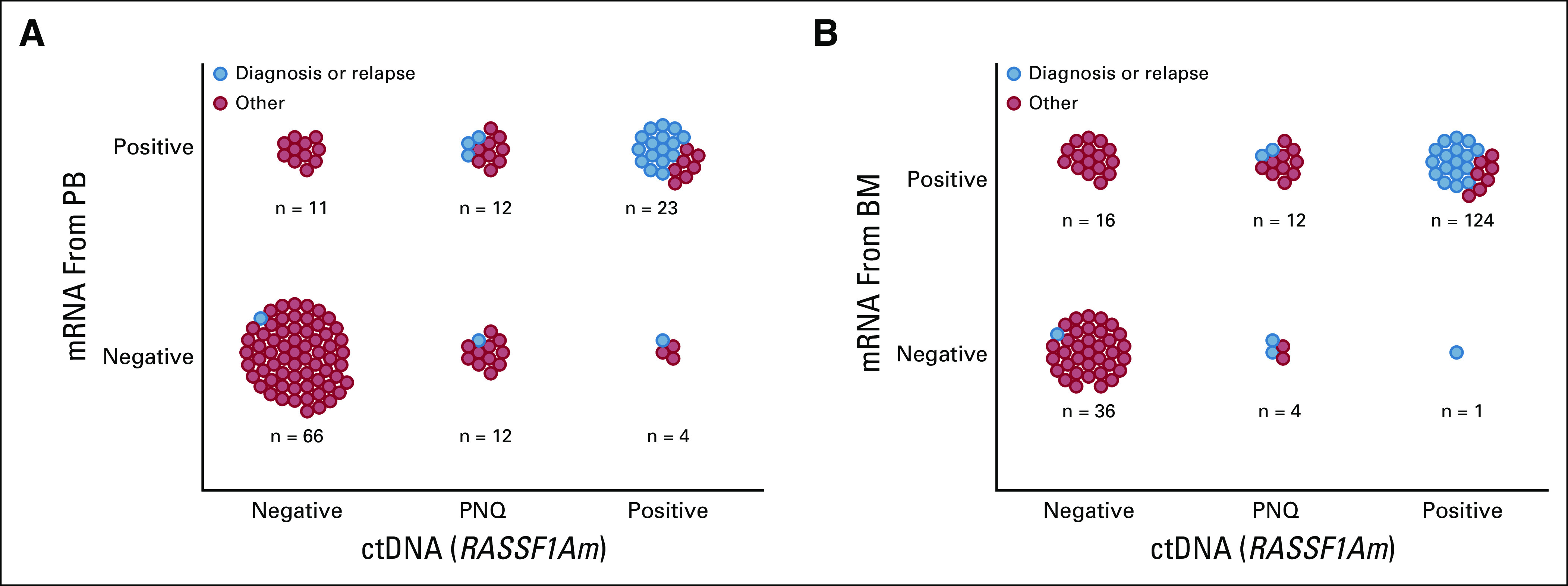
(A) Association between mRNA in peripheral blood (PB) samples and circulating tumor (ctDNA). (B) Association between mRNA in bone marrow (BM) samples and ctDNA. PNQ, positive not quantifiable; RASSF1Am, hypermethylated RASSF1A.
In 93 matched BM mRNA and ctDNA (PB) samples, double-negative or double-positive results were found in 77% (Fig 3B). In contrast to PB, the BM mRNA panel identified more positive samples than ctDNA, and the individual markers PHOX2B and TH correlated best with RASSF1Am (Appendix Table A4).
Discrepant Findings Between ctDNA and PB or BM mRNA MRD
Discrepant results between ctDNA and mRNA were detected in 27 PB and 21 BM samples, respectively, and listed in Table 3 and Appendix Table A4. Total cfDNA levels in the ctDNA-positive/mRNA-negative samples were relatively high, with a median of 38.92 and 11.09 ng/mL for the BM mRNA-negative and PB mRNA-negative samples, respectively. From 3 of 5 BM mRNA-negative/ctDNA-positive samples, cryopreserved BM cells were available and tested all negative for RASSF1Am. In some patients (N850, N865, N732), the high levels of ctDNA probably correlated with the large primary or local relapse tumors, and these patients had no or very little BM infiltration.
TABLE 3.
Assessment of Discrepant Samples

In the ctDNA-negative samples, in general, the cfDNA levels were lower, with a median of 6.1 ng/mL for the BM mRNA-positive/ctDNA-negative and 1.52 ng/mL for the PB mRNA-positive/ctDNA-negative samples. In this group, the mRNA levels (in both BM and PB) were very low, mostly < 0.1%. From 15 of the 16 BM mRNA-positive/ctDNA-negative samples, cryopreserved cells were available and tested for RASSF1Am; of the 5 positive samples, 4 were not in the quantitative range, which indicated low levels of BM infiltration. In the samples from patients N777, N798, N2011, N2014, and N802, no ctDNA was detected. Apart from the very-low mRNA levels (only used for research purposes), N777 and N798 were considered to be in complete remission at that time. Subsequent samples (if available) showed negative MRD results. At the time of sampling for patients N2011 and N2014, the MIBG score was very low. Therefore, it is likely that the (biologically active) tumor load in these patients was very low. Patient N802 was treated for an isolated CNS relapse.
In the samples from patients N2012, N2013, N2016, N2024, N2029, and N2031, no ctDNA was detected, while low amounts of mRNA were detected in the BM. In the case of restricted, minimal BM disease, mRNA detection was more sensitive than ctDNA (Appendix Table A4). However, in some patients, a primary tumor was still present (median, 50 mm) while ctDNA was negative (Table 3).
DISCUSSION
ctDNA in plasma is a powerful source for the detection of tumor-derived aberrations in a minimally invasive setting. Many ctDNA studies in adults for the detection of MRD are based on detection of tumor-specific mutations by targeted sequencing or digital droplet PCR (ddPCR).12,16,17 Because recurrent mutations are not common in neuroblastoma,32 tumor-specific aberrations need to be characterized before they can be used as an MRD marker. However, temporospatial heterogeneity has been reported in neuroblastoma by several studies,20,26 which raises the question of whether we should only use the small part of the tumor that is derived from the biopsy to design tumor-specific MRD markers. In the current study, we show that RASSF1Am is a universal marker for detecting ctDNA in patients with neuroblastoma. The use of RASSF1Am as an MRD marker has several potential benefits. First, it is a sensitive marker, with a sensitivity of 1 tumor cell in 105 mononuclear cells.28 Second, RASSF1Am qPCR can be used in all patients with stage M neuroblastoma because it has been shown that RASSF1A is hypermethylated in all previously tested stage M neuroblastoma tumors.28 Third, detection of RASSF1Am is less costly compared with WES and even sWGS (approximately 40- and 10-fold less expensive, respectively). Finally, we show in this report that RASSF1Am in plasma is tumor specific. Hypermethylation of RASSF1A has been described in several types of cancer and in physiologic circumstances in placental cells.33 RASSF1A is not methylated in normal hematologic cells.28,33,34 However, in 2 of 61 samples from healthy individuals, we detected very low, nonquantifiable levels of RASSF1Am. In addition, in other studies, infrequent detection of RASSF1Am has been observed in plasma samples from healthy control participants.35,36 Therefore, when detecting very-low levels of RASSF1Am in patients with neuroblastoma (indicated as PNQ range), results should be analyzed with caution.
It has been shown that neuroblastoma tumors shed high amounts of ctDNA in the plasma.25,26,37 In the current study, we found a median cfDNA concentration of 73.1 ng/mL at diagnosis for patients with stage M disease. This study confirms that cfDNA levels of patients with neuroblastoma are significantly higher than that of healthy donors, with patients with stage M disease having the highest levels. However, the levels we found are lower compared with previously published studies.25,26,37 This inconsistency may be due to differences in isolation of cfDNA because we did not use a circulating nucleic acid kit or to differences in quantification methods. We used qPCR, whereas Chicard and colleagues25,26 used the Qubit fluorometric assay (Thermo Fisher Scientific, Waltham, MA). We found the majority of the cfDNA (94% at diagnosis) to be tumor derived in patients with stage M or HR disease, which is also supported by previous research.26,37
We tested 143 samples from 54 patients with neuroblastoma and detected ctDNA in 57 samples. ctDNA was detected at diagnosis in all 14 patients with stage M and 4 of 8 patients with localized and stage MS neuroblastoma. Misawa et al29 described detection of RASSF1Am at diagnosis in the serum of 17 of 68 patients (all stages) and in 11 of 18 patients with stage M disease. There are two likely causes for the increased ctDNA detection in our study. First, we used plasma, whereas Misawa et al tested serum, which is known to be more contaminated by genomic DNA originated from leukocytes during ex vivo clotting.38 Second, Misawa et al used conventional PCR, which is less sensitive than qPCR. In the current study, ctDNA levels decreased during induction chemotherapy and were high again at relapse. This suggests that with increasing tumor burden, ctDNA levels also increase. Our group has previously described that hypermethylation of RASSF1A is variable in tumors of patients with stage MS (median, 65%) and localized (median, 30%) disease28; therefore, the level of ctDNA can be slightly underestimated in these patients when using RASSF1Am as marker.
We compared the performance of ctDNA with PB and BM mRNA in 128 and 93 samples, respectively. There was a strong correlation between ctDNA and BM mRNA when tumor burden was high or no tumor was detected. However, in some samples, discrepancies were observed for which additional clinical data about tumor response status were retrieved. Most patients in whom we detected relatively high levels of ctDNA compared with PB or BM mRNAs still had considerable tumor volumes or negative or low MIBG scores (data not shown); therefore, it is likely that the ctDNA in these patients originated from the primary tumor. No ctDNA was detected in 17 samples with very low PB or BM mRNA levels (< 1%). Two of these patients were in complete remission but in the other 15 patients, considerable tumor volumes were detected on imaging or urine catecholamines were still positive, which indicate the need to optimize pre-analytic sample handling and prospective study of cfDNA kinetics in well-characterized patient cohorts with available paired (nuclear) imaging and BM assessment.
While the detection of ctDNA is very promising for future MRD studies, the current study has some limitations. Stored remains were used, which resulted in missing samples and paired clinical data. Prospective collaborative studies on the use of ctDNA in the new SIOPEN HR-2 (ClinicalTrials.gov identifier: NCT04221035) patient cohort are being initiated within the SIOPEN liquid biopsy group. For detection and quantification of low levels of ctDNA in the plasma, DNA extraction methods can be optimized with an isolation method specific for cfDNA, and ddPCR39 may be a more suited technique compared with qPCR.40 Moreover, large amounts of cfDNA (up to 96%) could be destroyed during bisulfite conversion41; therefore, we are investigating alternative methylation-specific ddPCR methods. Finally, Stutterheim et al28 showed that the percentage of RASSF1Am can be variable in neuroblastoma tumors, especially in tumors of patients with localized disease. Previous studies showed that RASSF1A was the most frequent hypermethylated tumor suppressor gene in neuroblastoma as well as identified other hypermethylated tumor suppressor genes, and inclusion of these genes as MRD markers might increase the sensitivity.42,43
In this study, we used RASSF1Am as a ctDNA marker. We analyzed 135 sequential samples at diagnosis, during treatment, and at follow-up for 46 patients with HR/stage M neuroblastoma. In conclusion, ctDNA can be used for monitoring disease in patients with neuroblastoma. In HR patients and all patients with stage M at diagnosis, ctDNA is present. Our data indicate that at low tumor load, the testing of both ctDNA and mRNA increases the sensitivity of molecular disease monitoring. It is likely that ctDNA can originate from both primary tumor and metastases and may be of special interest for disease monitoring in patients who experience relapse in other organs than the BM.
Appendix
Sample Collection
Clinical samples were collected in EDTA tubes and processed within 24 hours, and 2 mL peripheral blood (PB) or 0.5 mL bone marrow (BM) was transferred to PAXgene Blood RNA Tubes (QIAGEN, Venlo, the Netherlands) and stored at −20°C. The remainder of the blood samples were centrifuged to separate plasma from the PB cells (1,375 × g for 10 minutes without brake). Subsequently, the plasma was stored at −20°C. Mononuclear cells were isolated from the remaining BM sample by Ficoll density centrifugation and cryopreserved in 10% dimethyl sulfoxide.
DNA Isolation and Bisulfite Conversion
Dependent on the available plasma volume, DNA was extracted by using the QIAamp DNA Mini Blood Kit (QIAGEN) for 200 μL plasma, or the MagNA Pure 96 isolation robot (Roche, Basel, Switzerland) for 500-1,000 μL plasma and eluted in H2O. After DNA isolation, bisulfite conversion was performed using the EpiTect Bisulfite Kit (QIAGEN) according to the manufacturer’s instructions. Converted DNA samples were used directly or stored at −20°C.
Real-Time Quantitative Polymerase Chain Reaction
Real-time quantitative polymerase chain reaction (qPCR) was performed as previously described (van Wezel EM, et al: J Mol Diagn 17:43-52, 2015). Primers and probes were obtained from Eurogentec (Liege, Belgium). Primer and probe sequences have been described previously28 (Scheffer PG, et al: BJOG 118:1340-1348, 2011) and are listed in Appendix Table A2. To control for DNA input, Albumin (ALB) (before bisulfite conversion) and β-actin (ACTB; after bisulfite conversion) qPCRs were carried out. Subsequently, qPCRs for unmethylated and hypermethylated RASSF1A were performed. qPCR for ALB, ACTB, and unmethylated RASSF1A was performed in duplicate; hypermethylated RASSF1A was tested in triplicate.
PB and BM mRNA Reverse Transcriptase qPCR
Total RNA from whole blood and BM samples was extracted using the PAXgene Blood RNA kit (QIAGEN) according to the manufacturer’s instructions. cDNA was synthesized and reverse transcriptase (RT) qPCR, with a maximum of 50 cycles was performed for β-glucuronidase (GUSB), paired ctDNA in neuroblastoma like homeobox 2B (PHOX2B), tyrosine hydroxylase (TH), dopa decarboxylase (DDC), growth-associated protein 43 (GAP43), cholinergic receptor nicotinic α-3 (CHRNA3), and dopamine β-hydroxylase (DBH), as has been described previously.31 Expression was normalized to GUSB expression using the following equation: [normalized threshold cycle (ΔCT) = (CtGUSB − Ctmarker)]. All RT-qPCR reactions were performed in triplicate (except GUSB, which was performed in duplicate), and mean values were used for analysis. Samples were scored for positivity according to previously published thresholds9,31 (Stutterheim J, et al: J Clin Oncol 26:5443-5449, 2008). Samples with an insufficient CtGUSB value (Ct value > 25, corresponding to < 5,000 copies) were excluded (Stutterheim J, et al: J Clin Oncol 26:5442-5449, 2008; Beillard E, et al: Leukemia 17:2474-2486, 2003; Gabert J, et al: Leukemia 17:2318-2357, 2003).
FIG A1.

Overview of the DCOG NBL2009 treatment protocol. ASCT, autologous stem-cell transplantation and myeloablative therapy (carboplatin, etoposide, and melphalan); cis-RA, cis-retinoic acid; EBRT, external beam radiation therapy; N5, vindesine, etoposide, and cisplatin; N6, vincristine, dacarbazine, ifosfamide, and doxorubicin; S, surgery (was performed after vindesine, etoposide, and cisplatin and vincristine, dacarbazine, ifosfamide, and doxorubicin courses; optimal timing of surgery was discussed).
TABLE A1.
Pediatric Control Group Data
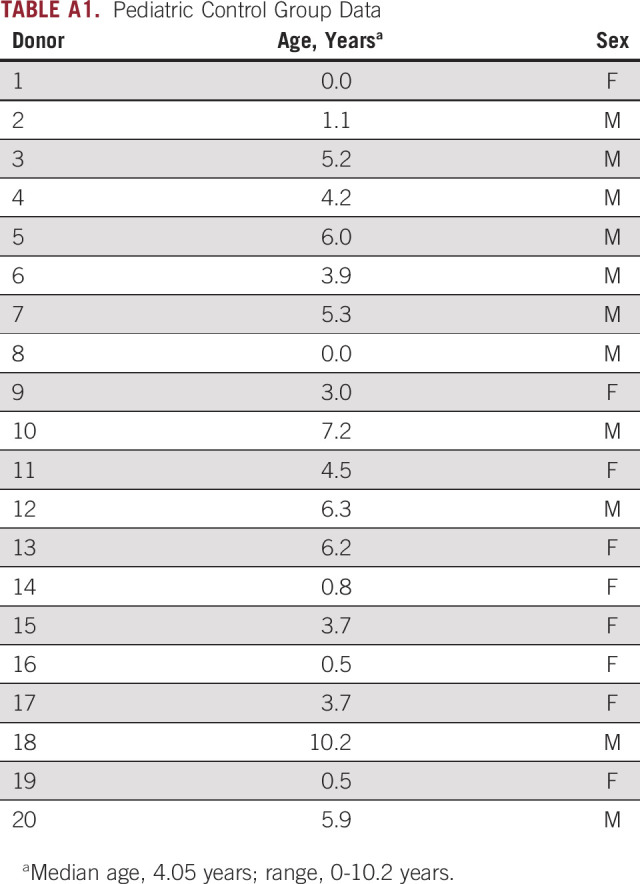
TABLE A2.
Primer and Probe Sequences

TABLE A3.
Clinical and Sample Data

TABLE A4.
RASSF1Am Results Versus mRNA Panel and Individual mRNA Markers
Presented at the Advances in Neuroblastoma Research 2016 Congress, Cairns, Queensland, Australia, June 19-23, 2016; 49th Congress of the International Society of Paediatric Oncology, Washington, DC, October 12-15, 2017; and White Nights St Petersburg International Oncology Forum, St Petersburg, Russia, June 22-25, 2019.
SUPPORT
Supported by Koningin Wilhelmina Fonds, Dutch Cancer Society (UVA 2010-4738), and AMeesing Foundation voor Mees.
AUTHOR CONTRIBUTIONS
Conception and design: Lieke M. J. van Zogchel, Esther M. van Wezel, C. Ellen van der Schoot, Godelieve A. M. Tytgat
Financial support: Godelieve A. M. Tytgat
Administrative support: Godelieve A. M. Tytgat
Provision of study material or patients: Janine Stutterheim, Roswitha Schumacher-Kuckelkorn, Godelieve A. M. Tytgat
Collection and assembly of data: Lieke M. J. van Zogchel, Esther M. van Wezel, Jalenka van Wijk, Wassilis S. C. Bruins, Lily Zappeij-Kannegieter, Tirza J. E. Slager, Roswitha Schumacher-Kuckelkorn, Iedan R. N. Verly, C. Ellen van der Schoot, Godelieve A. M. Tytgat
Data analysis and interpretation: Lieke M. J. van Zogchel, Esther M. van Wezel, Jalenka van Wijk, Janine Stutterheim, Wassilis S. C. Bruins, Roswitha Schumacher-Kuckelkorn, Iedan R. N. Verly, C. Ellen van der Schoot, Godelieve A. M. Tytgat
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
No potential conflicts of interest were reported.
REFERENCES
- 1.Pinto NR, Applebaum MA, Volchenboum SL, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. 2015;33:3008–3017. doi: 10.1200/JCO.2014.59.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park JR, Kreissman SG, London WB, et al. Effect of tandem autologous stem cell transplant vs single transplant on event-free survival in patients with high-risk neuroblastoma: A randomized clinical trial. JAMA. 2019;322:746–755. doi: 10.1001/jama.2019.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JR, Bagatell R, Cohn SL, et al. Revisions to the International Neuroblastoma Response Criteria: A consensus statement from the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2017;35:2580–2587. doi: 10.1200/JCO.2016.72.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.London WB, Castleberry RP, Matthay KK, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol. 2005;23:6459–6465. doi: 10.1200/JCO.2005.05.571. [DOI] [PubMed] [Google Scholar]
- 5.Viprey VF, Gregory WM, Corrias MV, et al. Neuroblastoma mRNAs predict outcome in children with stage 4 neuroblastoma: A European HR-NBL1/SIOPEN study. J Clin Oncol. 2014;32:1074–1083. doi: 10.1200/JCO.2013.53.3604. [DOI] [PubMed] [Google Scholar]
- 6.Cheung NK, Ostrovnaya I, Kuk D, et al. Bone marrow minimal residual disease was an early response marker and a consistent independent predictor of survival after anti-GD2 immunotherapy. J Clin Oncol. 2015;33:755–763. doi: 10.1200/JCO.2014.57.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreissman SG, Seeger RC, Matthay KK, et al. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): A randomised phase 3 trial. Lancet Oncol. 2013;14:999–1008. doi: 10.1016/S1470-2045(13)70309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burchill SA, Beiske K, Shimada H, et al. Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer. 2017;123:1095–1105. doi: 10.1002/cncr.30380. [DOI] [PubMed] [Google Scholar]
- 9.Stutterheim J, Zappeij-Kannegieter L, Versteeg R, et al. The prognostic value of fast molecular response of marrow disease in patients aged over 1 year with stage 4 neuroblastoma. Eur J Cancer. 2011;47:1193–1202. doi: 10.1016/j.ejca.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 11.Gormally E, Caboux E, Vineis P, et al. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: Practical aspects and biological significance. Mutat Res. 2007;635:105–117. doi: 10.1016/j.mrrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 13.Leary RJ, Kinde I, Diehl F, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBride DJ, Orpana AK, Sotiriou C, et al. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes Chromosomes Cancer. 2010;49:1062–1069. doi: 10.1002/gcc.20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 17.Tsao SC, Weiss J, Hudson C, et al. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci Rep. 2015;5:11198. doi: 10.1038/srep11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tie J, Kinde I, Wang Y, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26:1715–1722. doi: 10.1093/annonc/mdv177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buono G, Gerratana L, Bulfoni M, et al. Circulating tumor DNA analysis in breast cancer: Is it ready for prime-time? Cancer Treat Rev. 2019;73:73–83. doi: 10.1016/j.ctrv.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Van Roy N, Van Der Linden M, Menten B, et al. Shallow whole genome sequencing on circulating cell-free DNA allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clin Cancer Res. 2017;23:6305–6314. doi: 10.1158/1078-0432.CCR-17-0675. [DOI] [PubMed] [Google Scholar]
- 21.Combaret V, Audoynaud C, Iacono I, et al. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res. 2002;62:3646–3648. [PubMed] [Google Scholar]
- 22. doi: 10.1200/JCO.2005.04.0170. Combaret V, Bergeron C, Noguera R, et al: Circulating MYCN DNA predicts MYCN-amplification in neuroblastoma. J Clin Oncol 23:8919-8920, 2005; author reply 8920. [DOI] [PubMed] [Google Scholar]
- 23.Combaret V, Bréjon S, Iacono I, et al. Determination of 17q gain in patients with neuroblastoma by analysis of circulating DNA. Pediatr Blood Cancer. 2011;56:757–761. doi: 10.1002/pbc.22816. [DOI] [PubMed] [Google Scholar]
- 24.Combaret V, Iacono I, Bellini A, et al. Detection of tumor ALK status in neuroblastoma patients using peripheral blood. Cancer Med. 2015;4:540–550. doi: 10.1002/cam4.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chicard M, Boyault S, Colmet Daage L, et al. Genomic copy number profiling using circulating free tumor DNA highlights heterogeneity in neuroblastoma. Clin Cancer Res. 2016;22:5564–5573. doi: 10.1158/1078-0432.CCR-16-0500. [DOI] [PubMed] [Google Scholar]
- 26.Chicard M, Colmet-Daage L, Clement N, et al. Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin Cancer Res. 2018;24:939–949. doi: 10.1158/1078-0432.CCR-17-1586. [DOI] [PubMed] [Google Scholar]
- 27.Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10:eaat4921. doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stutterheim J, Ichou FA, den Ouden E, et al. Methylated RASSF1a is the first specific DNA marker for minimal residual disease testing in neuroblastoma. Clin Cancer Res. 2012;18:808–814. doi: 10.1158/1078-0432.CCR-11-0849. [DOI] [PubMed] [Google Scholar]
- 29.Misawa A, Tanaka S, Yagyu S, et al. RASSF1A hypermethylation in pretreatment serum DNA of neuroblastoma patients: A prognostic marker. Br J Cancer. 2009;100:399–404. doi: 10.1038/sj.bjc.6604887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanik GA, Parisi MT, Shulkin BL, et al. Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: A report from the Children’s Oncology Group. J Nucl Med. 2013;54:541–548. doi: 10.2967/jnumed.112.112334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stutterheim J, Gerritsen A, Zappeij-Kannegieter L, et al. Detecting minimal residual disease in neuroblastoma: The superiority of a panel of real-time quantitative PCR markers. Clin Chem. 2009;55:1316–1326. doi: 10.1373/clinchem.2008.117945. [DOI] [PubMed] [Google Scholar]
- 32.Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan KC, Ding C, Gerovassili A, et al. Hypermethylated RASSF1A in maternal plasma: A universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin Chem. 2006;52:2211–2218. doi: 10.1373/clinchem.2006.074997. [DOI] [PubMed] [Google Scholar]
- 34.Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers. 2007;23:73–87. doi: 10.1155/2007/291538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheffer PG, de Haas M, van der Schoot CE. The controversy about controls for fetal blood group genotyping by cell-free fetal DNA in maternal plasma. Curr Opin Hematol. 2011;18:467–473. doi: 10.1097/MOH.0b013e32834bab2d. [DOI] [PubMed] [Google Scholar]
- 36.White HE, Dent CL, Hall VJ, et al. Evaluation of a novel assay for detection of the fetal marker RASSF1A: Facilitating improved diagnostic reliability of noninvasive prenatal diagnosis. PLoS One. 2012;7:e45073. doi: 10.1371/journal.pone.0045073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong FC, Sun K, Jiang P, et al. Cell-free DNA in maternal plasma and serum: A comparison of quantity, quality and tissue origin using genomic and epigenomic approaches. Clin Biochem. 2016;49:1379–1386. doi: 10.1016/j.clinbiochem.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunetti C, Anelli L, Zagaria A, et al. Droplet digital PCR is a reliable tool for monitoring minimal residual disease in acute promyelocytic leukemia. J Mol Diagn. 2017;19:437–444. doi: 10.1016/j.jmoldx.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29:e65. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoebeeck J, Michels E, Pattyn F, et al. Aberrant methylation of candidate tumor suppressor genes in neuroblastoma. Cancer Lett. 2009;273:336–346. doi: 10.1016/j.canlet.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Kiss NB, Kogner P, Johnsen JI, et al. Quantitative global and gene-specific promoter methylation in relation to biological properties of neuroblastomas. BMC Med Genet. 2012;13:83. doi: 10.1186/1471-2350-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]