

Abstract
Purpose
Urachal adenocarcinoma is a rare type of primary bladder adenocarcinoma that comprises less than 1% of all bladder cancers. The low incidence of urachal adenocarcinomas does not allow for an evidence-based approach to therapy. Transcriptome profiling of urachal adenocarcinomas has not been previously reported. We hypothesized that an in-depth molecular understanding of urachal adenocarcinoma would uncover rational therapeutic strategies.
Patients and Methods
We performed targeted exon sequencing and global transcriptome profiling of 12 urachal tumors to generate a comprehensive molecular portrait of urachal adenocarcinoma. A single patient with an MSH6 mutation was treated with the anti–programmed death-ligand 1 antibody, atezolizumab.
Results
Urachal adenocarcinoma closely resembles colorectal cancer at the level of RNA expression, which extends previous observations that urachal tumors harbor genomic alterations that are found in colorectal adenocarcinoma. A subset of tumors was found to have alterations in genes that are associated with microsatellite instability (MSH2 and MSH6) and hypermutation (POLE). A patient with an MSH6 mutation was treated with immune checkpoint blockade, which resulted in stable disease.
Conclusion
Because clinical trials are next to impossible for patients with rare tumors, precision oncology may be an important adjunct for treatment decisions. Our findings demonstrate that urachal adenocarcinomas molecularly resemble colorectal adenocarcinomas at the level of RNA expression, are the first report, to our knowledge, of MSH2 and MSH6 mutations in this disease, and support the consideration of immune checkpoint blockade as a rational therapeutic treatment of this exceedingly rare tumor.
INTRODUCTION
In the United States, bladder carcinoma is the 4th most common malignancy in men and the 9th most common in women.1 Overall, an estimated 16,000 people died of bladder cancer in 2015. Bladder cancer takes on a spectrum of histomorphologic appearances. The predominant histologic subtype is urothelial carcinoma (90% to 95%) and, less frequently, adenocarcinoma (2%) or squamous cell carcinoma (2.5%). Urachal carcinomas are a subtype of bladder adenocarcinoma that arises from the urachus, an embryologic remnant that connects the bladder and the allantois during fetal development.2 Postnatally, it fuses to become a fibrous cord known as the median umbilical ligament. Urachal tumors comprise approximately one third of all bladder adenocarcinomas and usually develop in the 5th to 6th decade of life, with a male predominance.3,4 Because of their frequent presentation at the dome of the bladder, urachal adenocarcinomas are clinically and pathologically grouped with bladder neoplasms and are typically seen by urologic oncologists and genitourinary pathologists. Evidence-based treatment of this disease is hindered by its rarity; thus, current treatment paradigms for urachal adenocarcinoma are primarily anecdotal in nature.
As a result of the complex embryology of the allantois and cloaca, the cellular origin and molecular pathogenesis that drive the development of urachal adenocarcinoma are speculative.2 Intestinal metaplasia or enteric rests are hypothesized as the histogenetic precursor of these tumors. Although the genetics of urachal adenocarcinoma have been investigated by multiple groups, no reports on the global gene expression patterns of urachal adenocarcinoma have been published. One study selectively examined the prevalence of KRAS and BRAF mutations in high-stage urachal adenocarcinomas and, although they found no BRAF mutations, 42% harbored KRAS mutations.5 The study also noted a high rate of loss of protein expression for a number of genes that are correlated with microsatellite instability. A more recent study found a high rate of NF1 mutations via whole-exome sequencing of seven urachal adenocarcinomas.6 Finally, another recent study showed that urachal adenocarcinomas harbor mutations in mitogen-activated protein kinase (MAPK) pathways, similar to colorectal adenocarcinoma, and showed the potential for treatment with the anti–epidermal growth factor receptor antibody cetuximab.7
Herein, we report, to our knowledge, the first transcriptome analysis of urachal adenocarcinoma using whole-transcriptome profiling by RNA sequencing. A pan-cancer transcriptomic analysis of urachal tumors comparing them with 12 cancers of different tissue origins suggest that their RNA expression patterns most closely resemble colorectal adenocarcinoma and glioblastoma (GBM). Our work also validates reports that urachal adenocarcinomas harbor alterations that are typically found in colorectal carcinoma—that is, APC, SMAD4, and KRAS mutation—but extends those observations to show that a subset of urachal cancers has inactivation of genes that are involved in microsatellite instability (MSH2, MSH6) or hypermutation (POLE), and that all urachal tumors invariably have mutations of TP53 (100%). One patient with an MSH6 mutation was treated with the anti–programmed death-ligand 1 (PD-L1) antibody atezolizumab, which resulted in stable disease. In aggregate, our studies demonstrate that urachal tumors harbor a high molecular resemblance to colorectal adenocarcinoma and suggest a novel therapeutic option: immune checkpoint blockade.
PATIENTS AND METHODS
Sample and Data Acquisition
Samples and clinical data were obtained after approval by the University of North Carolina institutional review board. Thirteen primary urachal adenocarcinomas with formalin-fixed, paraffin-embedded (FFPE) tissue available at University of North Carolina, Chapel Hill, were identified by using CoPath Natural Language Search (Cerner Corporation, Kansas City, MO). Hematoxylin and eosin–stained slides and clinical history were reviewed by a board-certified pathologist (S.E.W.) to confirm the diagnosis on the basis of the following criteria: tumor in the dome or posterior wall of the bladder, sharp demarcation between tumor and surface epithelium, and exclusion of primary adenocarcinoma located elsewhere. The surgical procedure, tumor location within the bladder, histologic subtype, and tumor stage were all recorded from the accompanying pathology reports.
RNA Expression
For RNA sequencing, RNA was extracted from 10-µm-thick unstained sections of FFPE blocks that were isolated from urachal tumors. Macrodissection was used for tumor enrichment. RNA was extracted by using the High-Pure FFPE RNA Extraction Protocol (Roche, Indianapolis, IN). A minimum of 2 µg of total RNA was isolated from FFPE tissues. Extracted RNA was converted to double-stranded cDNA, and sequencing adapters were ligated by using the Illumina RNA Access Library Prep Protocol (Illumina, San Diego, CA). Samples were sequenced by paired-end, 100-bp sequencing on an Illumina HiSEquation 2000 at the High Throughput Sequencing Core Facility at the University of North Carolina. Sequence reads were aligned to the human reference transcriptome, and gene expression was generated as reads per kilobase of exon model per million mapped reads per gene by using MapSplice (University of Kentucky Bioinformatics Labs, Lexington, KY). RNA sequencing data were normalized for variations in read counts, log2 transformed, and median centered before analysis. When combining data sets, we adjusted for batch effects using the surrogate variable analysis R package (version 3.12.0; R Foundation, Vienna, Austria).
Clustering with the combined Urachal (sequenced urachal tumors) and PanCan (TCGA Pan-Cancer Dataset13) data set was performed by using average linkage clustering with a centered correlation similarity metric with Cluster 3.0 (Human Genome Center, Tokyo, Japan) software on the top 10% most differentially expressed genes (as determined by standard deviation) across the combined PanCan and Urachal data set. PanCan subtype centroids were derived by determining the median expression of each gene in the transcriptome across each of the PanCan tumor types. A Pearson correlation was calculated between each of the PanCan tumor type centroids and each Urachal tumor. Clustering between The Cancer Genome Atlas (TCGA) bladder urothelial carcinoma (BLCA), TCGA colorectal adenocarcinoma (COADREAD), and Urachal samples was performed using average linkage clustering with a centered correlation similarity metric with Cluster 3.0 software on the top 10% most differentially expressed genes after adjusting for batch effects as described above.
Targeted Exon Sequencing
Targeted exon sequencing was conducted through the UNCseq pipeline as previously described.8 Twelve of 13 urachal samples had both tumor and tumor-adjacent normal tissue submitted to the UNCseq pipeline that passed quality control standards and were included in the DNA analysis. Analysis to identify significantly mutated genes, altered pathways, and clustering was confined to mutations that were classified as either having a moderate or high impact on protein function through UNCseq. Clustering of TCGA BLCA, TCGA COADREAD, and UNCseq Urachal samples was performed on the basis of a compilation of the mutation frequency of the previously identified significantly mutated genes in the TCGA BLCA and TCGA COADREAD data sets that were present in the UNCseq targeted regions.9,10 Pathway mutation frequency was calculated on the basis of the number of samples in each cohort that contained at least one mutation in the gene list associated with that pathway. The transforming growth factor (TGF)-β pathway was represented by the SMAD2, SMAD3, SMAD4, and TGFBR1 genes. The β-catenin pathway was represented by the APC, CTNNB1, and AMER1 genes. The MAPK pathway was represented by the NF1, KRAS, BRAF, HRAS, NRAS, RAF1, MEK1, MEK2, ERK1, and ERK2 genes. Copy number alteration on a cohort level was derived by running Genomic Identification of Significant Targets in Cancer (GISTIC) 2.0 on the Gene Pattern online platform.11 The DNA mismatch repair (MMR) pathway was represented by the MLH1, MLH3, MSH2, MSH3, MSH6, and PMS2 genes, with the POLE gene included and separately identified. Mutation frequency was calculated using all identified mutations in each sample and dividing it by the total Megabase region of 30× coverage within each sample. The insertion-to-deletion ratio was calculated by identifying the mutations that were identified as either nucleotide insertion or deletion events and dividing it by the total number of insertion and deletion events and single-nucleotide variant events (total mutations) in each sample.
Statistics
A Pearson correlation was performed in R between the PanCan transcriptome centroids and the Urachal sample expression values as detailed above. All clustering was performed in Cluster 3.0 using linkage and similarity metrics as described above. Copy number alteration significance values were calculated using the GISTIC q-value metric.
RESULTS
Urachal Adenocarcinomas Molecularly Resemble Colorectal Adenocarcinoma and Glioblastoma in a Pan-Cancer Analysis
Thirteen urachal adenocarcinomas were identified from a search of the University of North Carolina surgical pathology database (Table 1). All were confirmed to be urachal adenocarcinomas on the basis of standard criteria (Patients and Methods). We first performed global transcriptome profiling of 13 urachal adenocarcinomas using RNA sequencing. Transcript abundance was estimated by RNA-seq by expected maximization on the basis of University of California, Santa Cruz, known genes annotation (GAF2.1).12 To assess the similarity of urachal adenocarcinomas to other cancers, after normalization and correction for batch effect by using surrogate variable analysis, we performed hierarchical clustering using the top 10% of differentially expressed genes within the previously described TCGA Pan-Cancer data set, which includes tumors from 12 different tissues of origin.13 Five of 13 urachal tumors clustered with the TCGA colon and rectal (COADREAD) cancers, while four clustered most closely with the TCGA GBM tumors, which suggests that urachal tumors have global gene expression patterns that significantly resemble these two tumor types (Fig 1A). Next, we more quantitatively assessed the level of similarity between each urachal tumor and the TCGA Pan-Cancer tumor types across all genes. To this end, using all expressed genes, we derived centroid values for each gene within a TCGA Pan-Cancer tumor type and determined the correlation between each TCGA Pan-Cancer tumor type and each individual urachal sample (Fig 1B). Similar to hierarchical clustering results (Fig 1A), we observed that a subset of urachal tumors had high similarity (R = 0.45 to 0.65) to the TCGA COADREAD tumors, whereas others had more moderate similarity to the TCGA GBM samples (R = 0.15 to 0.35). In aggregate, these results support the notion that subsets of urachal tumors are molecularly similar to either colorectal cancer or GBM.
Table 1.
Clinical Characteristics of Urachal Tumors

Fig 1.
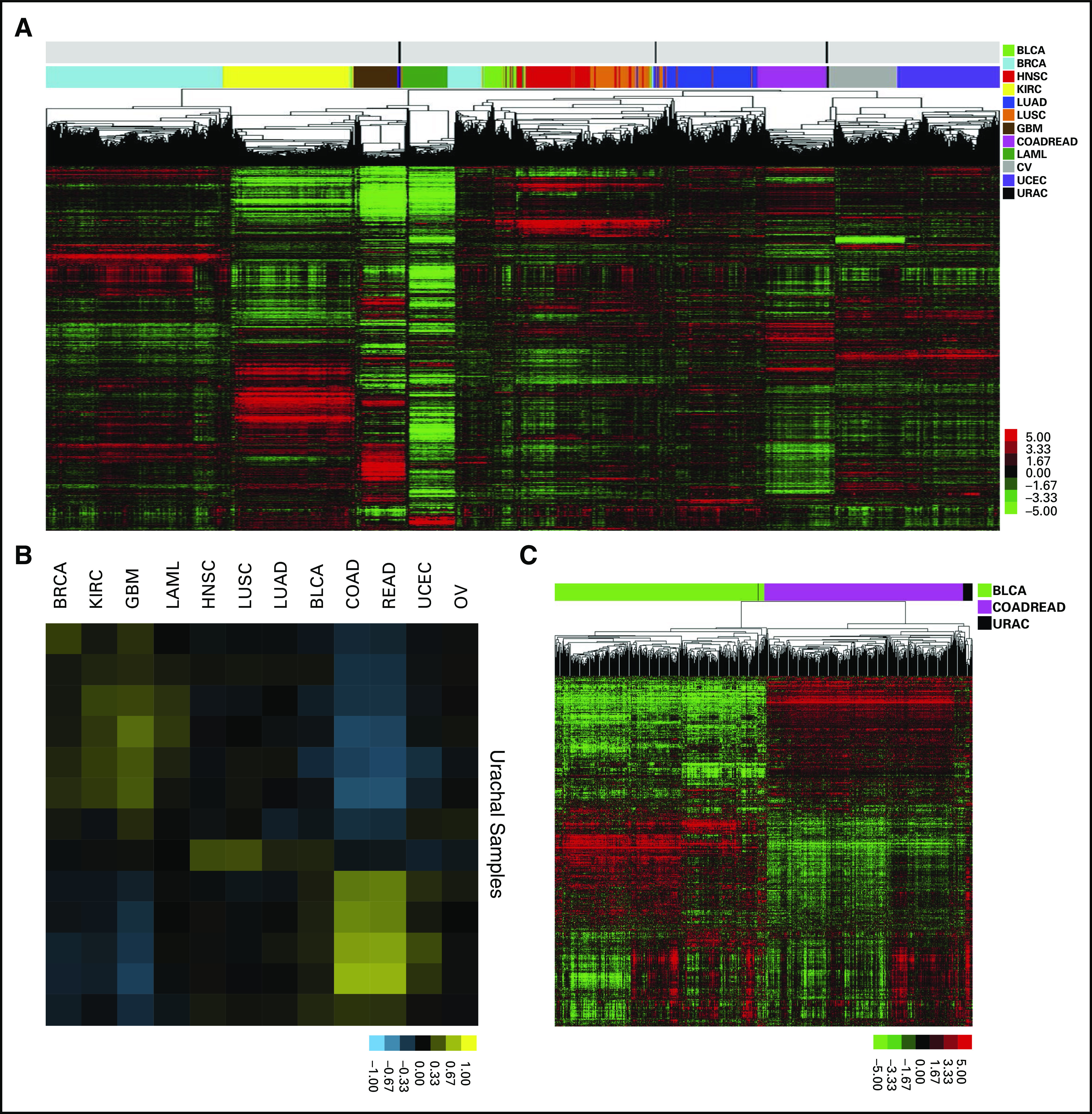
Pan-cancer analysis reveals that urachal adenocarcinomas molecularly resemble colorectal adenocarcinoma. (A) Unsupervised clustering of The Cancer Genome Atlas (TCGA) Pan-Cancer tumors and UNC urachal adenocarcinomas. (B) Correlation matrix of urachal adenocarcinoma samples to TCGA Pan-Cancer transcriptome centroids. (C) Unsupervised clustering of TCGA colorectal adenocarcinomas, TCGA urothelial carcinomas, and UNC urachal carcinomas. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; GBM, glioblastoma; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; LAML, acute myeloid leukemia; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma; READ, rectum adenocarcinoma; UCEC, uterine corpus endometrial carcinoma; URAC, urachal adenocarcinoma.
Urachal adenocarcinomas arise from an embryologic remnant of the allantois that is formed when the cloaca divides into an anterior and posterior portion. Whereas the anterior portion becomes the urogenital sinus, the posterior portion goes on to form the rectum.3 Of note, urachal remnants are lined by urothelium with varying numbers of columnar and/or mucus-secreting cells14; therefore, to more specifically compare urachal tumors with bladder and colorectal tumors, we hierarchically clustered the urachal tumors using the top 10% of the most differentially expressed genes between TCGA COADREAD and TCGA BLCA tumors.9,10 The large majority of the urachal tumors (n = 12) clustered with the COADREAD tumors (Fig 1C), which suggests a higher molecular similarity with colorectal cancer than with bladder cancer and further supports our findings from the TCGA Pan-Cancer analysis.
Targeted Exon Sequencing Reveals Genomic Alterations That Parallel Colorectal Cancer
We next performed targeted exon capture sequencing of approximately 800 genes using the UNCseq panel of genes (Data Supplement) for 11 urachal tumors. There was universal inactivation of TP53 by mutation (Fig 2A). Of interest, other genes that mutated at a high frequency included APC (25%), ARID4B (25%), MLL3 (25%), NF1 (25%), and MTOR (33%). APC mutations were of particular interest, given it is uniquely mutated in colorectal cancers.13 Although none of the MTOR mutations has been previously reported, of interest, two of four of these mutations occur in the focal adhesion kinase targeting domain, where activating mutations have been previously described and shown to impart sensitivity to rapamycin.15 Moreover, colorectal cancers seem to have one of the highest rate of MTOR mutations across the published TCGA data sets (Appendix Fig A1). In contrast, there did not seem to be a significant number of mutations in genes that are typically altered in bladder cancer, such as FGFR3, ARID1A, KDM6A, CDKN1A, or E2F3.10 Moreover, hierarchical clustering of TCGA BLCA (n = 127), TCGA COADREAD (n = 224), and UNCseq urachal tumors, on the basis of the percentage of mutation of each gene using significantly mutated gene lists from the TCGA BLCA and COADREAD data sets, demonstrated that urachal tumors clustered more closely with colorectal tumors (Fig 2B).
Fig 2.
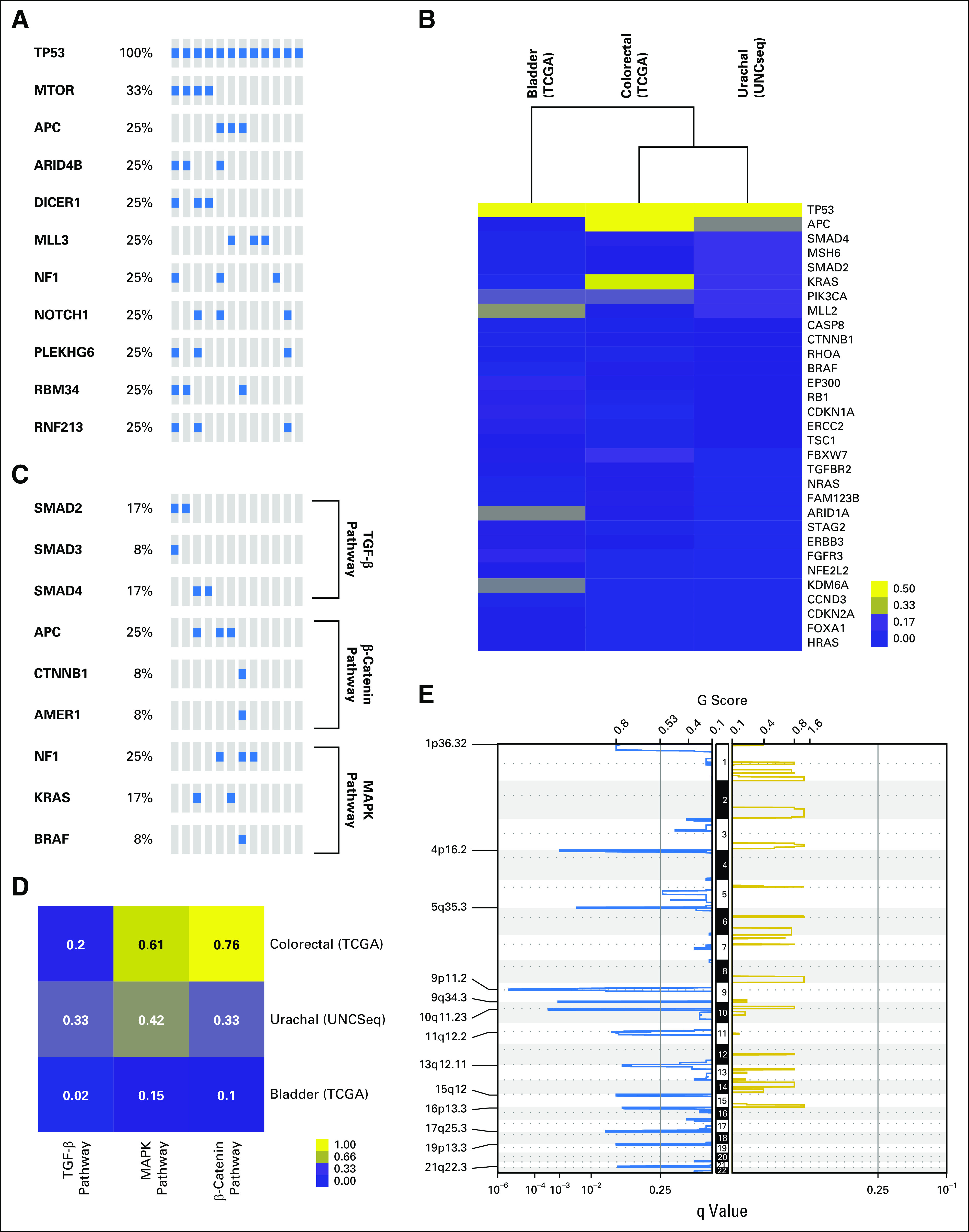
Targeted exon sequencing reveals that genomic alterations of urachal adenocarcinoma parallel those of colorectal adenocarcinoma. (A) Oncoprint of significantly mutated genes (> 10%) in urachal adenocarcinoma samples. (B) Unsupervised clustering of urachal, colorectal, and bladder mutation frequency across BLCA and COADREAD significantly mutated genes as defined by the The Cancer Genome Atlas (TCGA). (C) Oncoprints of transforming growth factor (TGF)-β, mitogen-activated protein kinase (MAPK), and β-catenin pathway mutations in colorectal (TCGA), bladder (TCGA), and urachal (UNCSeq) tumors. (D) A supervised heatmap of the frequency of TFG-β, MAPK, and β-catenin pathway mutations in colorectal (TCGA), bladder (TCGA), and urachal (UNCSeq) tumors. (E) Genomic Identification of Significant Targets in Cancer (GISTIC) plot identifies significant DNA copy number alterations. Gains and losses are depicted in gold and blue, respectively, ordered by genomic position, and a significance threshold (false discovery rate < 0.25) is indicated.
Colorectal cancers are typified by alterations in several pathways, including β-catenin—by APC loss—as well as activation of the RAS/MAPK signaling pathway—typically by KRAS mutation—and TGFB (by SMAD4 inactivation) pathways. We noted that urachal tumors harbored high levels of genomic alterations of all three of these canonical colorectal cancer pathways, including β-catenin activation by APC mutation as well as mutations in CTNNB1 and AMER1 (APC membrane recruitment protein 1), MAPK activation by KRAS mutation or NF1 loss, and TGFB activation by SMAD2, SMAD3, or SMAD4 mutations (Fig 2C). Indeed, urachal tumors had mutational frequencies of these pathways that were near that of the TCGA COADREAD data set (Fig 2D), which reinforces the notion that a subset of urachal adenocarcinomas genomically resembles colorectal cancer.
To analyze the genomic landscape of urachal adenocarcinomas, we performed cohort-level copy number alteration analysis by using GISTIC 2.0.11 No regions of the genome had significant copy number amplification; however, several regions showed significant focal deletions (Fig 2E). The 16p13.3 and 19p13.3 cytoband deletions were the only significant genomic alterations that urachal adenocarcinoma shares with either bladder or colorectal cancer, and both regions are significantly deleted across all three cancer types (Data Supplement); however, no other regions of the genome show similarities in copy number alteration between urachal adenocarcinomas and the other two cancers, which indicates that, whereas transcriptomic profiling indicates that urachal adenocarcinomas are similar to colorectal adenocarcinomas, the genomic landscape of urachal adenocarcinoma is distinct. Of interest, the 17q25.3 cytoband, which is significantly deleted in urothelial adenocarcinomas, contains RNF213, one of the genes mutated in 25% of urachal adenocarcinomas and that is involved in the inhibition of noncanonical Wnt/calcium signaling, which further supports similarities with colorectal adenocarcinoma.16
Urachal Adenocarcinomas Have Inactivation of Genes Associated With Microsatellite Instability and Hypermutation
Microsatellite instability is a hypermutable phenotype that is caused by the loss of DNA MMR activity and is detected in approximately 15% of all colorectal cancers.17 We detected inactivating mutations in MSH2 and MSH6 as well as mutation of the catalytic subunit of the DNA polymerase epsilon complex (POLE), which has been demonstrated to result in a hypermutable phenotype in 25% (3 of 12) of urachal tumors18 (Fig 3A). A comparison of both the mutational burden and the number of indels across UNCseq urachal tumors, UNCseq colorectal (n = 67), and UNCseq bladder tumors (n = 51) demonstrated that urachal tumors with inactivation of MSH2 or MSH6 had high mutation burdens and indel (insertion/deletion) rates (Figs 3B and 3C); therefore, a subset of urachal tumors harbors mutations in either DNA MMR genes or DNA polymerases that are associated with hypermutation. These urachal tumors have a hypermutable phenotype and are associated with increased mutational loads and indel rates comparable to those observed in DNA MMR–deficient colorectal and bladder cancers.
Fig 3.
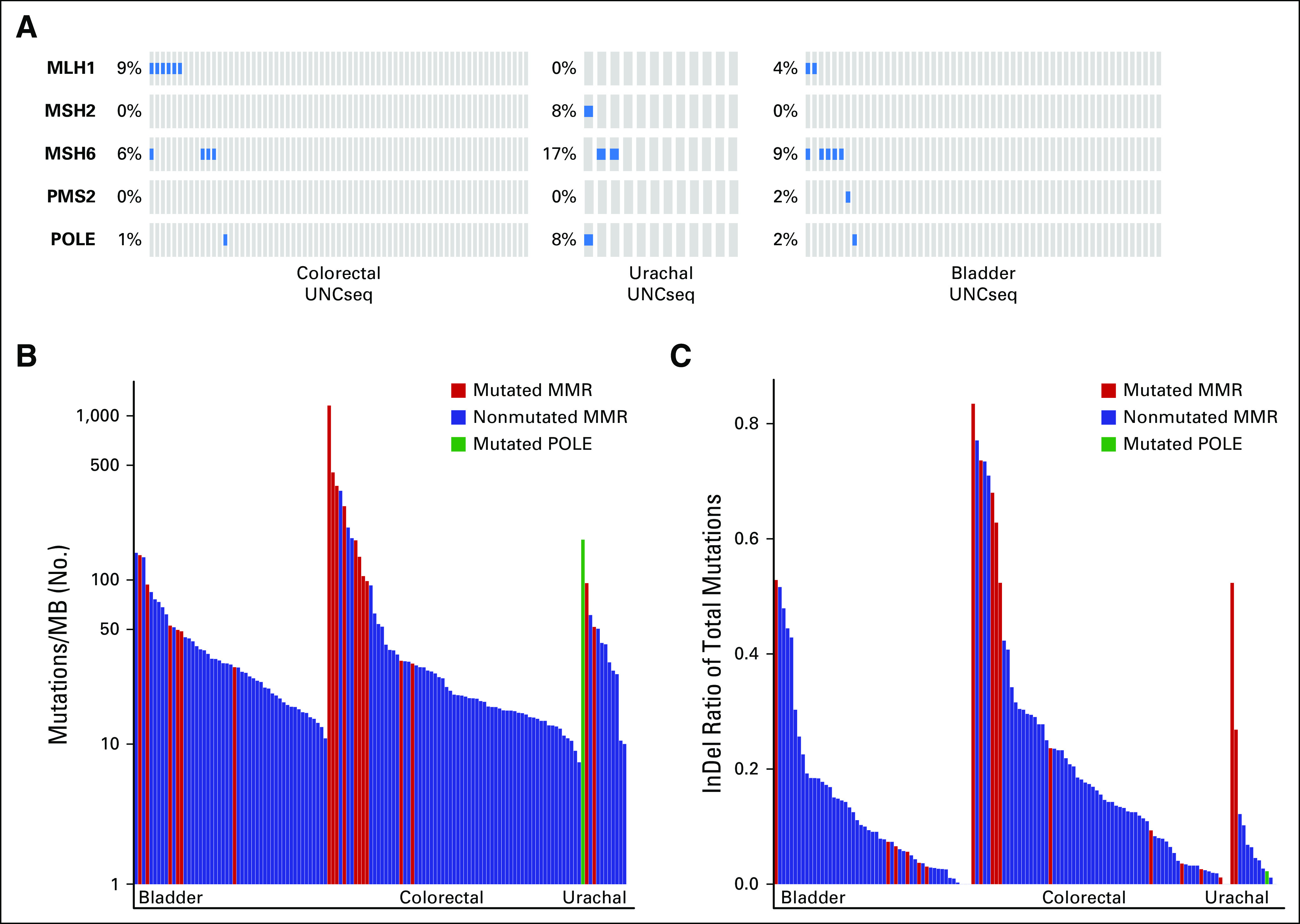
Urachal adenocarcinomas have inactivation of genes that are associated with microsatellite instability and hypermutation. (A) Oncoprints of DNA mismatch repair gene pathways across UNCSeq colorectal, urachal, and bladder samples. (B) Supervised bar plot of the mutations/Megabase (MB) across UNCSeq bladder, colorectal, and urachal samples, with samples that contain mutations in the DNA mismatch repair (MMR) pathway indicated. (C) Supervised bar plot of the ratio of insertions to deletions (InDel)/mutation for UNCSeq bladder, colorectal, and urachal samples, with samples that contain mutations in the DNA mismatch repair pathway indicated.
Atezolizumab Treatment Results in Stable Disease
Programmed death-1 (PD-1) blockade has resulted in significant responses in tumors with MMR deficiency.19 One of the patients whose urachal tumor was found to harbor an MSH6 mutation was treated with the anti–PD-L1 antibody, atezolizumab. The patient received atezolizumab every 3 weeks with initial progression in target lesions (two lung nodules and one hilar lymph node), followed by regression in the two lung nodules and an increase in the left hilar node associated with necrosis at second assessment (Figs 4A and 4B). This pattern of initial growth followed by response is consistent with immune-related responses—that is, flare—observed during immune-checkpoint inhibitor therapy.20 Given the rarity of urachal adenocarcinoma, clinical trials of immune checkpoint inhibition are unlikely; however, the successful treatment with atezolizumab of a patient who harbors a DNA MMR pathway mutation provides anecdotal evidence of the efficacy of this therapy in urachal adenocarcinoma.
Fig 4.

Evaluation of a patient with urachal adenocarcinoma who was treated with atezolizumab. (A) Spider plot of individual lesions and combined tumor burden of the atezolizumab-treated patient. Arrow indicates start of atezolizumab treatment. (B) Images of target lesions at imaging time points.
DISCUSSION
Here, we describe the comprehensive genomic characterization of urachal adenocarcinoma and the first report, to our knowledge, of global RNA expression profiling of urachal tumors. We find that urachal tumors molecularly resemble colorectal cancer at the level of gene expression and validate previous reports that have shown that urachal tumors harbor genomic alterations—that is, KRAS, APC, and SMAD2/SMAD4 mutations—found in colorectal cancer.5-7 In aggregate, this work strengthens the links between these two seemingly disparate cancers.
A major novelty of our work is the finding that 25% (3 of 12) of urachal tumors harbor inactivating mutations of genes that are involved in DNA MMR, MSH6 and MSH2, or the DNA polymerase, POLE. These mutations are particularly interesting given their potential to predict response to immune checkpoint blockade. Perhaps most importantly, because clinical trials are next to impossible for rare tumors, patients with these tumors are most likely to benefit from precision oncology. Much like colorectal cancers with inactivating mutations in DNA MMR genes or POLE, we demonstrate that urachal adenocarcinomas with inactivation of these genes harbor a higher mutational burden and a higher rate of indels than those with an intact DNA MMR pathway. Emerging evidence suggests that tumors with higher mutational load have enhanced response to immune checkpoint blockade, likely because mutational load correlates robustly with predicted neoantigen burden.21-24 Along these lines, tumors with defective DNA MMR have heightened clinical benefit from anti–PD-1 therapy.19 Successful treatment of our patient with an MSH6 mutation with atezolizumab demonstrates the potential utility of precision oncology in this rare tumor type with a lack of clearly defined therapeutic options. Nonetheless, our case report remains anecdotal.
Whereas we demonstrate that there are striking similarities between urachal adenocarcinomas and colorectal cancers, we also note that some urachal adenocarcinomas seemed to have gene expression patterns that also more closely resembled GBMs. Of interest, the mesenchymal subtype of GBM seems to be enriched for inactivation of NF1 as well as gene expression of mesenchymal markers, such as MET.25 Additional exploration of whether urachal tumors truly resemble GBM should be considered.
In summary, to our knowledge, our study is the first to perform global transcriptome profiling of urachal adenocarcinomas. When placed in the context of a Pan-Cancer data set, urachal adenocarcinomas seem to most highly resemble colorectal cancer. Our transcriptome studies therefore reinforce the notion from genomic studies that urachal adenocarcinomas resemble colorectal cancer; however, our studies report that these rare tumors have mutations in DNA MMR proteins and POLE and describe the successful treatment of a patient by using the anti–PD-L1 antibody atezolizumab. Overall, our studies and case report highlight the potential utility of precision oncology in rare tumor types that have no clear standard of care therapy and are unlikely to have sufficient numbers of patients to complete large clinical trials.
ACKNOWLEDGMENT
We thank the members of the Kim laboratory for useful discussions. We also thank the University of North Carolina Translational Pathology Laboratory for expert technical assistance.
Appendix
Fig A1.

Pan-Cancer mechanistic target of rapamycin (MTOR) mutation analysis shows colorectal tumors have the highest mutation frequency among characterized tumor types. ACC, adenoid cystic carcinoma; AML, acute myeloid leukemia; ccRCC, clear cell renal cell carcinoma; chRCC, chromophobe renal cell carcinoma; GBM, glioblastoma; pRCC, papillary renal cell carcinoma; TCGA, The Cancer Genome Atlas.
Footnotes
Supported by TPL, TPF, UNCseq, and the University of North Carolina (UNC) University Cancer Research Fund (UCRF); by the UNC Network Group Integrated Translational Science Center award (National Cancer Institute Grant No. 5U10-CA181009; to D.N.H.); American Cancer Society Grant No. RSG-14-219-01-TBG (to W.Y.K.); and the Bladder Cancer Advocacy Network (to W.Y.K.). The UNC Translational Pathology Laboratory is supported, in part, by National Cancer Institute Grant No. 2-P30-CA016086-40 and the UNC UCRF.
AUTHOR CONTRIBUTIONS
Conception and design: Sara E. Wobker, Angela B. Smith, Matthew I. Milowsky, D. Neil Hayes, William Y. Kim
Administrative support: D. Neil Hayes
Provision of study materials or patients: Michael E. Woods, Peter Black, D. Neil Hayes
Collection and assembly of data: Sara E. Wobker, Michael E. Woods, Michele C. Hayward, Juneko E. Grilley-Olson, Nirali M. Patel, Peter Black, Joel S. Parker, D. Neil Hayes, William Y. Kim
Data analysis and interpretation: Jordan Kardos, Matthew E. Nielsen, Katrina A. McGinty, Nirali M. Patel, Peter Black, Matthew I. Milowsky, D. Neil Hayes, William Y. Kim
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Comprehensive Molecular Characterization of Urachal Adenocarcinoma Reveals Commonalities With Colorectal Cancer, Including a Hypermutable Phenotype
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Jordan Kardos
No relationship to disclose
Sara E. Wobker
No relationship to disclose
Michael E. Woods
No relationship to disclose
Matthew E. Nielsen
Stock and Other Ownership Interests: Grand Rounds Health
Angela B. Smith
No relationship to disclose
Eric M. Wallen
Stock and Other Ownership Interests: MDxHealth
Raj S. Pruthi
No relationship to disclose
Michele C. Hayward
No relationship to disclose
Katrina A. McGinty
No relationship to disclose
Juneko E. Grilley-Olson
Consulting or Advisory Role: Sanofi
Research Funding: Bayer (Inst), Novartis (Inst), Peregrine Pharmaceuticals (Inst), NanoCarrier (Inst), Genentech (Inst), Seattle Genetics (Inst), MedImmune (Inst), GlaxoSmithKline (Inst), Pfizer (Inst)
Nirali M. Patel
No relationship to disclose
Karen E. Weck
No relationship to disclose
Peter Black
Consulting or Advisory Role: AbbVie, Astellas Pharma, Janssen Oncology, Amgen, Novartis, BioCancell, Sitka, Cubist, Bayer, Merck, Sanofi, Biosyent, Ferring, Eli Lilly, Roche, Spectrum Pharmaceuticals, Allergan
Speakers' Bureau: Ferring, Red Leaf Medical, New B Innovation, iProgen
Patents, Royalties, Other Intellectual Property: 1. PCT/CA2014/000787. Canada. 2014-11-03 Cancer Biomarkers and Classifiers and uses thereof. 2. #61899648 United States. 2013-03-13 Bladder cancer signature.
Travel, Accommodations, Expenses: Janssen Pharmaceuticals, Bayer, Sanofi
Joel S. Parker
No relationship to disclose
Matthew I. Milowsky
Research Funding: Mirati Therapeutics (Inst), Pfizer (Inst), Cerulean Pharma (Inst), Merck (Inst), Seattle Genetics (Inst), Acerta Pharma (Inst), BioClin Therapeutics (Inst), Genentech (Inst), Bristol-Myers Squibb (Inst), X4 Pharma (Inst), Med-Immune (Inst), Incyte (Inst), Innocrin Pharma (Inst), Inovio Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: Genentech
D. Neil Hayes
Leadership: GeneCentric
Stock and Other Ownership Interests: GeneCentric
Consulting or Advisory Role: GeneCentric
Patents, Royalties, Other Intellectual Property: I hold several diagnostic patents or pending patents in the area of solid tumor diagnostics
William Y. Kim
Stock and Other Ownership Interests: Johnson & Johnson, Bristol-Myers Squibb, Medivation, Agios
Patents, Royalties, Other Intellectual Property: BASE47 bladder cancer subtype classifier
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A: Cancer statistics, 2015. CA Cancer J Clin 65:5-29, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Gopalan A, Sharp DS, Fine SW, et al. : Urachal carcinoma: A clinicopathologic analysis of 24 cases with outcome correlation. Am J Surg Pathol 33:659-668, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siefker-Radtke A: Urachal adenocarcinoma: A clinician’s guide for treatment. Semin Oncol 39:619-624, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Wright JL, Porter MP, Li CI, et al. : Differences in survival among patients with urachal and nonurachal adenocarcinomas of the bladder. Cancer 107:721-728, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Sirintrapun SJ, Ward M, Woo J, et al. : High-stage urachal adenocarcinoma can be associated with microsatellite instability and KRAS mutations. Hum Pathol 45:327-330, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Singh H, Liu Y, Xiao X, et al. : Whole exome sequencing of urachal adenocarcinoma reveals recurrent NF1 mutations. Oncotarget 7:29211-29215, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collazo-Lorduy A, Castillo-Martin M, Wang L, et al. : Urachal carcinoma shares genomic alterations with colorectal carcinoma and may respond to epidermal growth factor inhibition. Eur Urol 70:771-775, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X, Wang A, Walter V, et al. : Combined targeted DNA sequencing in non-small cell lung cancer (NSCLC) using UNCseq and NGScopy, and RNA sequencing using UNCqeR for the detection of genetic aberrations in NSCLC. PLoS One 10:e0129280, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muzny DM, Bainbridge MN, Chang K, et al. : Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330-337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinstein JM, Akbani R, Broom BM, et al. : Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507:315-322, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mermel CH, Schumacher SE, Hill B, et al. : GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12:R41, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B, Dewey CN: RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoadley KA, Yau C, Wolf DM, et al. : Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158:929-944, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhillon J, Liang Y, Kamat AM, et al. : Urachal carcinoma: A pathologic and clinical study of 46 cases. Hum Pathol 46:1808-1814, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grabiner BC, Nardi V, Birsoy K, et al. : A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4:554-563, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scholz B, Korn C, Wojtarowicz J, et al. : Endothelial RSPO3 controls vascular stability and pruning through non-canonical WNT/Ca2+/NFAT signaling. Dev Cell 36:79-93, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Dudley JC, Lin MT, Le DT, et al. : Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res 22:813-820, 2016 [DOI] [PubMed] [Google Scholar]
- 18.Rayner E, van Gool IC, Palles C, et al. : A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat Rev Cancer 16:71-81, 2016 [DOI] [PubMed] [Google Scholar]
- 19.Le DT, Uram JN, Wang H, et al. : PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372:2509-2520, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodi FS, Hwu W-J, Kefford R, et al. : Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol 34:1510-1517, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenberg JE, Hoffman-Censits J, Powles T, et al. : Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 387:1909-1920, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizvi NA, Hellmann MD, Snyder A, et al. : Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124-128, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kardos J, Chai S, Mose LE, et al. : Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 1:e85902, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Allen EM, Miao D, Schilling B, et al. : Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350:207-211, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhaak RGW, Hoadley KA, Purdom E, et al. : Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98-110, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]