

Abstract
PURPOSE
Endometrial cancer (EC) is not considered a component of the hereditary breast and ovarian cancer syndrome but can arise in patients with germline BRCA1/2 (gBRCA1/2) mutations. Biallelic BRCA1/2 alterations are associated with genomic features of homologous recombination DNA repair deficiency (HRD) in cancer. We sought to determine if ECs in gBRCA1/2 mutation carriers harbor biallelic alterations and/or features of HRD.
METHODS
Of 769 patients with EC who underwent germline panel testing, 10 pathogenic gBRCA1/2 mutation carriers were identified, and their tumor- and normal-derived DNA was subjected to massively parallel sequencing targeting at least 410 cancer-related genes. Three gBRCA1/2-associated ECs were identified in 232 ECs subjected to whole-exome sequencing by The Cancer Genome Atlas. Somatic mutations, copy number alterations, loss of heterozygosity, microsatellite instability (MSI), and genomic HRD features were assessed.
RESULTS
Of the 13 patients included who had EC, eight harbored pathogenic gBRCA1 mutations and five harbored gBRCA2 mutations. Eight (100%) and two (40%) ECs harbored biallelic BRCA1 and BRCA2 alterations through loss of heterozygosity of the wild-type allele. All ECs harbored somatic TP53 mutations. One monoallelic/sporadic gBRCA2-associated EC had MLH1 promoter methylation and was MSI high. High large-scale state transition scores, a genomic feature of HRD, were found only in ECs with bi- but not monoallelic BRCA1/2 alterations. The Signature Multivariate Analysis HRD signature Sig3 was enriched in biallelic gBRCA1/2 ECs, and the three ECs from The Cancer Genome Atlas with BRCA1 biallelic alterations subjected to whole-exome sequencing displayed a dominant HRD-related mutational signature 3.
CONCLUSION
A subset of gBRCA1/2-associated ECs harbor biallelic BRCA1/2 alterations and genomic features of HRD, which may benefit from homologous recombination–directed treatment regimens. ECs in BRCA2 mutation carriers might be sporadic and even MSI high, and may potentially benefit from immune-checkpoint inhibition.
INTRODUCTION
The tumor suppressor genes BRCA1 and BRCA2 (BRCA1/2) play central roles in DNA repair via the homologous recombination (HR) pathway.1 BRCA1/2 pathogenic germline mutations account for a large proportion of cases of hereditary breast and ovarian cancer (HBOC) syndrome, increasing the lifetime risk of developing breast, ovarian, prostate, and/or pancreatic cancers.2,3 Endometrial cancers (ECs) are currently not formally associated with HBOC but have been reported in patients with germline BRCA1/2 (gBRCA1/2) mutations.4,5
CONTEXT
Key Objective
To determine if endometrial cancers (ECs) that arise in patients with pathogenic BRCA1 or BRCA2 germline mutations are sporadic cancers or display genomic features of homologous recombination DNA repair deficiency (HRD), which may guide treatment strategies.
Knowledge Generated
ECs in patients with BRCA1 germline mutations showed biallelic BRCA1 alterations coupled with genomic features of HRD. In contrast, only a subset of ECs in BRCA2 germline mutation carriers harbored biallelic BRCA2 alterations; the remaining cases were likely sporadic cancers.
Relevance
Knowledge about the BRCA1/2 allele status and/or HRD features of ECs in patients with germline BRCA1/2 alterations may guide therapeutic decision making, given that tumors with biallelic alterations in BRCA1/2 have been associated with increased response to HR-targeted therapies such as platinum salts and PARP inhibitors.
Independent studies have suggested that ECs may be a component of HBOC5 and specifically highlighted a possible increased risk of serous carcinomas in women with germline BRCA1 (gBRCA1) mutations.5,6 Conversely, in a recent pan-cancer analysis of pathogenic germline variants by The Cancer Genome Atlas (TCGA), enrichment of gBRCA1/2 pathogenic variants in EC was not identified.7
Even if not formally accepted as part of the HBOC syndrome, the identification of ECs with defective HR DNA repair could have important treatment implications in the era of precision medicine. Biallelic inactivation of BRCA1/2, either through locus-specific loss of heterozygosity (LOH) of the BRCA1/2 wild-type allele or through a somatic BRCA1/2 pathogenic mutation, has been associated with genomic features of HR deficiency8,9 and increased survival after DNA damage–inducing platinum-based chemotherapy.9 Importantly, the frequency of LOH of the wild-type allele or somatic mutations of BRCA1/2 in the context of gBRCA1/2 mutation carriers varies according to tumor type.9
In this study, we sought to determine whether ECs arising in gBRCA1/2 mutation carriers harbor biallelic BRCA1/2 alterations and show features of HR deficiency, including the Catalog of Somatic Mutations in Cancer mutational signature 3, Signature Multivariate Analysis (SigMA) HR deficiency signature Sig3, or large-scale chromosomal breaks in the form of large-scale state transitions (LSTs). In addition, we assessed the clinicopathologic data of ECs in gBRCA1/2 mutation carriers and explored the repertoire of somatic mutations present in these tumors.
METHODS
Case Selection
All patients with EC and gBRCA1/2 mutations who consented to germline analysis under an institutional review board–approved protocol at Memorial Sloan Kettering Cancer Center and whose tumors were subjected to targeted massively parallel sequencing via Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT)10,11 from July 2015 to May 2019 were identified (n = 769 sequenced; n = 10 with pathogenic gBRCA1/2 mutation). All gBRCA1/2 variants were reviewed by a board-certified molecular pathologist (D.M.) and classified according to the American College of Medical Genetics and Genomics criteria12 as pathogenic. Clinicopathologic data, including age at diagnosis, cancer stage, and past cancer history, were obtained from medical records. All cases were centrally rereviewed by pathologists (A.P.M.S., R.A.S.) with experience and expertise in gynecologic pathology. In addition, ECs with pathogenic gBRCA1/2 alterations (n = 3) subjected to whole-exome sequencing (WES) as part of The Cancer Genome Atlas (TCGA; n = 232)13 were identified in a curated data set from Riaz et al,8 as previously described.14 WES-derived somatic mutation Multi-Center Mutation Calling in Multiple Cancers data of the cases were retrieved from TCGA Genomic Data Commons (https://gdc.cancer.gov/about-data/publications/mc3-2017),15 and clinicopathologic data were obtained from the TCGA data portal.
Massively Parallel Sequencing Analysis
Tumor and matched normal DNA samples were subjected to targeted massively parallel sequencing using MSK-IMPACT, which targets all exons and selected introns of 410 (n = 6) or 468 (n = 4) cancer genes, as previously described.10,11 The median depth of coverage was 613× (range, 406× to 992×) for tumor and 509× (range, 401× to 685×) for normal samples (Data Supplement). Analysis of sequencing data for the identification of single nucleotide variants (SNVs) and small insertions and deletions (indels) was performed as previously described.16,17 FACETS18 (fraction- and allele-specific copy number estimates from tumor sequencing) was used to determine copy number alterations (CNAs) and whether genes harboring somatic or germline mutations were targeted by loss of heterozygosity (LOH), as previously described.16,17 The cancer cell fractions of all somatic mutations were computed using ABSOLUTE, version 1.0.6,19 as previously described.16,17 A combination of mutation function predictors was used to define the potential functional impact of each missense SNV.20 Mutational hotspots were annotated according to Chang et al.21
Microsatellite Instability Score
The algorithm MSIsensor22 was used to assess microsatellite instability (MSI), as previously described.14 ECs subjected to MSK-IMPACT and WES (TCGA) with MSIsensor scores of greater than or equal to 1023 and greater than or equal to 3.522, respectively, were deemed MSI high.
Mutational Signatures
Mutational signatures were inferred from all synonymous and nonsynonymous SNVs using the algorithm deconstructSigs24 at default parameters, as previously described.14 deconstructSigs was only applied to samples with at least 20 somatic SNVs.14 In addition, Signature Multivariate Analysis (SigMA) was used for samples with at least five somatic SNVs; SigMA is a tool to detect the HR deficiency mutational signature Sig3 and other signatures from targeted gene panels.25
Large-Scale State Transitions
For the assessment of genomic features of HR deficiency, large-scale state transitions (LSTs) were defined, and a cutoff of greater than or equal to 15 was used for LST-high cases, as previously described.14,26
DNA Mismatch Repair Immunohistochemistry and MLH1 Promoter Methylation
Immunohistochemistry for the DNA mismatch repair (MMR) proteins MLH1, MSH2, MSH6, and PMS2 was performed in the clinical setting as previously described.27 Loss of DNA MMR protein expression was defined as the complete absence from all tumor cell nuclei. The presence of a positive internal control (ie, blood vessels, stromal cells, lymphocytes) was required for interpretation. MLH1 promoter methylation was assessed in the clinical setting on DNA obtained from formalin-fixed paraffin-embedded tumor samples, as previously described.28
Statistical Analyses
Fisher’s exact and Mann-Whitney U tests were used for comparison of categorical and continuous variables, respectively. Statistical analyses were performed using SPSS Statistics, version.25 (IBM, Armonk, NY). Two-tailed P < 0.05 was considered statistically significant.
RESULTS
Clinicopathologic Features of ECs Arising in gBRCA1/2 Carriers
Ten ECs from patients with pathogenic gBRCA1/2 mutations subjected to MSK-IMPACT sequencing (n = 769 ECs; 1.3%) and three subjected to WES by TCGA (n = 232 ECs; 1.3%)13 were included in this study. Of these 13 gBRCA1/2-associated ECs, eight and five patients with EC harbored gBRCA1 and germline BRCA2 (gBRCA2) mutations, respectively (Table 1). None of these gBRCA1/2-associated ECs displayed germline alterations in other genes associated with hereditary cancer susceptibility, including the DNA MMR genes.
TABLE 1.
Demographics and Baseline Characteristics of gBRCA1/2-Associated Endometrial Cancers
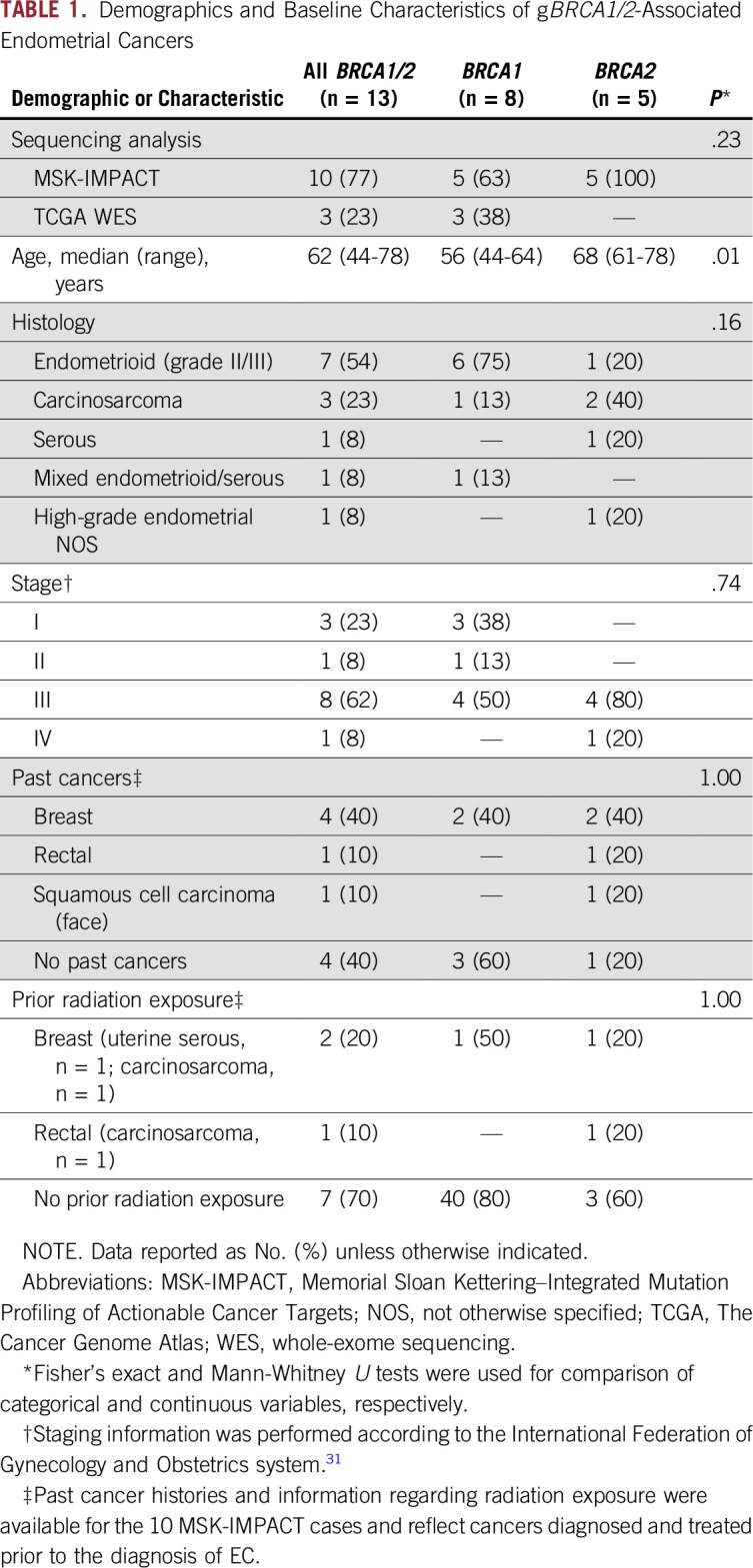
The median age at EC diagnosis was 62 (range, 44 to 78) years, with gBRCA1 patients (n = 8) being younger at EC diagnosis (median [range] age, 56 [44 to 64] years) than patients with EC harboring gBRCA2 (n = 5; median [range] age, 68 [61 to 78] years; Mann-Whitney U test P = .01; Tables 1 and 2).
TABLE 2.
BRCA1/2 Alterations, Clinicopathologic Information and Sequencing Results of gBRCA1/2-Associated Endometrial Cancers

The histologic types of the gBRCA1/2-associated ECs varied. The majority of gBRCA1/2-associated ECs (n = 6 of 8; 75%) displayed an endometrioid histology (n = 2 and 4 International Federation of Gynecology and Obstetrics [FIGO] grades II and III, respectively) but lacked a solid/pseudoendometrioid/transitional cell-like pattern of morphology, which has been associated with gBRCA1/2 mutations in high-grade serous ovarian cancers.29,30 In contrast, ECs from patients with pathogenic gBRCA2 mutations were more heterogeneous at the phenotypic level, and included endometrioid (n = 1; FIGO grade II), carcinosarcoma (n = 2), high-grade EC not otherwise specified (NOS; n = 1) and serous (n = 1) cancers (Tables 1 and 2). Of note, none of the gBRCA1/2-associated ECs included in this study were of serous histology.
gBRCA1/2-associated ECs presented at different clinical FIGO stages (2009 staging system31). Although a subset of the ECs occurring in pathogenic gBRCA1 mutation carriers were uterus- or cervix-confined cancers (stage I, n = 3; stage II, n = 1; stage III, n = 4), all EC patients with pathogenic gBRCA2 mutations presented at an advanced stage (stage III, n = 4; stage IV, n = 1; Tables 1 and 2).
Four of the patients with gBRCA1/2-associated EC had a breast cancer diagnosis prior to their diagnosis of EC (EC identifiers BEC-3, BEC-6, BEC-8, and BEC-12: BEC-3, gBRCA1/2, carcinosarcoma; BEC-6, gBRCA1/2, serous; BEC-8, gBRCA1/2, endometrioid; BEC-12, gBRCA1/2, high-grade EC NOS; Table 1). None of the patients received tamoxifen as part of their breast cancer treatment regimens. In addition, patient BEC-2 (gBRCA1/2, carcinosarcoma) had a rectal cancer 12 years before the EC diagnosis and had received pelvic radiation. Taken together, our data suggest the clinicopathologic features associated with ECs occurring in patients with pathogenic gBRCA1 or gBRCA2 mutations may be heterogeneous.
Biallelic BRCA1/2 Alterations
Allele-specific copy number analysis revealed that 77% (n = 10 of 13) of the ECs in patients with pathogenic gBRCA1/2 mutations displayed biallelic inactivation of BRCA1/2 uniformly through LOH of the wild-type allele (Table 2; Fig 1). No somatic BRCA1/2 mutations were identified in the ECs studied (Fig 1; Data Supplement). Not all ECs occurring in pathogenic gBRCA1/2 mutation carriers harbored biallelic BRCA1/2 alterations, which have previously been associated with HR deficiency.8 Although all ECs in pathogenic gBRCA1 mutation carriers (100%) harbored biallelic BRCA1 inactivation, only two of the five ECs (40%) from patients with pathogenic gBRCA2 mutations displayed biallelic BRCA2 inactivation (Table 2; Fig 1). Biallelic BRCA1/2 inactivation was found across all histologic subtypes and clinical stages of EC. ECs in BRCA2 mutation carriers lacking LOH of the wild-type allele are likely sporadic tumors; all three were stage III at diagnosis and were endometrioid grade II, serous, or high-grade ECs (Table 2; Fig 1). These data suggest that the vast majority of gBRCA1/2-associated ECs, but only a subset of gBRCA1/2-associated ECs, harbor biallelic inactivation of the respective wild-type allele, and that some gBRCA1/2-associated ECs may, in fact, be sporadic.
FIG 1.

Landscape of somatic mutations in endometrial cancers (ECs) in patients with pathogenic BRCA1/2 germline mutations. Nonsynonymous somatic mutations identified in 410 cancer-related cancer genes in ECs from germline BRCA1 (n = 8) and germline BRCA2 (n = 5) mutation carriers (left) are shown. The mutation types are color coded according to the legend. Selected amplifications and homozygous deletions are shown at the bottom of the figure. Information on the germline mutation status, somatic loss of heterozygosity of BRCA1/2, histologic type, microsatellite instability (MSI) status, and sequencing type is provided in the phenobar below the sample names. indel, insertions and deletions; MSK-IMPACT, Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets; NOS, not otherwise specified; SNV, single nucleotide variant; TCGA, The Cancer Genome Atlas; WES, whole-exome sequencing.
Repertoire of Somatic Mutations and CNAs
The median number of somatic mutations detected in the 410 MSK-IMPACT genes (smallest gene panel) in the 13 gBRCA1/2-associated ECs was six (range, 2 to 38), with a median of five (range, 2 to 32) nonsynonymous mutations (Data Supplement). We observed that all ECs analyzed, irrespective of the presence of mono- or biallelic BRCA1/2 alterations, harbored somatic TP53 mutations, of which 12 (92%) were hotspot mutations (Fig 1; Data Supplement). Alterations affecting the PI3K pathway were common, with PIK3CA mutations present in five ECs (38%), PIK3R1 mutations in two ECs (15%), and PTEN mutations/homozygous deletions in three ECs (23%; Figs 1 and 2). Other recurrently altered genes included FAT1, PTCH1, KRAS, and MAP3K1 (each n = 2; Fig 1, Data Supplement).
FIG 2.
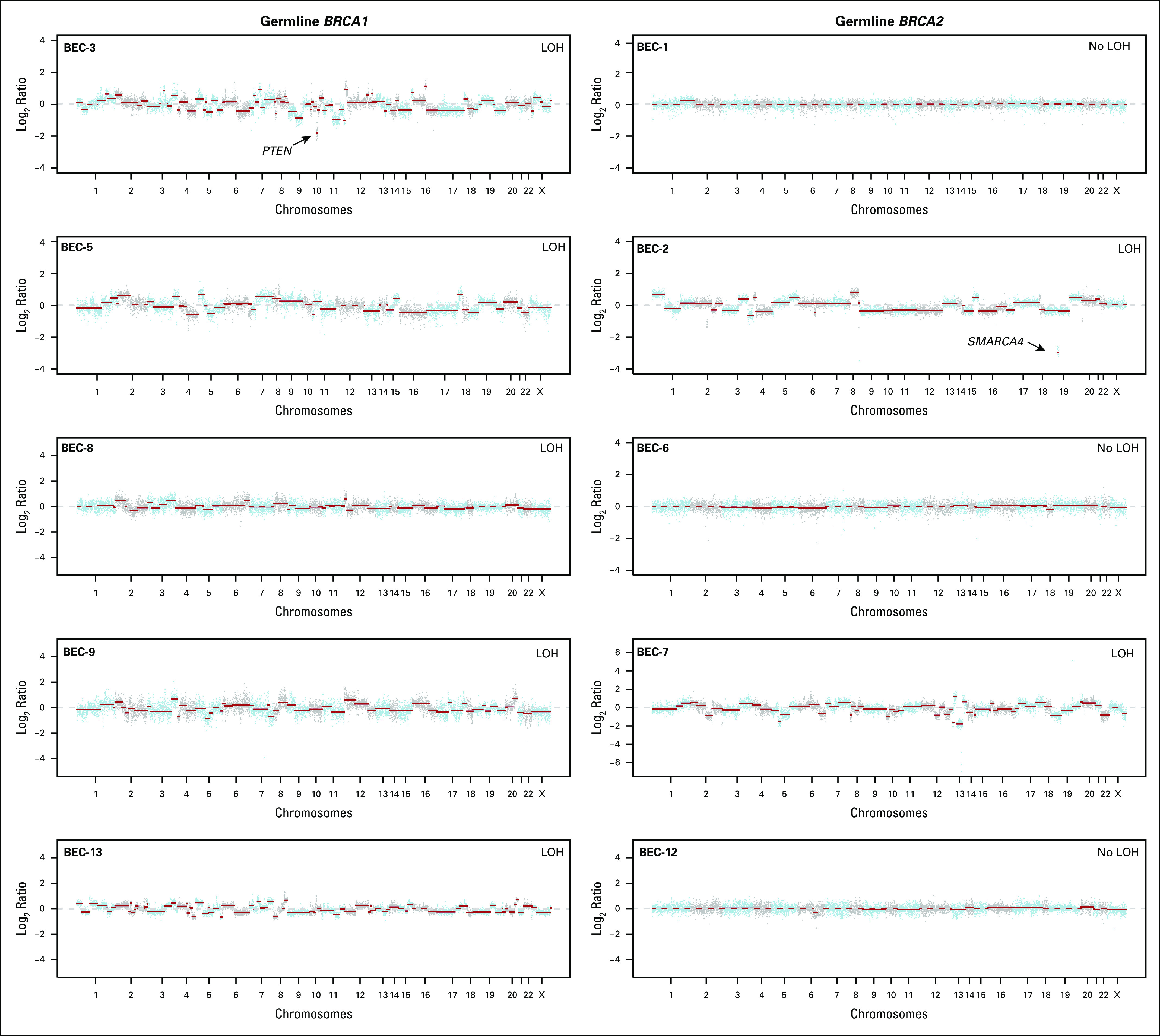

Copy number alterations in germline (gBRCA1/2)-associated endometrial cancers. Chromosome plots of the gBRCA1/2- and gBRCA1/2-associated endometrial cancers included in this study are presented. Copy number log2-ratios are depicted on the y-axis with the chromosome location on the x-axis. The presence of somatic loss of heterozygosity (LOH) of BRCA1/2 is displayed in the top right corner within each plot. Black arrows point to selected cancer gene amplifications and homozygous deletions. TCGA, The Cancer Genome Atlas.
We noted that BEC-1, a likely sporadic EC from a gBRCA2 mutation carrier with a monoallelic BRCA2 alteration, had a high mutational burden with 32 nonsynonymous somatic mutations. In addition, this case was MSI high with a high MSIsensor score (≥ 10; Fig 1, Table 2). None of the DNA MMR genes in BEC-1 harbored any somatic or germline mutations; however, consistent with the protein loss of MLH1 and PMS2 as assessed by immunohistochemistry (Fig 3), this case displayed MLH1 promoter methylation.
FIG 3.
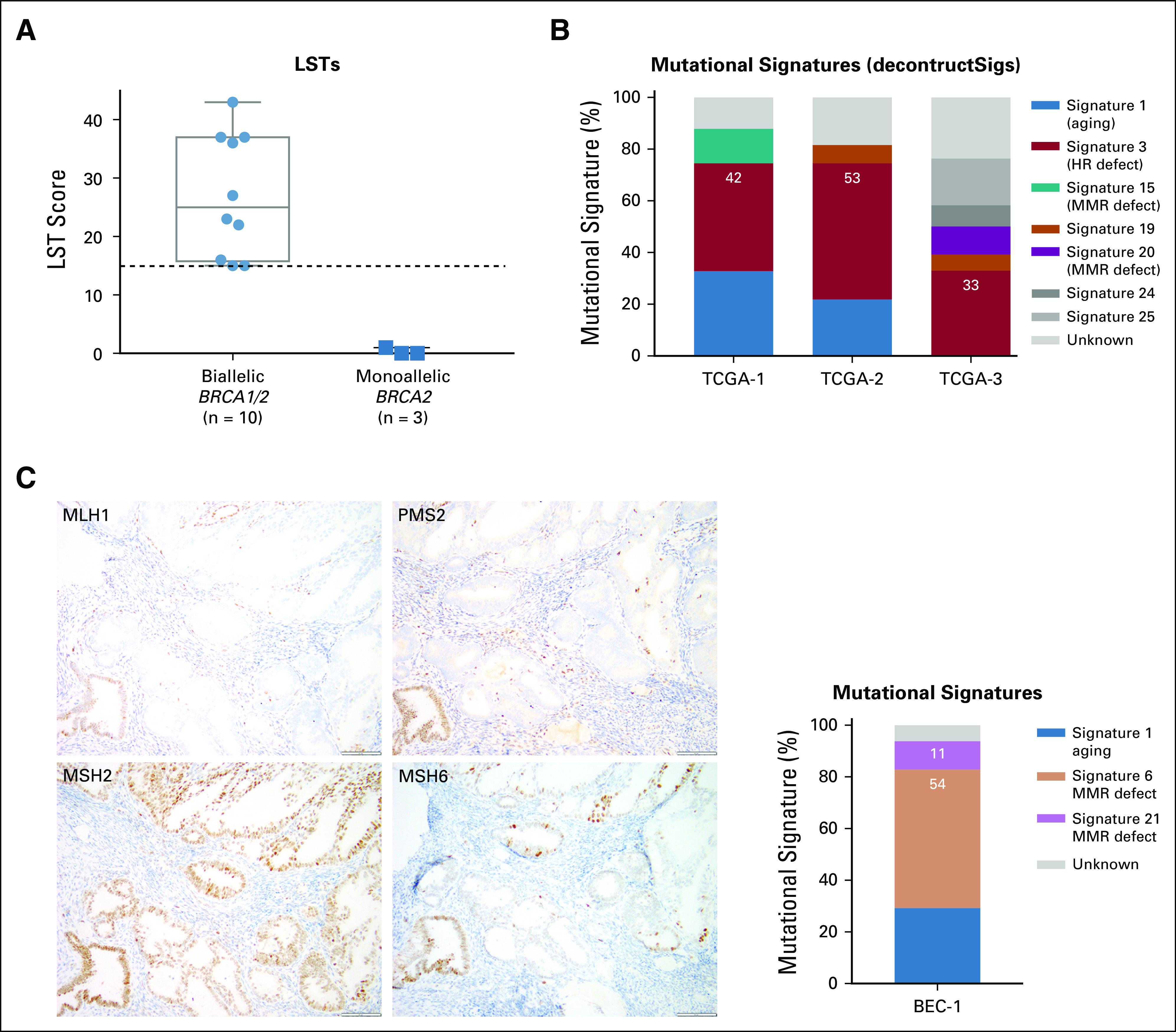
Genomic features of homologous recombination (HR) DNA repair deficiency. (A) Large-scale state transition (LST) scores in germline (gBRCA1/2)-associated endometrial cancers with biallelic (n = 10) and monoallelic (n = 3) BRCA1/2 alterations. Dashed line indicates the cutoff (≥ 15) for high LST scores.26 Biallelic BRCA1/2 alterations are associated with high LST scores. (B) Mutational signatures defined by deconstructSigs24 in three gBRCA1/2-associated endometrial cancers from The Cancer Genome Atlas (TCGA) subjected to whole-exome sequencing.13,14 Mutational signatures are color coded according to the legend on the right. All three germline gBRCA1/2-associated endometrial cancers have a dominant homologous recombination DNA repair deficiency–related mutational signature 3. (C) Immunohistochemical analysis of the (left) DNA mismatch repair (MMR) proteins MLH1, PMS2, MSH2, and MSH6 and (right) mutational signatures of case BEC-1, a monoallelic/ sporadic gBRCA1/2-associated endometrial cancer. Mutational signatures defined by deconstructSigs24 are color coded according to the legend on the right. BEC-1 shows loss of MLH1 and PMS2 expression, MLH1 promoter hypermethylation (not shown), and dominant mutational signatures 6 and 21 associated with defective DNA MMR.
The levels of copy number alterations (CNAs) varied across the gBRCA1/2-associated ECs studied, with some having very few CNAs and others displaying high levels of genomic instability (Fig 2). The ECs with very low levels of gene CNAs (ie, BEC-1, BEC-6, and BEC-12) had monoallelic BRCA2 alterations and were likely sporadic, with one (BEC-1) being MSI high, as mentioned. On the other hand, ECs with the highest levels of CNAs had biallelic BRCA1/2 alterations (ie, BEC-2, BEC-3, BEC-5, BEC-7, and TCGA-3; Fig 2). When assessing amplifications and deletions, PTEN was the only recurrent homozygous deletion identified in two biallelic BRCA1 ECs (BEC-3 and TCGA-2). In addition, we found an NF1 homozygous deletion in the biallelic BRCA1 grade II endometrioid TCGA-2 and a SMARCA4 homozygous deletion in the biallelic BRCA2 carcinosarcoma BEC-2. Of note, SMARCA4 (BRG1) loss is commonly found in undifferentiated carcinoma of the endometrium,32,33 and BEC-2 was a carcinosarcoma with heterologous elements, which, in addition to the carcinoma and sarcomatous components, also displayed an undifferentiated component. Finally, amplification of BCL6, TP63, and EED was found in the biallelic BRCA1 grade III endometrioid TCGA-1 EC (Fig 2).
Taken together, we found that ECs in patients with a gBRCA1/2 mutation are heterogeneous at the mutational and gene copy number levels. In addition, in germline carriers of pathogenic BRCA2 mutations, the ECs may be sporadic and even have a high mutational burden and be MSI high.
HR DNA Repair Deficiency in gBRCA1/2 ECs
We first assessed the presence of LSTs, a genomic feature of HR deficiency, in all cases. We observed that ECs with biallelic BRCA1/2 alterations (n = 10) had high LST scores, whereas ECs with monoallelic BRCA1/2 alterations did not (Table 2; Fig 3A). In addition, mutational signature analysis was performed using deconstructSigs24 for ECs with at least 20 SNVs (ie, n = 1 MSK-IMPACT EC; n = 3 TCGA ECs), as previously described.14 All biallelic, gBRCA1/2-associated, TCGA ECs had a dominant mutational signature 3 associated with HR deficiency (Fig 3B).14 Given the limited number of SNVs and indels in the ECs subjected to MSK-IMPACT sequencing, other genomic HR-deficiency features such as indel length, microhomology, or mutational signatures using decomposition algorithms could not be used. Therefore, we used SigMA, a recently described, likelihood-based measure signature analysis combined with machine-learning techniques, which can be used to detect the mutational signature Sig3 associated with HR deficiency from targeted gene panels including MSK-IMPACT (sensitivity range, 48% to 62% for uterine cancers).25 Consistent with the LST analysis, all six biallelic, BRCA1/2-associated ECs subjected to MSK-IMPACT and with at least five SNVs displayed either a dominant HR deficiency-related Sig3 (n = 2) or a dominant clock signature with secondary HR-deficiency signatures Sig3 (n = 3) or Sig834 (n = 1), in contrast to the two sporadic ECs with monoallelic BRCA2 alterations, which displayed MSI (SigMA)/signature 6 (deconstructSigs) and APOBEC signatures (Table 2; Fig 3C). The three biallelic gBRCA1/2-associated ECs from TCGA subjected to WES displayed a strong dominant SigMA Sig3 (Table 2). These data provide evidence to suggest that in some patients with gBRCA1/2 mutations, ECs may have biallelic BRCA1/2 alterations and be HR deficient.
DISCUSSION
The inclusion of EC as a component of HBOC syndrome could have substantial clinical impact on the screening, evaluation, and treatment of patients with gBRCA1/2 mutations. Previous reports of ECs in this population have acknowledged that the prevalence of EC in gBRCA1/2 mutation carriers is low.5 Here, through an in-depth analysis of ECs from patients with pathogenic gBRCA1/2 mutations, we demonstrate that the majority of these cases (77%) harbored biallelic BRCA1/2 alterations and displayed genomic features of HRD, providing evidence to suggest an etiological link between the pathogenic gBRCA1/2 mutations and the development of these ECs. In fact, this phenomenon was uniformly observed in patients with pathogenic gBRCA1 mutations; conversely, only two of the five patients with pathogenic gBRCA2 mutations had somatic inactivation of the BRCA2 wild-type allele and genomics features of HR deficiency. In ovarian cancer, patients with HR-deficient tumors have improved overall survival with platinum-based therapy and have better responses to HR deficiency–directed treatments such as PARP inhibitors than their wild-type counterparts.35,36
Knowledge of the allele status of BRCA1/2 alterations and/or HR-deficiency features could be beneficial in therapeutic decision making for patients with gBRCA1/2 mutations and EC. Our analysis has demonstrated that not all ECs arising in the context of pathogenic gBRCA2 mutations harbor LOH of the wild-type allele and genomics features of HR deficiency. In addition, it revealed that one of these cases was MSI high, likely representing a sporadic (ie, non-BRCA1/2) EC arising in a gBRCA2 mutation carrier. In this context, this patient would potentially benefit from immune checkpoint inhibitors, which have been approved for the management of recurrent MSI-high/DNA MMR–deficient cancers.37 In fact, a recent case report highlighted a complete remission after PD-1 blockade in a patient with MMR-deficient EC and a pathogenic monoallelic gBRCA1 mutation.38
The majority (75%) of biallelic gBRCA1/2-associated ECs were of intermediate/high-grade endometrioid as opposed to serous histology in previous reports.5,6 All six biallelic gBRCA1/2-associated endometrioid ECs harbored TP53 mutations, had high LST scores, and relatively high levels of CNAs (Table 2; Fig 2), and would likely be of copy-number high (serous-like) molecular subtype, as described by TCGA.13 It should be noted, however, that of these six endometrioid ECs with biallelic BRCA1 alterations, four harbored somatic mutations characteristic of endometrioid ECs, including PIK3CA mutations, PIK3R1 mutations, and PTEN mutations/homozygous deletions. Conversely, two of these ECs lacked alterations affecting genes recurrently altered in endometrioid ECs (Fig 1), suggesting that at the genetic level, these latter two cases resembled serous rather than endometrioid carcinomas, and that there is heterogeneity in gBRCA1/2-associated ECs. Carcinosarcomas of the uterus are rare, representing less than 5% of all uterine tumors.39 We noted a high frequency (30%) of carcinosarcomas in the 10 gBRCA1/2-associated ECs subjected to MSK-IMPACT sequencing, whereas overall, only 10% of the 769 ECs subjected to germline MSK-IMPACT sequencing were carcinosarcomas (remaining cases: 55% endometrioid, 15% serous, 10% endometrial cancer NOS, 4% mixed endometrial, 3% clear cell, 1% de- or undifferentiated, and 1% other). Interestingly, recurrent somatic BRCA1/2 mutations and BRCA2 deletions also have been reported in uterine carcinosarcomas.40-42 These findings suggest that ECs arising in gBRCA1/2 mutation carriers are heterogeneous at the histologic level and are potentially enriched for endometrioid tumors and carcinosarcomas.
The data presented here do provide insight into the genomics of ECs in patients with pathogenic gBRCA1/2 mutations, which may play a role in the tumorigenesis and/or progression of EC in some patients. This study has several limitations, however, primarily driven by the small sample size, and it does not resolve the controversy of EC as a feature of HBOC syndrome. The low incidence of these cases (1.3%; n = 10 of 769 ECs subjected to germline MSK-IMPACT testing) not only limits the sample size of this study but also, on its own, presents a challenge in defining EC as part of the HBOC syndrome spectrum of malignancies. In contrast, during the same time, 4.5% and 11.1% of breast and ovarian cancers tested by germline MSK-IMPACT were gBRCA1/2 associated, respectively. The MSK cohort during the time studied is enriched for advanced-stage disease,10,11 and neither the actual prevalence of pathogenic gBRCA1/2 mutations in patients with EC nor the impact of these mutations on the outcome of patients with EC could be fully defined. Although we have assessed the genomics features of HR deficiency, including LSTs and SigMA HR deficiency signature Sig3 in all cases, HR deficiency is more accurately assessed with whole-genome sequencing.43 Hence, additional studies testing ECs developing in the context of pathogenic gBRCA1/2 mutations but lacking loss of the wild-type allele by whole-genome sequencing are warranted.
Despite these limitations, here we demonstrate the importance of germline and somatic genetic characterization of ECs. In fact, ECs developing in the context of pathogenic gBRCA1/2 mutations may harbor genomics features of HR deficiency and be causally linked to the loss of function of these tumor suppressor genes. However, the presence of a pathogenic gBRCA1/2 mutation does not rule out the possibility of an EC being sporadic and displaying DNA MMR deficiencies.
ACKNOWLEGEMENT
We thank the members of the Molecular Diagnostics Service in the Department of Pathology, Memorial Sloan Kettering Cancer Center.
Footnotes
Funded in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology and the National Cancer Institute Cancer Center Core Grant No. P30-CA008748. J.S.R.-F. is funded in part by the Breast Cancer Research Foundation, and B.W. by Cycle for Survival.
Presented in part as posters at the Society of Gynecologic Oncology Annual Meeting, Honolulu, HI, March 16-19, 2019; and the US and Canadian Academy of Pathology Annual Meeting, National Harbor, MD, March 16-21, 2019.
AUTHOR CONTRIBUTIONS
Conception and design: Evan S. Smith, Diana Mandelker, Britta Weigelt
Collection and assembly of data: Evan S. Smith, Arnaud Da Cruz Paula, Ana Paula Martins Sebastiao, Nadeem Riaz, Mark E. Robson, Robert A. Soslow, Diana Mandelker, Britta Weigelt
Data analysis and interpretation: Evan S. Smith, Arnaud Da Cruz Paula, Karen A. Cadoo, Nadeem R. Abu-Rustum, Xin Pei, David N. Brown, Lorenzo Ferrando, Nadeem Riaz, Mark E. Robson, Jorge S. Reis-Filho, Diana Mandelker, Britta Weigelt
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Karen A. Cadoo
Research Funding: AstraZeneca (Inst), Syndax (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Nadeem R. Abu-Rustum
Honoraria: Prime Oncology
Research Funding: Stryker/Novadaq (Inst), Olympus (Inst), GRAIL (Inst)
Travel, Accommodations, Expenses: Prime Oncology
Nadeem Riaz
Honoraria: PeerView
Speakers' Bureau: Illumina
Research Funding: Bristol-Myers Squibb, Pfizer
Travel, Accommodations, Expenses: Varian Medical Systems
Mark E. Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca
Research Funding: AstraZeneca (Inst), Myriad Genetics (Inst), InVitae (Inst), AbbVie (Inst), Tesaro (Inst), Medivation (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Pfizer
Other Relationship: Research to Practice, Clinical Care Options, Physician Education Resource
Robert A. Soslow
Speakers' Bureau: Ebix/Oakstone
Patents, Royalties, Other Intellectual Property: Methods for diagnosis and treatment of endometrial cancer. Assignee: 2015 The Regents of the University of California 8/18/2015 (Inst); Application: Compositions and methods for the diagnosis and treatment of ovarian cancers that are associated with reduced smarca4 gene expression or protein function (Inst); Royalties from published books: Cambridge University Press and Springer Publishing
Jorge S. Reis-Filho
Consulting or Advisory Role: Roche, Invicro, Ventana Medical Systems, Volition RX, Paige.AI, Goldman Sachs, Novartis
Britta Weigelt
Consulting or Advisory Role: Roche (I), Invicro (I), Ventana Medical Systems (I), Volition RX (I), Paige.AI (I), Goldman Sachs (I), Novartis (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 2.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 3.Foulkes WD, Knoppers BM, Turnbull C. Population genetic testing for cancer susceptibility: Founder mutations to genomes. Nat Rev Clin Oncol. 2016;13:41–54. doi: 10.1038/nrclinonc.2015.173. [DOI] [PubMed] [Google Scholar]
- 4.Segev Y, Iqbal J, Lubinski J, et al. The incidence of endometrial cancer in women with BRCA1 and BRCA2 mutations: An international prospective cohort study. Gynecol Oncol. 2013;130:127–131. doi: 10.1016/j.ygyno.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 5.Shu CA, Pike MC, Jotwani AR, et al. Uterine cancer after risk-reducing salpingo-oophorectomy without hysterectomy in women with BRCA mutations. JAMA Oncol. 2016;2:1434–1440. doi: 10.1001/jamaoncol.2016.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pennington KP, Walsh T, Lee M, et al. BRCA1, TP53, and CHEK2 germline mutations in uterine serous carcinoma. Cancer. 2013;119:332–338. doi: 10.1002/cncr.27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. doi: 10.1016/j.cell.2018.03.039. Huang KL, Mashl RJ, Wu Y, et al: Pathogenic germline variants in 10,389 adult cancers. Cell 173:355-370.e14, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riaz N, Blecua P, Lim RS, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8:857. doi: 10.1038/s41467-017-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maxwell KN, Wubbenhorst B, Wenz BM, et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat Commun. 2017;8:319. doi: 10.1038/s41467-017-00388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. doi: 10.1038/nm.4333. Zehir A, Benayed R, Shah RH, et al: Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23:703-713, 2017 [Erratum: Nat Med. 23:1004, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. doi: 10.1038/nature12113. Cancer Genome Atlas Research Network, Kandoth C, Schultz N, et al: Integrated genomic characterization of endometrial carcinoma. Nature 497:67-73, 2013 [Erratum: Nature 500:242, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashley CW, Da Cruz Paula A, Kumar R, et al. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol Oncol. 2019;152:11–19. doi: 10.1016/j.ygyno.2018.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. doi: 10.1016/j.cell.2018.02.060. Bailey MH, Tokheim C, Porta-Pardo E, et al: Comprehensive characterization of cancer driver genes and mutations. Cell 173:371-385.e18, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geyer FC, Li A, Papanastasiou AD, et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun. 2018;9:1816. doi: 10.1038/s41467-018-04128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pareja F, Lee JY, Brown DN, et al. The genomic landscape of mucinous breast cancer. J Natl Cancer Inst. 2019;111:737–741. doi: 10.1093/jnci/djy216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martelotto LG, De Filippo MR, Ng CK, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. 2015;237:179–189. doi: 10.1002/path.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8:174–183. doi: 10.1158/2159-8290.CD-17-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu B, Ye K, Zhang Q, et al. MSIsensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–1016. doi: 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. doi: 10.1200/PO.17.00084. Middha S, Zhang L, Nafa K, et al: Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis Oncol 10.1200/PO.17.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulhan DC, Lee JJ, Melloni GEM, et al. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51:912–919. doi: 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 26.Popova T, Manié E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 27.Modica I, Soslow RA, Black D, et al. Utility of immunohistochemistry in predicting microsatellite instability in endometrial carcinoma. Am J Surg Pathol. 2007;31:744–751. doi: 10.1097/01.pas.0000213428.61374.06. [DOI] [PubMed] [Google Scholar]
- 28.Xiong Z, Laird PW. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritterhouse LL, Nowak JA, Strickland KC, et al. Morphologic correlates of molecular alterations in extrauterine Müllerian carcinomas. Mod Pathol. 2016;29:893–903. doi: 10.1038/modpathol.2016.82. [DOI] [PubMed] [Google Scholar]
- 30.Soslow RA, Han G, Park KJ, et al. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod Pathol. 2012;25:625–636. doi: 10.1038/modpathol.2011.183. [DOI] [PubMed] [Google Scholar]
- 31.Amant F, Mirza MR, Koskas M, et al. Cancer of the corpus uteri. Int J Gynaecol Obstet. 2018;143(suppl 2):37–50. doi: 10.1002/ijgo.12612. [DOI] [PubMed] [Google Scholar]
- 32.Karnezis AN, Hoang LN, Coatham M, et al. Loss of switch/sucrose non-fermenting complex protein expression is associated with dedifferentiation in endometrial carcinomas. Mod Pathol. 2016;29:302–314. doi: 10.1038/modpathol.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramalingam P, Croce S, McCluggage WG. Loss of expression of SMARCA4 (BRG1), SMARCA2 (BRM) and SMARCB1 (INI1) in undifferentiated carcinoma of the endometrium is not uncommon and is not always associated with rhabdoid morphology. Histopathology. 2017;70:359–366. doi: 10.1111/his.13091. [DOI] [PubMed] [Google Scholar]
- 34.Nik-Zainal S, Morganella S. Mutational signatures in breast cancer: The problem at the DNA level. Clin Cancer Res. 2017;23:2617–2629. doi: 10.1158/1078-0432.CCR-16-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20:764–775. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franzese E, Centonze S, Diana A, et al. PARP inhibitors in ovarian cancer. Cancer Treat Rev. 2019;73:1–9. doi: 10.1016/j.ctrv.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site - when a biomarker defines the indication. N Engl J Med. 2017;377:1409–1412. doi: 10.1056/NEJMp1709968. [DOI] [PubMed] [Google Scholar]
- 38.Dizon DS, Dias-Santagata D, Bregar A, et al. Complete remission following pembrolizumab in a woman with mismatch repair-deficient endometrial cancer and a germline BRCA1 mutation. Oncologist. 2018;23:650–653. doi: 10.1634/theoncologist.2017-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cantrell LA, Blank SV, Duska LR. Uterine carcinosarcoma: A review of the literature. Gynecol Oncol. 2015;137:581–588. doi: 10.1016/j.ygyno.2015.03.041. [DOI] [PubMed] [Google Scholar]
- 40.Jones S, Stransky N, McCord CL, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun. 2014;5:5006. doi: 10.1038/ncomms6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao S, Bellone S, Lopez S, et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc Natl Acad Sci USA. 2016;113:12238–12243. doi: 10.1073/pnas.1614120113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones NL, Xiu J, Chatterjee-Paer S, et al. Distinct molecular landscapes between endometrioid and nonendometrioid uterine carcinomas. Int J Cancer. 2017;140:1396–1404. doi: 10.1002/ijc.30537. [DOI] [PubMed] [Google Scholar]
- 43.Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]