

Abstract
PURPOSE
BRCA1 or BRCA2 loss of function results in homologous recombination deficiency (HRD), which is targetable by poly (ADP-ribose) polymerase (PARP) inhibitors and other DNA-damaging agents. In cancers associated with germline BRCA1/2 alterations (BRCA1/2-associated cancers: breast, ovarian, pancreatic, prostate), BRCA1/2 alterations result in HRD and are biomarkers for PARP inhibitor use. In other (non–BRCA1/2-associated) cancer types, the association between BRCA1/2 alteration and HRD is less clear.
METHODS
A total of 234,154 tumor samples were sequenced by hybrid capture-based comprehensive genomic profiling. Somatic, germline, and zygosity status was determined computationally. BRCA1/2 alterations were classified as predicted germline/somatic and biallelic/monoallelic. Genome-wide loss of heterozygosity (gLOH) was evaluated as a marker of HRD.
RESULTS
BRCA1/2 alterations were observed at a 4.7% frequency. BRCA1/2 mutations were predicted germline in 57.4% of BRCA1/2-associated and 37.2% of non–BRCA1/2-associated cancers. The fraction of BRCA1/2-altered cases that were biallelic was 68.7%, with a higher biallelic fraction in BRCA1/2-associated (89.9%) versus non–BRCA1/2-associated cancers (43.6%). Differences in tissue distribution of biallelic BRCA1 versus BRCA2 alterations were noted, including a higher rate of biallelic BRCA2 alteration in prostate cancer. Biallelic BRCA1/2 alteration was observed at a 3.2% frequency (BRCA1/2-associated cancers, 8.9%; non–BRCA1/2-associated cancers, 1.3%) and > 1% frequency in at least 13 cancer types. Across cancer types, biallelic BRCA1/2 alteration was associated with increased gLOH versus monoallelic or wild-type BRCA1/2; predicted germline or somatic mutations were both associated with elevated gLOH.
CONCLUSION
Biallelic BRCA1/2 alterations were associated with elevated gLOH in diverse cancer types, including those not traditionally associated with BRCA1/2 cancer syndromes. Biomarker development for PARP inhibitors should integrate methods to distinguish biallelic from monoallelic BRCA1/2 status, and biallelic BRCA1/2 alteration should be broadly evaluated across cancer types as a biomarker for underlying HRD and PARP inhibitor sensitivity.
INTRODUCTION
BRCA1 and BRCA2 encode critical components of the homologous recombination (HR) DNA repair pathway that maintains genomic stability.1 Germline BRCA1/2 (gBRCA1/2) alterations are associated with elevated risk for breast, ovarian, pancreatic, and prostate cancer (BRCA1/2-associated cancers),2,3 and tumors that arise in BRCA1/2 mutation carriers have often lost the wild-type allele.4 Synthetic lethal interactions between BRCA1/2 loss of function and poly (ADP-ribose) polymerase inhibitors (PARPi) underlie development and regulatory approval of PARPi targeted therapy for ovarian, breast, and pancreatic cancer.1,5 Companion diagnostic testing for gBRCA1/2 and somatic BRCA1/2 (sBRCA1/2) alteration1 can guide PARPi therapy selection.
CONTEXT
Key Objective
BRCA1/2 loss-of-function alterations result in homologous recombination deficiency (HRD) and are biomarkers for poly (ADP-ribose) polymerase (PARP) inhibitor sensitivity in breast, ovarian, prostate, and pancreatic cancer. To determine the relevance of BRCA1/2 alterations across cancer types, we evaluated the pan-cancer landscape of BRCA1/2 alterations and their association with the genome-wide loss-of-heterozygosity (gLOH) marker of HRD.
Knowledge Generated
The fraction of BRCA1/2 alterations that were biallelic differed by cancer type and predicted germline/somatic status. BRCA1/2 alterations were most frequently biallelic in breast, ovarian, prostate, and pancreatic cancer; in other cancer types, 44% of BRCA1/2 alterations were biallelic. Across cancer types, biallelic BRCA1/2 alteration was associated with elevated gLOH compared with monoallelic or wild-type BRCA1/2; this association with HRD was observed irrespective of predicted germline or somatic status.
Relevance
BRCA1/2 biallelic alteration is associated with HRD across tumor types and should be broadly evaluated as a biomarker in trials of PARP inhibitors and other agents that target HRD.
BRCA1 or BRCA2 loss-of-function results in HR deficiency (HRD) and accumulation of chromosomal rearrangements and copy number alterations. BRCA1 and BRCA2 have distinct functions in the homology-mediated repair process, and their inactivation leads to different patterns of rearrangements, with BRCA1 loss of function associated with tandem duplications and BRCA2 loss of function associated with deletions.6,7 However, the genomic impact of either BRCA1 or BRCA2 loss of function can be measured using the genome-wide loss-of-heterozygosity (gLOH) signature of HRD.1,8 In clinical trials of ovarian cancer, high gLOH (gLOH-high) was associated with improved benefit from the PARPi rucaparib; therefore, gLOH measurement may guide therapeutic decision making.9,10
Emerging data from clinical trials suggest that BRCA1/2 genomic alteration status may also be a predictive biomarker for PARPi in prostate cancer.11-13 However, PARPi has limited activity in other cancer types with BRCA1/2 alteration.14-16 Here, we assessed a genomic data set of 234,154 tumor specimens to determine the landscape of BRCA1/2 biallelic alterations and their association with gLOH to understand the potential clinical relevance of BRCA1/2 alterations across cancer types.
METHODS
Approval for this study, including a waiver of informed consent and Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (protocol #20152817). Comprehensive genomic profiling (CGP) using hybrid capture-based next-generation sequencing (NGS) was performed on tumor tissue specimens (N = 234,154) submitted to Foundation Medicine during routine clinical care17 (December 2013-March 2019; Data Supplement). BRCA1/2 genomic alterations were defined (Appendix) as likely pathogenic alterations or variants of unknown significance (VUSs; not counted as BRCA1/2 alterations). Zygosity and somatic/germline status for mutations was computationally predicted without matched normal tissue; in validation testing of 480 tumor-only predictions against matched normal specimens, accuracy was 95% for somatic and 99% for germline predictions.18 BRCA1/2 alterations were categorized as biallelic (mutations with LOH of the wild-type allele,18 homozygous deletion, or two or more BRCA1 or BRCA2 alterations in a sample), monoallelic (heterozygous mutations with retained wild-type allele),18 or unknown (alterations where zygosity status could not be determined). Percent gLOH was calculated as a signature of HRD as previously described.9,10 See the Appendix for full methods.
RESULTS
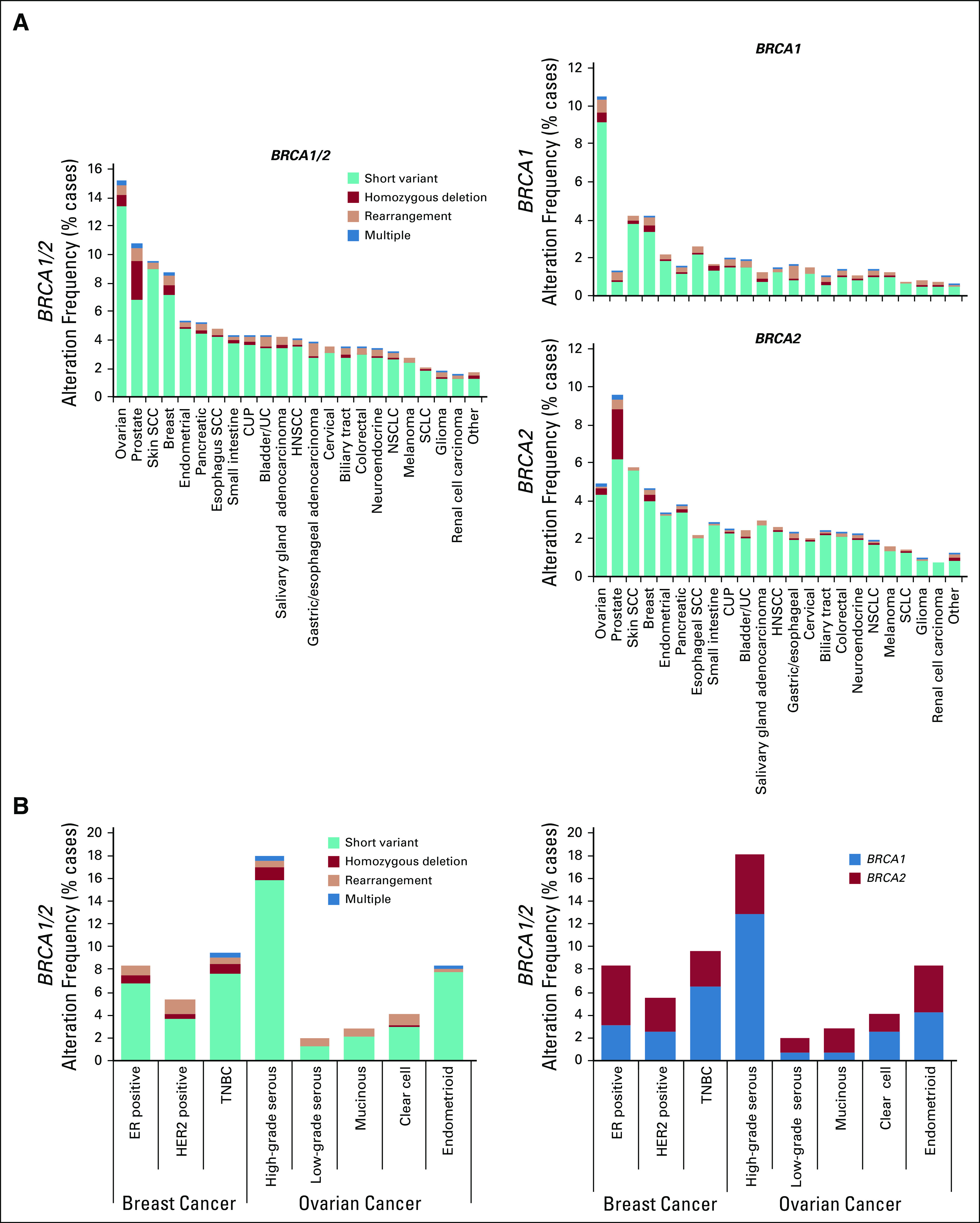
To assess the prevalence of BRCA1/2 genomic alterations across cancer types, we examined CGP results from 234,154 tumors sequenced as part of routine clinical care. Overall, BRCA1/2 alterations were observed in 4.7% of cases (BRCA1, 2.1%; BRCA2, 2.7%; Fig 1A; Appendix Fig A1). As expected, BRCA1/2 alterations were most frequently identified in BRCA1/2-associated cancers (BRCA1/2-associated group, 9.9%; ovarian, 15.2%; prostate, 10.7%; breast, 8.8%; pancreatic, 5.2%). BRCA1 and BRCA2 alterations were most frequent in ovarian (10.5%) and prostate cancer (9.6%), respectively; unlike BRCA2, BRCA1 alterations were infrequent in prostate cancer. BRCA1/2 homozygous deletions were infrequent except in prostate cancer, where BRCA2 deletions were observed at a 2.6% frequency and accounted for 25% of BRCA1/2-altered cases. Across non–BRCA1/2-associated cancers, BRCA1/2 alterations were observed at a 3.0% frequency overall and > 1% frequency in each individual cancer type assessed. BRCA1/2 mutations were distributed throughout the length of each gene, and most were truncating events (Appendix Fig A2).
FIG 1.

Pan-cancer landscape of BRCA1/2 alterations. (A) Frequency of BRCA1 and BRCA2 alterations across multiple cancer types. Cancer types with ≥ 40 BRCA1/2-altered cases are shown, including ovarian cancer (n = 14,256), prostate cancer (n = 7,185), skin squamous cell carcinoma (SCC; n = 661), breast cancer (n = 21,164), endometrial cancer (n = 7,182), pancreatic cancer (n = 12,248), esophageal SCC (n = 836), small intestine cancer (n = 1,145), cancer of unknown primary (CUP; n = 11,130), bladder/urothelial cancer (UC; n = 4,718), salivary gland adenocarcinoma (n = 1,075), head and neck SCC (HNSCC; n = 2,921), gastric/esophageal adenocarcinoma (n = 8,061), cervical cancer (n = 1,694), biliary tract cancer (n = 6,003), colorectal cancer (n = 25,784), neuroendocrine cancer (n = 4,573), non–small-cell lung cancer (NSCLC; n = 43,242), melanoma (n = 6,016), small-cell lung cancer (SCLC; n = 2,262), glioma (n = 8,635), and renal cell carcinoma (n = 3,330); all other cancer types were analyzed as a group labeled other (n = 40,033). See the Data Supplement for detailed data. (B and C) Predicted germline/somatic status was determined computationally for BRCA1/2 short variant mutations. Fraction (%) of (B) BRCA1 or (C) BRCA2 mutations predicted to be germline v somatic was determined for each cancer type. See the Data Supplement for detailed data.
Germline/somatic status for BRCA1/2 short variant mutations was predicted using validated computational methods.18 We also evaluated performance of germline/somatic predictions in this study. First, in a subset of 23 tumor samples from Rutgers Cancer Institute of New Jersey that arose in patients with gBRCA1/2 variants identified by genetic testing, computational methods correctly identified 21 (91%) as predicted germline variants. Second, because cell-free DNA (cfDNA) sequencing can often distinguish germline from somatic variants,19 we evaluated 52 BRCA1/2 germline/somatic predictions from tissue samples and evaluated patient-matched cfDNA NGS results. Overall, 98.1% of mutations (51 of 52) were observed in cfDNA at allele frequencies consistent with germline/somatic predictions from tumor-only sequencing (Appendix Figs A3A and A3B).
Overall, 47.8% of BRCA1/2 mutations (BRCA1, 51.6%; BRCA2, 45.3%) were predicted to be germline (Figs 1B and 1C; Appendix Figs A3C-A3E). As expected, the majority of mutations were predicted to be germline in BRCA1/2-associated cancers (BRCA1, 58.1%; BRCA2, 56.8%), but predicted sBRCA1/2 mutations comprised an appreciable proportion of BRCA1/2 mutations. In prostate cancer, 51.7% of BRCA2 v 34.4% of BRCA1 mutations were predicted to be germline. In non–BRCA1/2-associated cancer types, BRCA1/2 mutations were less frequently predicted to be germline (37.2%). Predicted somatic mutations were frequent in skin squamous cell carcinoma (SCC) and melanoma, which accounted for 90.7% and 75.8% of BRCA1/2 mutations, respectively. As expected, predicted sBRCA1/2 mutations in these cancer types were often found in a mutational context of ultraviolet light exposure (skin SCC, 45% [14 of 31]; melanoma, 81% [50 of 62]).
We determined whether BRCA1/2 alterations were likely to affect a single allele (monoallelic) or both alleles (biallelic; Fig 2A; Appendix Fig A4). For cases where biallelic/monoallelic status could be determined, we estimated the fraction of BRCA1/2-altered cases with biallelic alteration (biallelic fraction). BRCA1/2 biallelic fraction was 68.7% overall and highest in BRCA1/2-associated cancers (BRCA1/2-associated group, 89.9%; ovarian, 94.9%; prostate, 87.3%; breast, 87.9%; pancreatic, 79.4%). Although biallelic fraction was lower in non–BRCA1/2-associated cancers (P < .0001, Fisher’s exact test), biallelic alterations nonetheless comprised 43.6% of BRCA1/2-altered cases: Biallelic fraction was > 50% for esophageal SCC, cancer of unknown primary, and biliary tract cancer.
FIG 2.
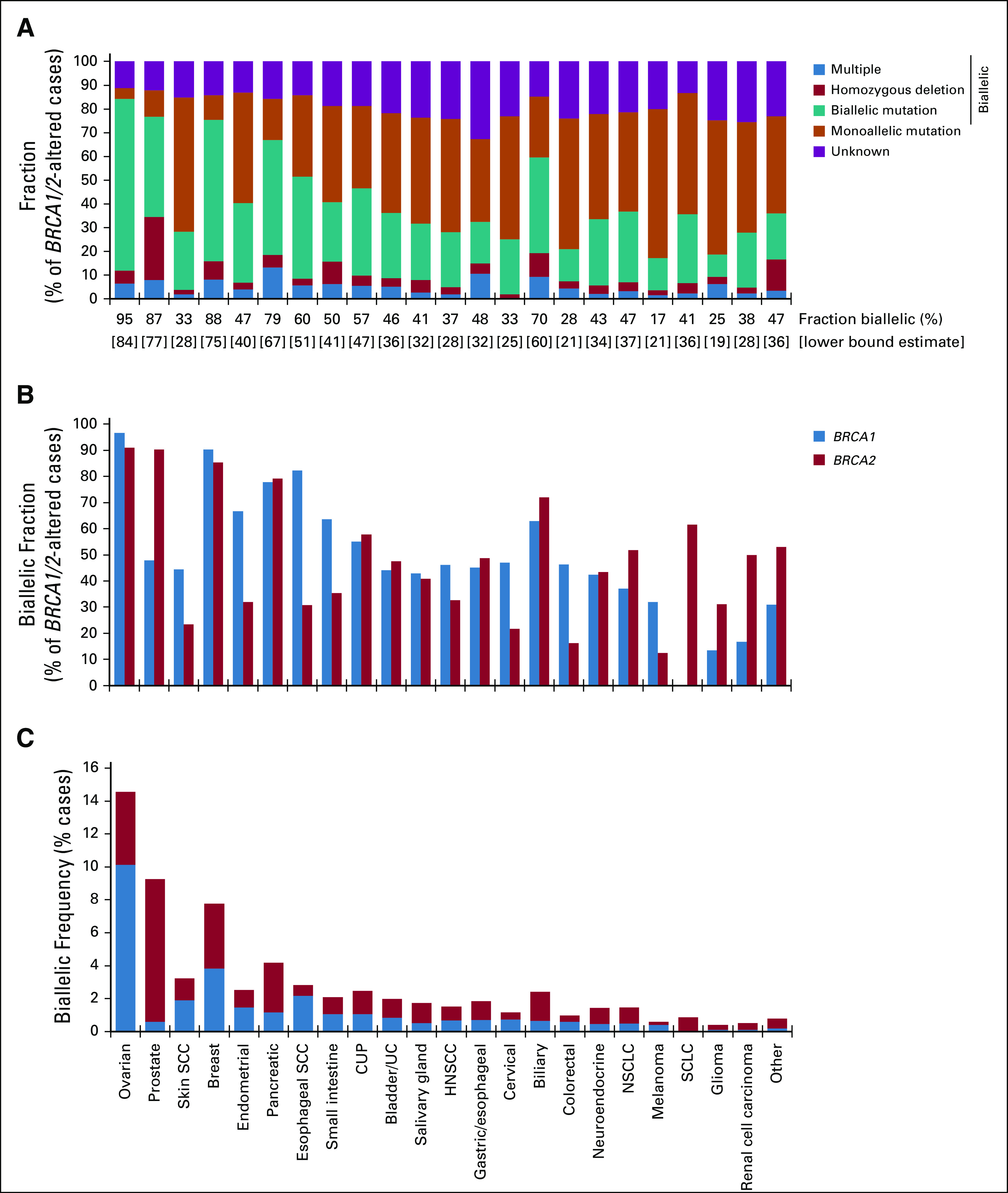
Assessment of BRCA1 and BRCA2 biallelic status. (A) Fraction of BRCA1/2-altered cases with biallelic or monoallelic alteration was determined. BRCA1/2-altered cases were evaluated for class of alteration identified and classified as biallelic (multiple BRCA1 or multiple BRCA2 alterations in the same sample, homozygous deletion, biallelic short variant mutation [loss of heterozygosity of the wild-type allele]), monoallelic (heterozygous mutation), or unknown (zygosity status could not be determined). Biallelic fraction (percentage of BRCA1/2-altered cases with biallelic alteration) was determined for cases where biallelic/monoallelic status could be called. A lower-bound estimate was established by assessing biallelic cases as a fraction of all BRCA1/2-altered cases, including those with unknown biallelic/monoallelic status (see Data Supplement). (B) Biallelic fraction was compared for BRCA1- v BRCA2-altered cases for each cancer type (see Data Supplement). (C) Overall frequency of biallelic BRCA1 and BRCA2 alterations across multiple cancer types (see Data Supplement). CUP, cancer of unknown primary; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; UC, urothelial cancer.
FIG 4.
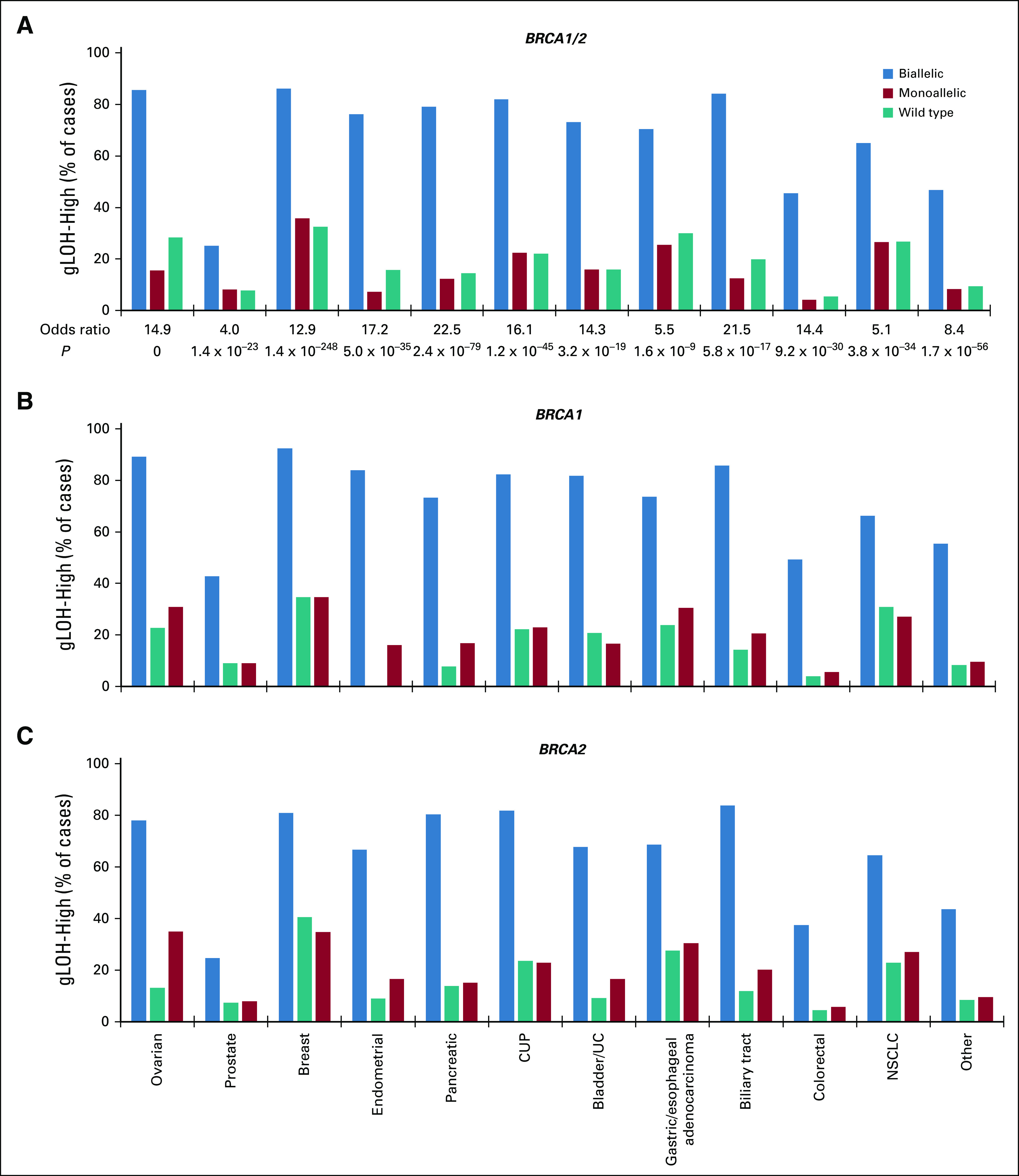
Association between BRCA1/2 alteration status and high genome-wide loss of heterozygosity (gLOH-high). The frequency of (A) BRCA1/2, (B) BRCA1, or (C) BRCA2 biallelic, monoallelic, and wild-type cases that were gLOH-high was compared across cancer types. Cancer types with > 50 biallelic BRCA1/2-altered samples were assessed individually, and all other cancer types were grouped together and analyzed as a single group (other). Odds ratios and P values (Fisher’s exact test) were for comparisons between biallelic and wild-type BRCA1/2 cases (see Data Supplement). gLOH-high, was defined as ≥ 16% genome-wide loss of heterozygosity cutoff. CUP, cancers of unknown primary; NSCLC, non–small-cell lung cancer; UC, urothelial cancer.
Biallelic fraction was compared for BRCA1 versus BRCA2 (Fig 2B). BRCA2 biallelic fraction was greater than BRCA1 in prostate cancer (odds ratio [OR], 10.1; fold difference, 1.9; P = 1.5 × 10−11) and small-cell lung cancer (biallelic fraction, 61.5% v 0% for BRCA2 v BRCA1, respectively; P = 2.8 × 10−4), whereas BRCA1 biallelic fraction was greater than BRCA2 in endometrial cancer (OR, 4.2; fold difference, 2.1; P = 8.4 × 10−8), esophageal SCC (OR, 10.5; fold difference, 2.7; P = .008), colorectal cancer (OR, 4.5; fold difference, 2.9; P = 7.1 × 10−13), and melanoma (OR, 3.3; fold difference, 2.6; P = .02).
To determine the frequency of BRCA1/2 loss of function across cancer types, we evaluated biallelic BRCA1/2-altered cases as a percentage of all cases (Fig 2C; Appendix Figs A4C-A4F). Biallelic BRCA1/2 alteration was found in 3.2% of all cases and greatest in BRCA1/2-associated cancers (8.9%). Although occurring at a lower frequency in non–BRCA1/2-associated cancers (1.3%), biallelic BRCA1/2 alteration was observed at > 1% frequency in at least 13 cancer types.
Predicted gBRCA1/2 and sBRCA1/2 mutations were separately assessed for biallelic fraction (Fig 3; Appendix Fig A5). Overall, 75.4% of predicted gBRCA1/2 mutations were biallelic v 48.5% of sBRCA1/2 mutations. For BRCA1/2-associated cancers, both predicted gBRCA1/2 (90.8%) and sBRCA1/2 (81.2%) mutations were frequently biallelic, whereas in non–BRCA1/2-associated cancers, fewer predicted gBRCA1/2 (46.4%) and sBRCA1/2 (25.4%) mutations were biallelic. Among BRCA1/2-associated cancers, cancer type–specific differences were observed. In ovarian and breast cancer, the majority of BRCA1 and BRCA2 mutations were biallelic, both for predicted germline and somatic mutations. In prostate cancer, the majority of BRCA2 (gBRCA2, 87.6%; sBRCA2, 75.0%) mutations were biallelic, but BRCA1 (gBRCA1, 40.0%; sBRCA1, 22.2%) mutations were less frequently biallelic. In pancreatic cancer, predicted gBRCA1/2 mutations were frequently biallelic (gBRCA1, 79.2%; gBRCA2, 79.7%) compared with sBRCA1/2 mutations (sBRCA1, 52.9%; sBRCA2, 46.0%). Among non–BRCA1/2-associated cancers, predicted gBRCA1 mutations were most frequently biallelic in endometrial (87.1%), unknown primary (66.7%), bladder/urothelial (63.6%), and neuroendocrine (60.0%) cancer; predicted gBRCA2 mutations were most frequently biallelic in salivary gland (85.7%), unknown primary (71.7%), biliary tract (65.0%), and endometrial (58.1%) cancer; and predicted sBRCA1/2 mutations were infrequently biallelic except for biliary tract cancer (sBRCA1, 50.0% [2 of 4]; sBRCA2, 61.5% [24 of 39]), endometrial cancer (sBRCA1, 48.7% [19 of 39]), and esophageal SCC (sBRCA1, 80% [4 of 5]; Appendix Figs A5B and A5C).
FIG 3.
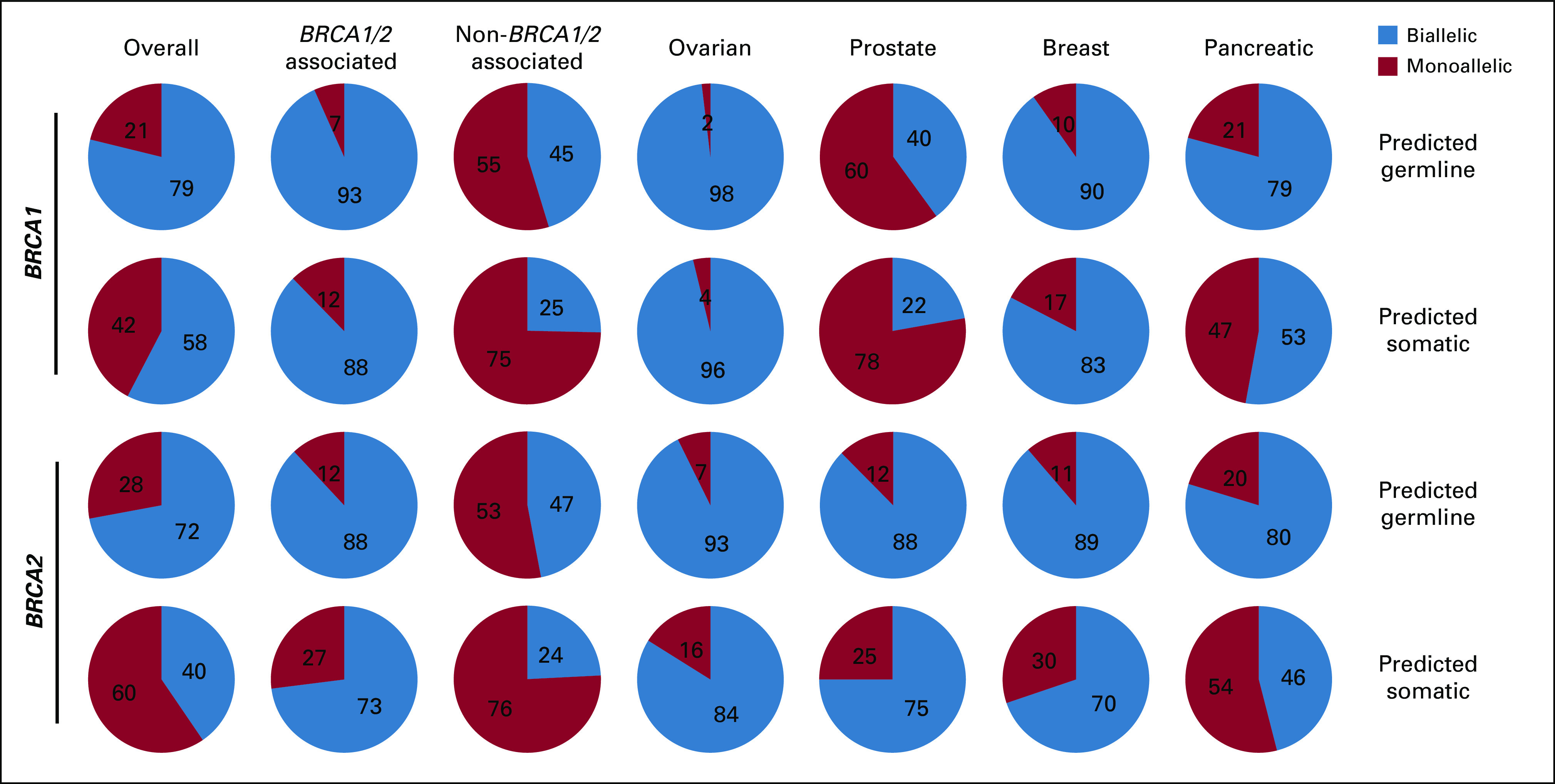
Biallelic and monoallelic distribution for germline BRCA1/2 and somatic BRCA1/2 mutations. BRCA1 and BRCA2 mutations with a germline or somatic prediction were evaluated for biallelic/monoallelic status for all cancers, BRCA1/2-associated cancers (as a group and as individual cancer types), and non–BRCA1/2-associated cancers (see Data Supplement).
We next determined whether BRCA1/2 status broadly associated with the gLOH signature of HRD. Cases with ≥ 16% gLOH were classified as gLOH-high on the basis of the cutoff established in the ARIEL3 trial of rucaparib in ovarian cancer.10 Biallelic BRCA1/2 alterations were associated with gLOH-high across every cancer type examined. The fraction of cases that were gLOH-high was significantly increased for biallelic BRCA1/2-altered compared with wild-type cases (Fig 4A, Appendix Fig A6A), whereas the fraction of monoallelic BRCA1/2 cases that were gLOH-high was similar to wild type. Significant association of biallelic but not monoallelic alterations with gLOH-high was observed both for BRCA1 and for BRCA2 when assessed individually (Figs 4B and 4C). Biallelic BRCA1/2 mutations were associated with gLOH-high irrespective of predicted germline or somatic status (Appendix Figs A6B and A6C). In some cancer types, significant but modest elevation in tumor mutational burden (TMB) was observed for biallelic BRCA1/2-mutated cases (predicted germline or somatic) versus wild type; however, the association with TMB was not consistent across cancer types (Appendix Fig A7). Monoallelic sBRCA1/2-mutated (but not gBRCA1/2) cases were commonly associated with elevated TMB versus wild type, and such mutations may be a consequence of increased mutation rate.
Although biallelic BRCA1/2 status was consistently associated with gLOH-high, the magnitude was variable for each cancer type, with the greatest association observed in pancreatic (OR, 22.5), biliary (OR, 21.5), endometrial (OR, 17.2), unknown primary (OR, 16.1), and ovarian (OR, 14.9) cancer (Fig 4A). More than 75% of cases with biallelic BRCA1/2 alterations were gLOH-high for ovarian, breast, pancreatic, unknown primary, and endometrial cancer, whereas fewer than half were gLOH-high for prostate and colorectal cancer. Conversely, > 25% of BRCA1/2 wild-type cases were gLOH-high for ovarian, breast, lung, and gastric/esophageal cancer.
The ≥ 16% gLOH-high cutoff was clinically validated in ovarian cancer9,10 and requires optimization for other cancer types. Therefore, gLOH was also assessed as a continuous variable (Figs 5A-5C). Consistent with the findings using a gLOH cutoff-based approach, gLOH scores were higher in BRCA1/2 biallelic versus wild-type cases across all cancer types evaluated; increased gLOH score was also observed when biallelic BRCA1 and BRCA2 were evaluated independently.
FIG 5.
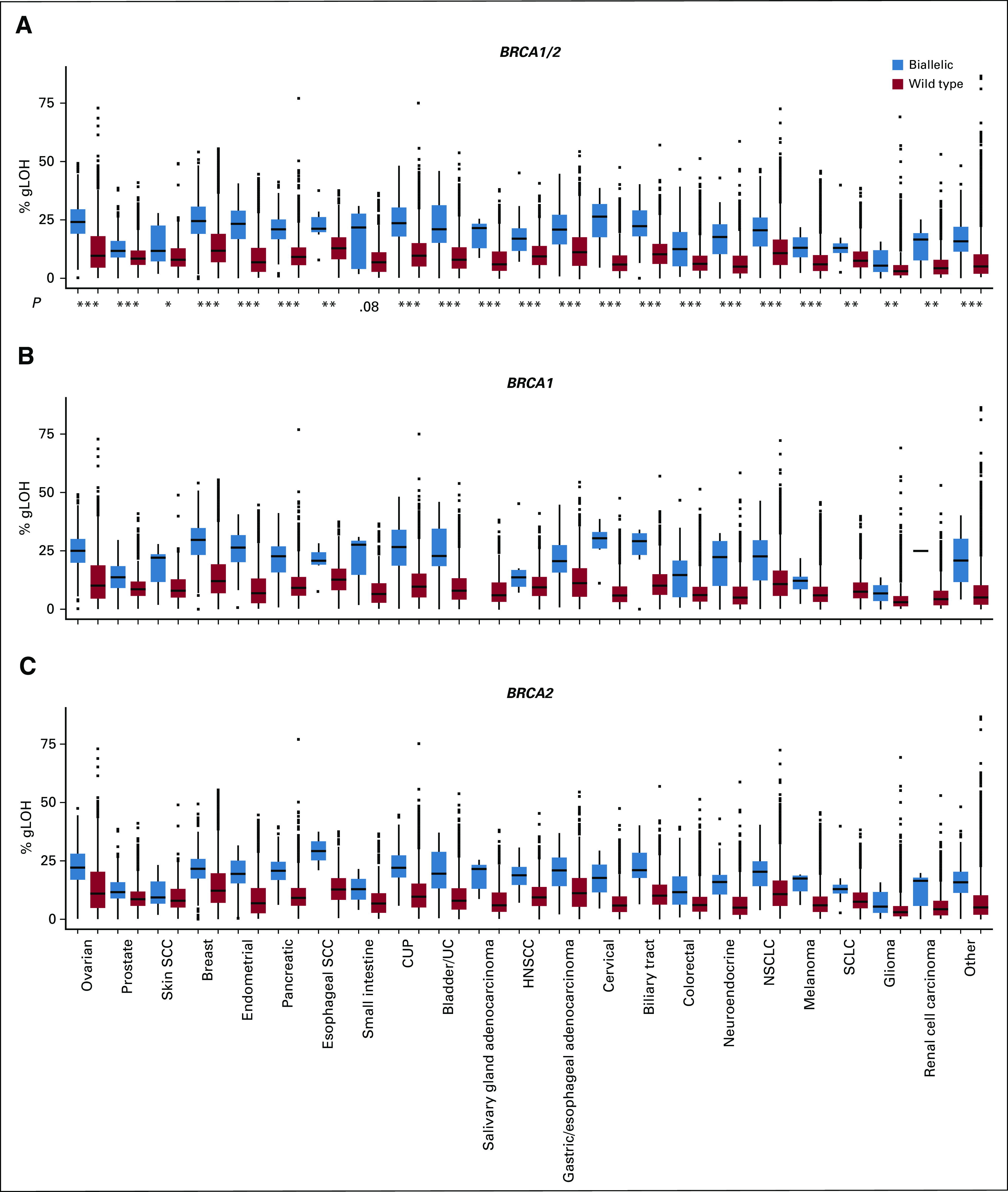
Association between BRCA1/2 biallelic alteration and genome-wide loss of heterozygosity (gLOH) score as a continuous variable. gLOH score was assessed in (A) BRCA1/2, (B) BRCA1, and (C) BRCA2 biallelic v wild-type cases across cancer types. Boxes span the first and third quartiles, and the median is denoted by the horizontal line in the box; whiskers indicate maximum and minimum values within 1.5× the interquartile range; black dots indicate outlier events. P values are by Mann-Whitney U test for comparison between biallelic and wild-type BRCA1/2 cases. *P < .05, **P < .01, ***P < .001 (see Data Supplement). CUP, cancer of unknown primary; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; UC, urothelial cancer.
HRD signatures (including gLOH-high) may identify additional BRCA1/2 wild-type tumors potentially suitable for PARPi.9,10,20 Overall, 19.3% of cases were gLOH-high compared with 3.2% of cases with biallelic BRCA1/2 alteration, and for most cancer types, the frequency of gLOH-high was greater than biallelic BRCA1/2 alterations (Data Supplement); distribution of gLOH scores varied between cancer types. To inform rational cancer type–specific gLOH-high cutoffs, we assessed the performance of different gLOH-high thresholds to classify biallelic BRCA1/2 compared with wild-type cases. Plotting sensitivity to detect cases with biallelic BRCA1/2 alteration and specificity (percentage of BRCA1/2 wild-type cases negative for the gLOH-high biomarker), we identified a cutoff that maximized the combined sensitivity and specificity score (Fig 6). For most cancers, the gLOH-high cutoff ranged between 14% and 16%, except for prostate (8.8% cutoff), breast (16.6% cutoff), biliary tract (17.6% cutoff), and gastric (16.7% cutoff) cancer. The identified cutoff for ovarian cancer was 15.1%, which was consistent with the 14% and 16% cutoffs identified in clinical trials.9,10
FIG 6.
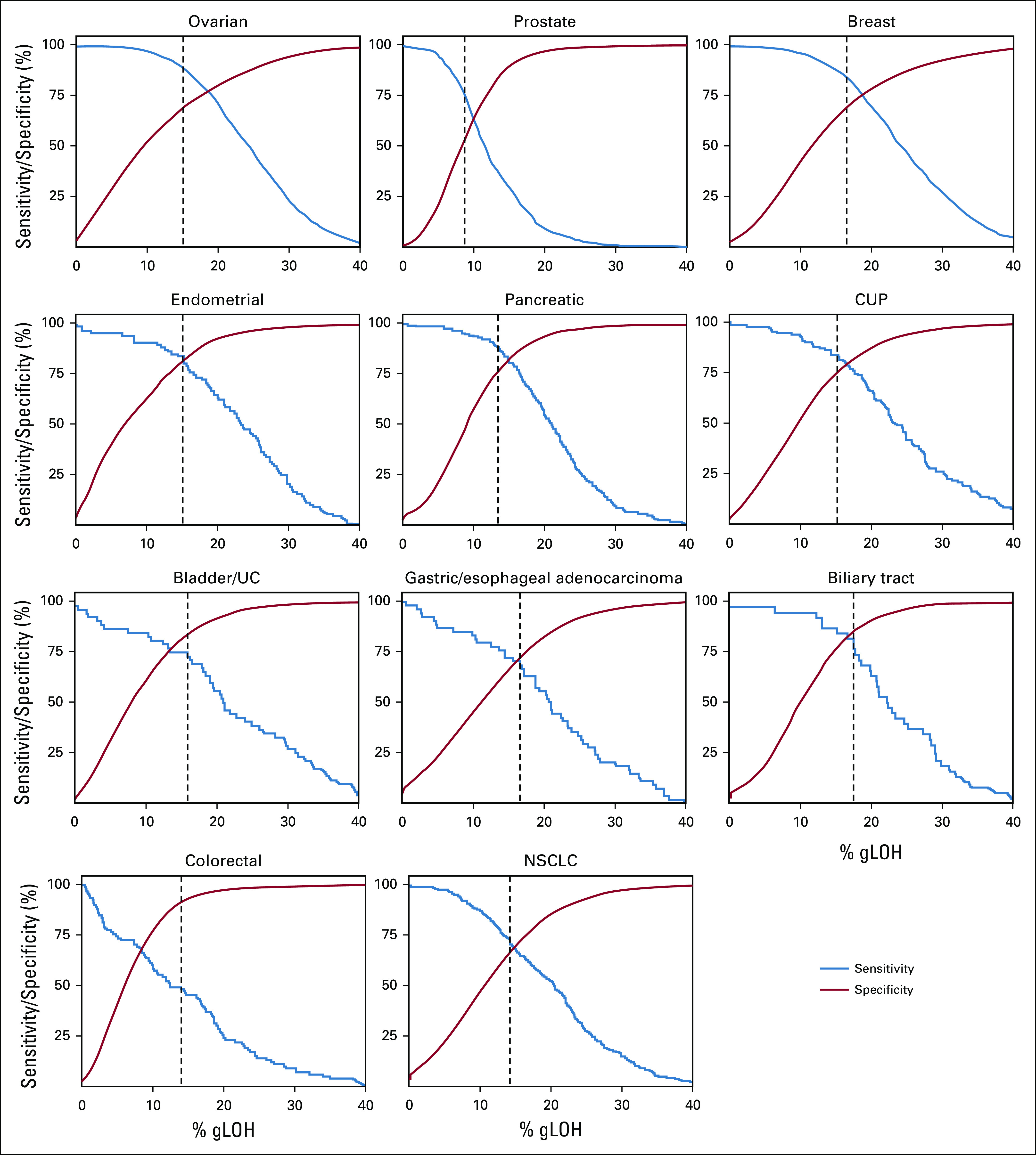
Sensitivity and specificity to classify BRCA1/2 biallelic alterations v wild type were evaluated for different genome-wide loss of heterozygosity (gLOH) cutoffs. Sensitivity was defined as percentage of biallelic BRCA1/2 cases that were of high gLOH (gLOH-high). Specificity was defined as percentage of BRCA1/2 wild-type cases that were negative for the gLOH-high biomarker. A cutoff that maximized the combined sensitivity and specificity score was identified (dotted black line; see Data Supplement). CUP, cancer of unknown primary; NSCLC, non–small-cell lung cancer.
DISCUSSION
The development of PARPi for BRCA1/2 altered ovarian, breast, and pancreatic cancer1,5 is predicated on synthetic lethality interactions between BRCA1/2 loss of function and PARPi/trapping. Emerging clinical trial data suggest that BRCA1/2 alteration may also be predictive of PARPi response in prostate cancer.1,11-13 Although responses to PARPi have been documented in other cancer types,14,21-23 the relevance of BRCA1/2 alterations in non–BRCA1/2-associated cancers remains unclear.4
To understand the landscape and phenotypic consequence of BRCA1/2 alterations, we assessed our data set of 234,154 cancer specimens sequenced using a clinical-grade CGP assay. BRCA1/2 alterations were frequent in BRCA1/2-associated cancers but also observed in a significant fraction (3%) of non–BRCA1/2-associated cancers. Predicted germline mutations comprised the majority of BRCA1/2 mutations in BRCA1/2-associated cancers; however, it is notable that 43% were predicted somatic (of which 81% were biallelic) given data that support sBRCA1/2 alteration as a biomarker for PARPi in ovarian and prostate cancer.1,11-13
BRCA1/2 alterations were assessed for biallelic versus monoallelic status to distinguish likely loss of function from biologically neutral alterations. Of note, although monoallelic alteration in BRCA1/2 mutation carriers may have a subtle haploinsufficient phenotype,24 it does not lead to severe HRD or sensitivity to platinum-based chemotherapy.25 Consistent with the established role of BRCA1/2 in the pathogenesis of BRCA1/2-associated cancers, 90% of BRCA1/2-altered cases were biallelic with high biallelic fraction both for predicted gBRCA1/2 and sBRCA1/2. In non–BRCA1/2-associated cancers, biallelic inactivation still occurred in a significant portion (44%) of BRCA1/2-altered cases. Differences in BRCA1 and BRCA2 biallelic fraction were observed: BRCA2 was more frequently biallelic in prostate and small-cell lung cancer, and BRCA1 was more frequently biallelic in endometrial cancer, esophageal SCC, colorectal cancer, and melanoma. These differences suggest that BRCA1 and BRCA2 may have different tissue specificities outside breast and ovarian cancer.
To determine whether BRCA1/2 alterations lead to HRD in both BRCA1/2-associated and non–BRCA1/2-associated cancers, we evaluated gLOH in BRCA1/2-altered versus wild-type cases. Across every cancer type evaluated, biallelic BRCA1/2 alteration was associated with increased gLOH. Therefore, biallelic BRCA1/2 alterations broadly result in the gLOH signature for HRD and may represent a therapeutic vulnerability targetable by PARPi. In contrast, monoallelic BRCA1/2 alterations were not associated with elevated gLOH and are likely biologically neutral for HR. Monoallelic alterations may be found in sporadic cancers from gBRCA1/2 carriers or as somatic passenger mutations.
Distinguishing biallelic from monoallelic status may be an important consideration for refining BRCA1/2 alteration as a predictive biomarker for PARPi. In BRCA1/2-associated cancers, BRCA1/2 alteration status alone has proven sufficient as a predictive biomarker,1 likely explained by the majority of BRCA1/2 alterations in this context being biallelic. Nevertheless, PARPi trials in prostate cancer have incorporated BRCA1/2 biallelic status into biomarker development,13 and our finding of significantly lower biallelic fraction for BRCA1 (v BRCA2) in prostate cancer suggests that integrating biallelic status could refine predictive biomarkers in this setting.
The lack of PARPi clinical activity in non–BRCA1/2-associated cancers reported previously could be explained by grouped analysis of biallelic and monoallelic BRCA1/2 alterations.4 Because of lower rates of biallelic alteration in non–BRCA1/2-associated cancers, PARPi clinical trials will likely require patient selection strategies that incorporate methods to discriminate biallelic from monoallelic BRCA1/2 alterations.
Our findings are consistent with a recent study that demonstrated an elevated HRD composite score in biallelic but not heterozygous BRCA1/2 cases relative to wild type in an aggregate set of non–BRCA1/2-associated cancers.4 A strength of the current study was that the large data set size enabled analysis of biallelic BRCA1/2 separately from monoallelic alterations, independent assessment of BRCA1 and BRCA2, and evaluation of non–BRCA1/2-associated cancers as individual cancer types rather than in aggregate, which may have enabled identification of associations between biallelic BRCA1/2 alterations and an HRD signature across cancer types not previously described.4
If BRCA1/2 biallelic alterations functionally result in HRD, they should represent a targetable vulnerability to PARPi and platinum-based chemotherapy, irrespective of whether they are drivers of disease pathogenesis or bystander passenger alterations.26 Our data demonstrate that biallelic BRCA1/2 alteration in non–BRCA1/2-associated cancers are associated with the gLOH-high signature of HRD and, therefore, warrant investigation in PARPi trials. Biallelic BRCA1/2 alteration was observed in 1.6% of non–BRCA1/2-associated cancer and at > 1% frequency in at least 13 cancer types. Although biallelic BRCA1/2 alterations occur at low prevalence in non–BRCA1/2-associated cancers, basket trials that led to the tumor-agnostic approvals of NTRK inhibitors for NTRK fusion-positive tumors and pembrolizumab for microsatellite instability–high tumors27 demonstrate feasibility of therapeutic development for rare pan-cancer biomarkers. Clinical trials such as the TAPUR study (ClinicalTrials.gov identifier: NCT02693535) that includes a PARPi treatment arm for BRCA1/2-altered cancers will inform PARPi development in non–BRCA1/2-associated cancers, and analyses may benefit from consideration of monoallelic versus biallelic status.
Although gLOH-high is associated with clinical benefit from rucaparib in ovarian cancer,9,10 understanding of the gLOH biomarker in other cancer types is required. Evaluation of sensitivity and specificity of varying gLOH thresholds to distinguish biallelic BRCA1/2-altered and wild-type cases may inform development of disease-specific gLOH-high cutoffs. In future studies, analysis of BRCA1/2 wild-type, gLOH-high cases may be a discovery tool for characterizing BRCA1/2 VUSs and for prioritizing candidate non-BRCA1/2 HR pathway gene biomarkers.9 Although detection of gLOH-high in BRCA1/2 wild-type cases potentially expands the patient population addressable by PARPi, the utility of gLOH-high requires validation in prospective trials for nonovarian cancers.
Limitations of this study should be acknowledged. First, we focus on gLOH as a biomarker for HRD; other HRD markers were not evaluated, including telomeric allelic imbalance (TAI), large-scale transition (LST), myChoice HRD (combination LOH/TAI/LST score; Myriad Genetics, Salt Lake City, UT), Signature 3, and HRDetect.7,8,28-33 In PARPi trials, HRD biomarkers have focused on approaches that are compatible with targeted NGS assays (gLOH, myChoice HRD), which are used in routine clinical practice.1,9,10 In contrast, Signature 3 and HRDetect signatures have been evaluated in the research setting using whole-exome or whole-genome sequencing; novel methods may enable future clinical assessment of Signature 3 with targeted assays.34 Second, germline/somatic status predictions using tumor-only sequencing and computational methods are less definitive compared with matched normal sequencing used in other studies.4,18 Third, using BRCA1/2 biallelic versus wild-type status to refine gLOH-high cutoffs is confounded by some BRCA1/2 wild-type gLOH-high cases that are HRD because of BRCA1/2 alteration–independent mechanisms, such as BRCA1/2 methylation or alteration in other HR genes. Another study evaluated a group of 102 HR pathway genes and found that biallelic alterations were associated with HRD.35 Other HR pathway genes sequenced here could inform gLOH-high thresholds in the future; however, we focused on BRCA1/2 because other HR pathway genes have not been consistently predictive of clinical response to PARPi,11-13,36 and robust clinical evidence supporting predictive biomarker genes beyond BRCA1/2 is lacking. Finally, clinical outcomes data that associate PARPi response with biallelic BRCA1/2 alteration and gLOH were not available and require evaluation in clinical trials.
We demonstrate that biallelic BRCA1/2 alterations are associated with elevated gLOH across all cancer types evaluated and may therefore represent a therapeutic vulnerability targetable by PARPi. Biomarker development for PARPi should reliably distinguish biallelic/monoallelic BRCA1/2 status, and biallelic BRCA1/2 alteration should be broadly evaluated as a pan-cancer biomarker in PARPi clinical trials.
Appendix
Supplementary Methods
Approval for this study, including a waiver of informed consent and Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (IRB; protocol #20152817). Comprehensive genomic profiling (CGP) was performed using hybrid capture-based next-generation sequencing (NGS) (median coverage, > 790×; Data Supplement) in a Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited, New York State–approved laboratory (Foundation Medicine; Frampton GM, et al: Nat. Biotechnol 31:1023-1031, 2013). CGP results included in this study were from tumor tissue specimens (N = 234,154) submitted as part of routine clinical care (December 2013-March 2019); for patient characteristics, see the Data Supplement. Results were analyzed for base substitutions, short insertions/deletions, rearrangements, and copy number alterations. BRCA1/2 genomic alterations were defined as likely pathogenic alterations (protein-truncating mutations/rearrangements [except for BRCA2 truncations that occur at K3326 or downstream], homozygous deletions, or characterized missense mutations [mutations designated as pathogenic in BRCA Exchange/ENIGMA consortium (Cline MS, et al: PLoS Genet doi:10.1371/journal.pgen.1007752, 2018) or consensus pathogenic in ClinVar, mutations included in the ARIEL3 trial (Coleman RL, et al: Lancet 390:1949-1961, 2017), and functionally characterized mutations]); other alterations that were classified as variants of unknown significance (VUSs) were not included as BRCA1/2 genomic alterations in the analysis. Zygosity and somatic/germline status for mutations were computationally predicted without matched normal tissue. In validation testing of 480 tumor-only sequencing calls against matched normal specimens, accuracy was 95% for somatic and 99% for germline calls (Sun JX, et al: PLOS Comput Biol 14:e1005965, 2018); in assessment of zygosity calls, significant enrichment in mutations with loss of heterozygosity (LOH) was observed for tumor suppressor genes (Sun JX, et al: PLOS Comput Biol 14:e1005965, 2018). To evaluate performance of germline/somatic computational predictions in this series, we compared predictions against a subset of cases with available results from patient-matched germline testing or cell-free DNA (cfDNA) NGS that were performed as previously described (Clark TA, et al: J Mol Diagnostics 20:686-702, 2018; Khiabanian H, et al: JCO Precis Oncol 2:1-15, 2018); BRCA1/2 genetic testing on a subset of patients at the Rutgers Cancer Institute of New Jersey were analyzed under the auspices of an IRB-approved protocol. For cfDNA analysis, somatic-like allele frequency (AF) was defined as mutations observed in cfDNA at 0%-20% AF. Germline-like AF was defined as mutations observed in cfDNA at 40%-60% AF (except for cfDNA samples with high circulating tumor DNA fraction [> 20%] that were excluded from the analysis as ambiguous). BRCA1/2 alterations were categorized as biallelic, monoallelic, or unknown. Biallelic alterations were mutations with LOH of the wild-type allele, as determined by zygosity status (Sun JX, et al: PLOS Comput Biol 14:e1005965, 2018); homozygous deletion; or ≥ 2 BRCA1 or ≥ 2 BRCA2 alterations in a sample. Monoallelic alterations were heterozygous mutations (retained wild-type allele; Sun JX, et al: PLOS Comput Biol 14:e1005965, 2018). Alterations where zygosity status could not be determined were classified as unknown. Percent genome-wide LOH (gLOH) was calculated as a signature of HRD as previously described (Coleman RL, et al: Lancet 390:1949-1961, 2017; Swisher EM, et al: Lancet Oncol 18:75-87, 2017). In brief, LOH segments were inferred across the 22 autosomal chromosomes using the genome-wide aneuploidy/copy number profile and minor AFs of the > 3,500 polymorphic single nucleotide polymorphisms (SNPs) sequenced in the FoundationOne assay (Foundation Medicine, Cambridge, MA). Using a comparative genomic hybridization-like method, we obtained a log-ratio profile of the sample by normalizing the sequence coverage obtained at all exons and genome-wide SNPs against a process-matched normal control (Frampton GM, et al: Nat. Biotechnol 31:1023-1031, 2013). This profile was segmented and interpreted using AFs of sequenced SNPs to estimate copy number (Ci) and minor allele count (Mi) at each segment (i). A segment was determined to have LOH if Ci ≠ 0 and Mi = 0. Low tumor content or low aneuploidy were the most common reasons for failure to pass the quality control to perform gLOH inference. Two types of LOH segments were excluded from the calculation of percent gLOH: LOH segments that spanned ≥ 90% of a whole chromosome or chromosome arm because these LOH events usually arise through non-HRD mechanisms (eg, mitotic nondisjunction) and regions in which LOH inference was ambiguous. For each tumor, the percent gLOH was computed as 100× the total length of nonexcluded LOH regions (xi) divided by the total length of nonexcluded regions of the genome. An ultraviolet signature trinucleotide context was defined as the top-weighted alteration classes in COSMIC Signature 7 (A[C>T]C, C[C>T]A, C[C>T]C, C[C>T]G, C[C>T]T, G[C>T]C, G[C>T]T, T[C>T]A, T[C>T]C, T[C>T]G, T[C>T]T; https://cancer.sanger.ac.uk/cosmic/signatures_v2; Alexandrov LB, et al: Nature 500:415-421, 2013). Tumor mutational burden was calculated by counting the number of synonymous and nonsynonymous mutations across a 0.8- to 1.2-Mb region, with computational germline status filtering, and reporting the result as mutations/Mb; this method has been previously validated for accuracy against whole-exome sequencing (Chalmers ZR, et al: Genome Med 9:34, 2017).
FIG A1.
(A) Frequency of BRCA1 and BRCA2 alterations across multiple cancer types. Multiple indicates samples with two or more concurrent BRCA alteration types. (B) Frequency of BRCA1/2 alterations in the subset of ovarian and breast cancer cases where molecular/histologic subtype information was available. For breast cancer, estrogen receptor (ER) status was available for a subset of samples; human epidermal (continued on following page) growth factor receptor 2 (HER2) status was determined on the basis of the presence or absence of a copy number amplification; triple-negative breast cancer (TNBC) was defined as ER-negative, HER2-negative samples. CUP, cancer of unknown primary; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; UC, urothelial cancer.
FIG A2.
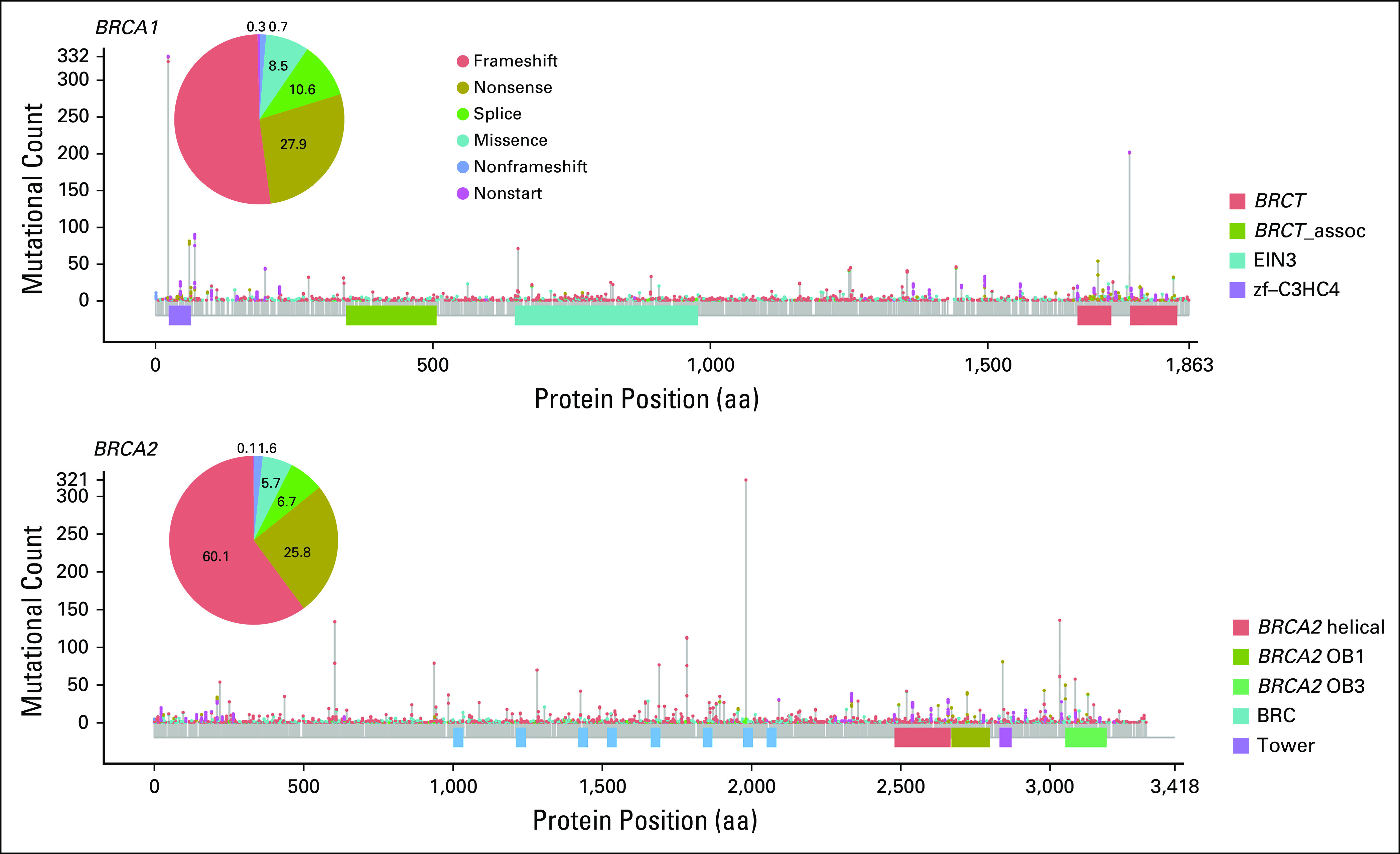
Summary of all BRCA1 and BRCA2 mutations included in the study by location and type. aa, amino acid.
FIG A3.
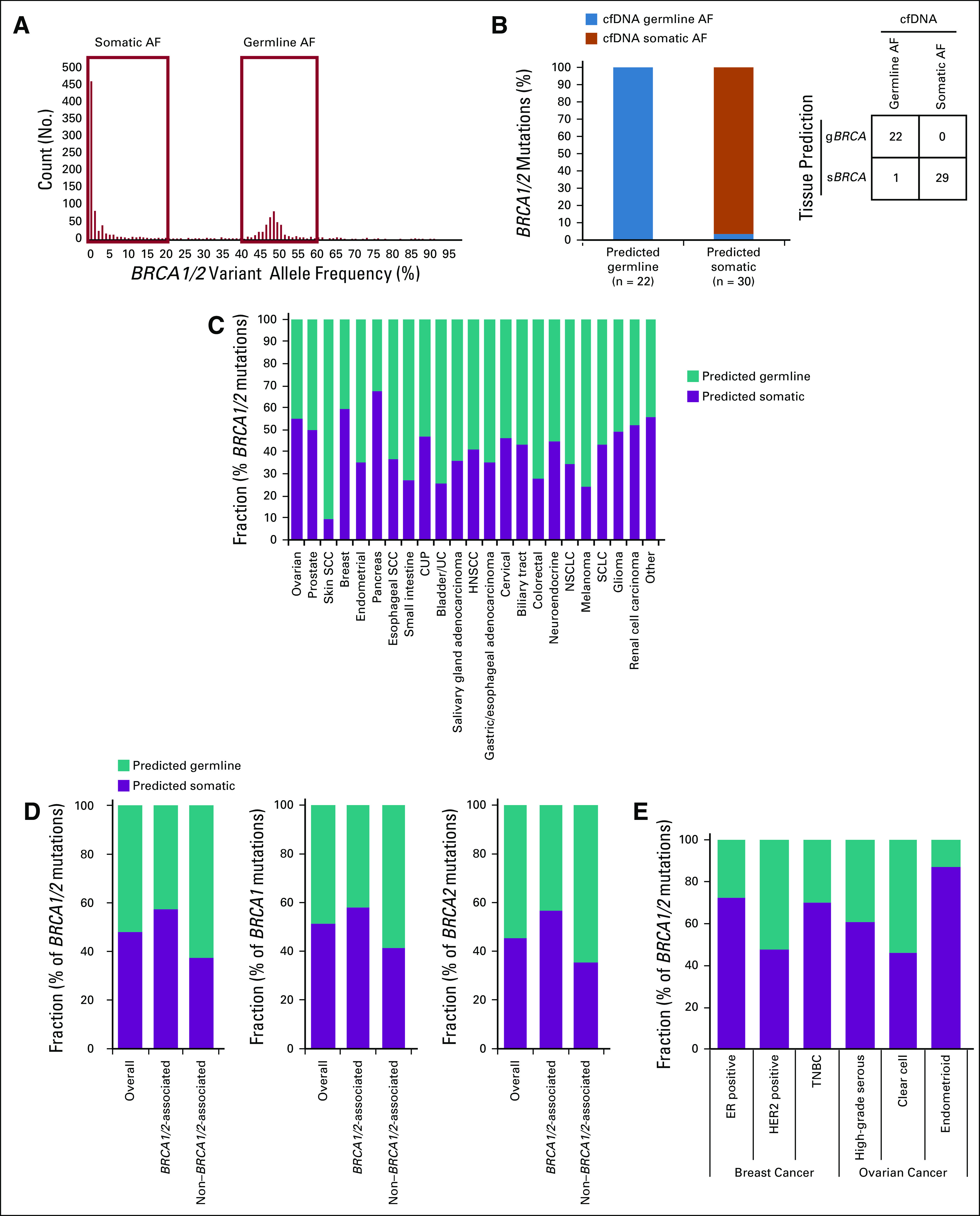
(A and B) Comparison of tissue-based germline/somatic predictions to BRCA1/2 allele frequencies (AFs) in patient-matched cell-free DNA (cfDNA). (A) Distribution of AFs for 1,207 cfDNA samples with BRCA1/2 mutation. Mutations with somatic AFs were defined as those identified in cfDNA at 0%-20% AF. Mutations with germline AF were defined as those identified in cfDNA at 40%-60% AF, except for cases with high circulating tumor DNA fraction (20%) that were excluded from the analysis as ambiguous AF. (B) Fifty-two tissue-derived germline BRCA1/2 (gBRCA1/2) and somatic BRCA1/2 (sBRCA1/2) mutation predictions (from 48 tissue samples) were compared with patient-matched (continued on following page) cfDNA AFs; 100.0% of germline predictions (22 of 22) were observed at germline-like AF and 96.7% of somatic predictions (29 of 30) were observed at somatic-like AF. (C-E) Predicted germline/somatic status calls were made for each BRCA1 or BRCA2 short variant mutation. For mutations yielding a successful call, frequency of predicted germline v somatic mutation was determined for (C) BRCA1/2 across cancer types, (D) BRCA1/2 (grouped and individually) overall (n = 5,845) for BRCA1/2-associated cancers (breast, ovarian, pancreatic, prostate; n = 3,061) and non–BRCA1/2-associated cancers (n = 2,784), and (E) BRCA1/2 for the subset of ovarian and breast cancer cases where molecular/histologic subtype information was available. CUP, cancer of unknown primary; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; TNBC, triple-negative breast cancer; UC, urothelial cancer.
FIG A4.
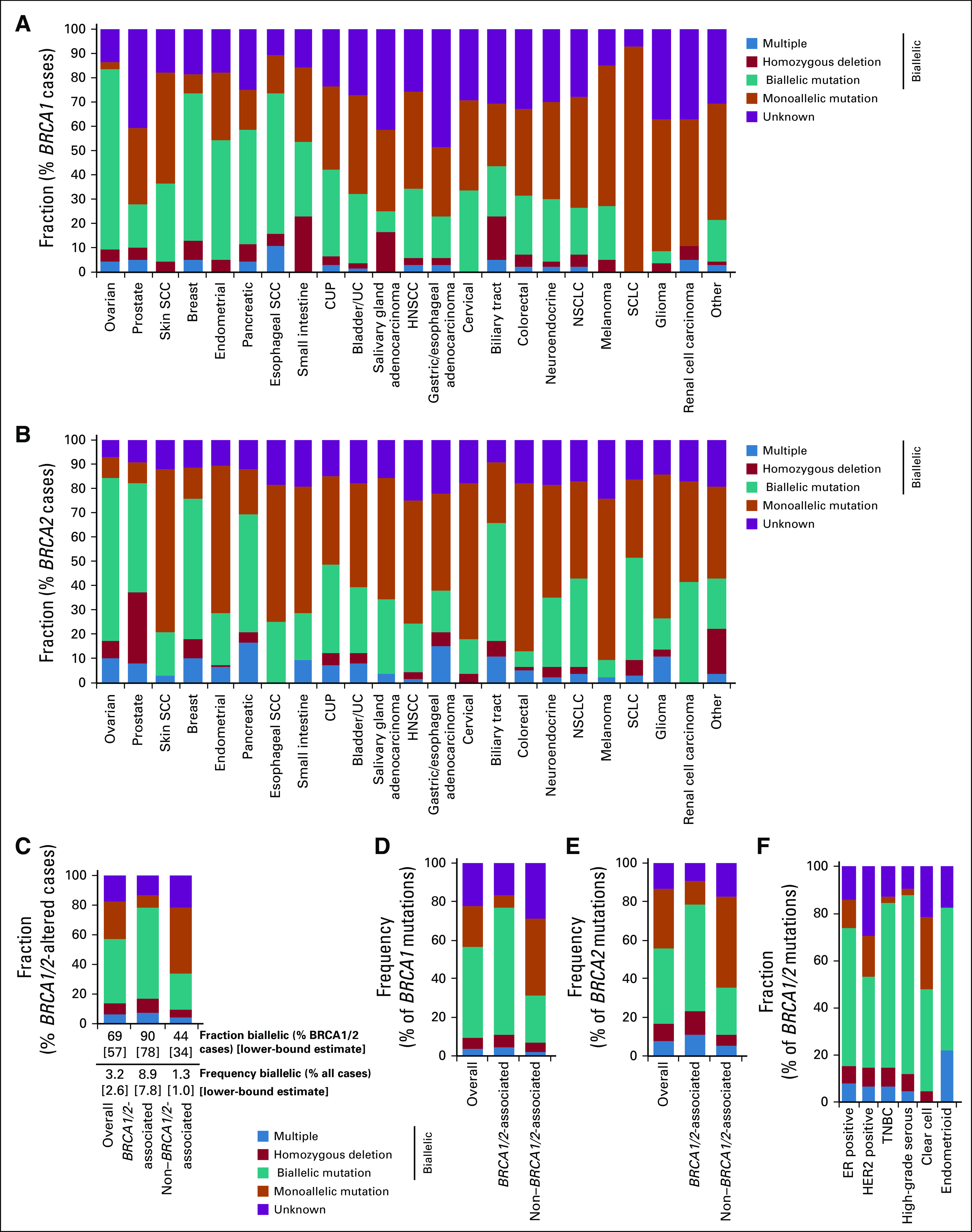
Relative fraction of BRCA1/2-altered cases with biallelic or monoallelic alteration was determined for (A) BRCA1 across cancer types and (B) BRCA2 across cancer types. (C) BRCA1/2; (D) BRCA1; (E) BRCA2 for overall, BRCA1/2-associated cancers, and non–BRCA1/2-associated cancers; and (F) BRCA1/2 for the subset of ovarian and breast cancer cases where molecular/histologic subtype information was available. CUP, cancer of unknown primary; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; TNBC, triple-negative breast cancer; UC, urothelial cancer.
FIG A5.
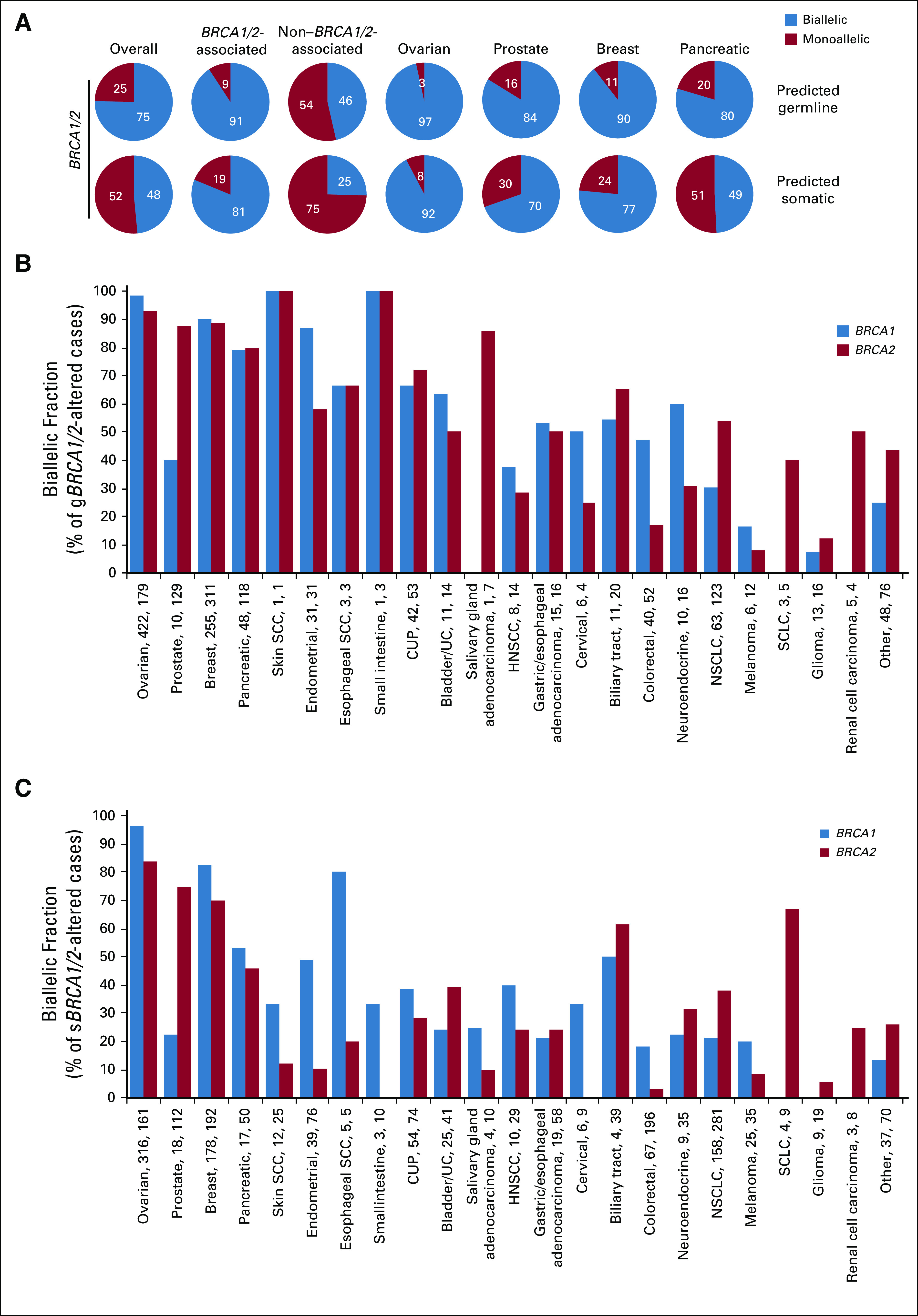
(A) BRCA1/2 mutations (grouped) with a germline (gBRCA1/2) or somatic (sBRCA1/2) prediction were evaluated for biallelic/monoallelic status for all cancers, BRCA1/2-associated cancers (as a group and as individual cancer types), and non–BRCA1/2-associated cancers (see Data Supplement). Biallelic fraction was assessed for (B) gBRCA1/2- and (C) sBRCA1/2-mutated cases. Numbers on the x-axis indicate number of cases assessed for biallelic status gBRCA1 or gBRCA2. CUP, cancer of unknown primary; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; UC, urothelial cancer.
FIG A6.

(A) The frequency of BRCA1/2 biallelic, monoallelic, and wild-type cases that were high genome-wide loss of heterozygosity (gLOH-high) was compared in the subset of ovarian and breast cancer cases where molecular/histologic subtype information was available (see Data Supplement). (B) Frequency of predicted germline BRCA1/2 (gBRCA1/2) biallelic, monoallelic, and wild-type cases that were gLOH-high (see Data Supplement). (C) Frequency of predicted somatic BRCA1/2 (sBRCA1/2) biallelic, monoallelic, and wild-type cases that were gLOH-high (see Data Supplement). CUP, cancer of unknown primary; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; NA, not available (no assessable alterations); NSCLC, non–small-cell lung cancer; TNBC, triple-negative breast cancer; UC, urothelial cancer.
FIG A7.
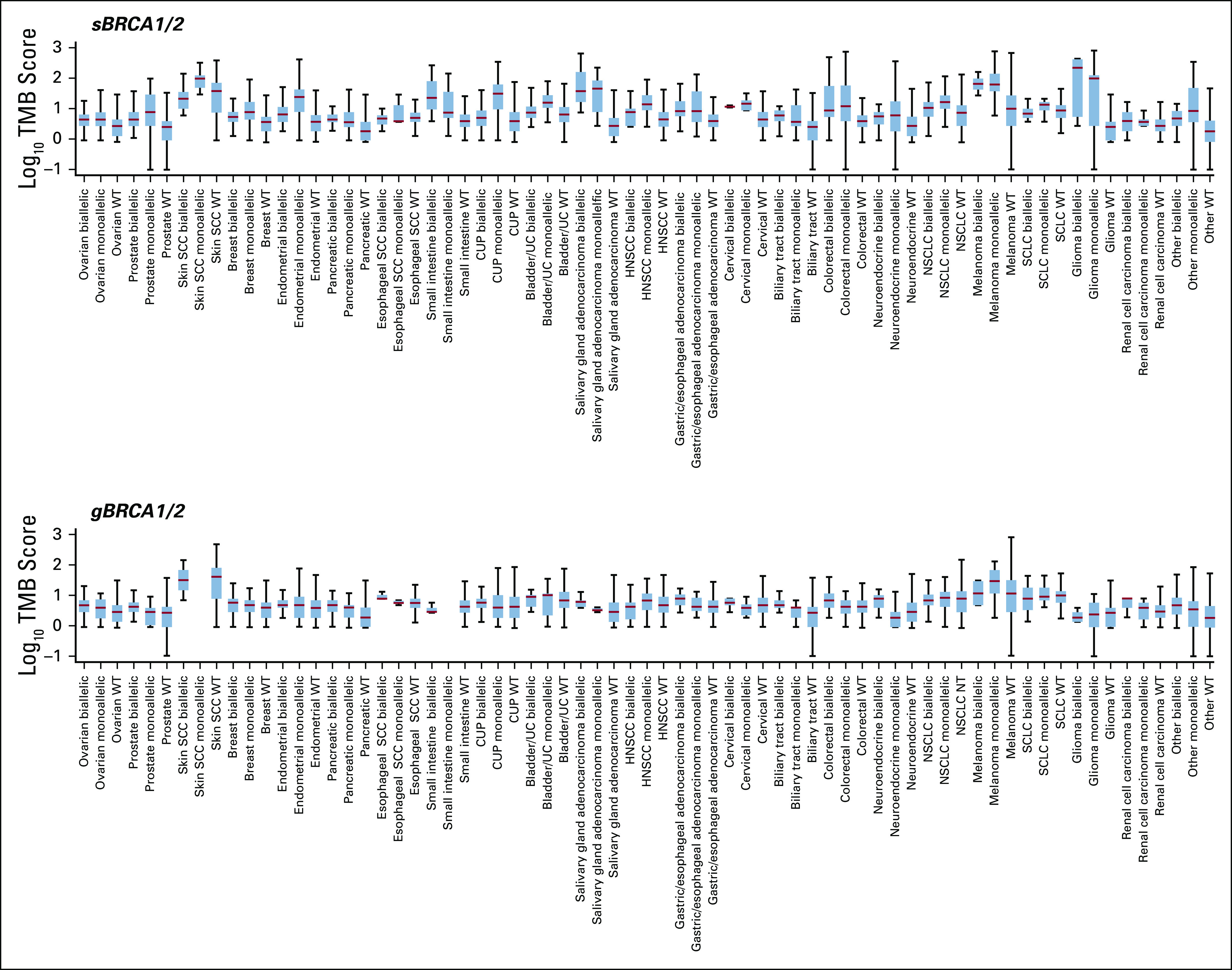
Cases with somatic BRCA1/2 (sBRCA1/2) or germline BRCA1/2 (gBRCA1/2) biallelic mutation, monoallelic mutation, or wild-type (WT) BRCA1/2 were plotted against tumor mutational burden (TMB in mutations/Mb; log10 score). Box and whisker plot where the box spans the first and third quartiles, the median is denoted by the horizontal line in the box, and whiskers indicate maximum and minimum values within 1.5× the interquartile range (see Data Supplement). CUP, cancer of unknown primary; HNSCC, head and neck squamous cell carcinoma; NSCLC, non–small-cell lung cancer; SCC, squamous cell carcinoma; SCLC, small-cell lung cancer; UC, urothelial cancer.
Footnotes
Presented at the European Society of Medical Oncology 2018 Congress, Munich, Germany, October 19-23, 2018.
AUTHOR CONTRIBUTIONS
Conception and design: Ethan S. Sokol, Jeffrey S. Ross, Steffi Oesterreich, Neeraj Agarwal, Vincent A. Miller, Siraj M. Ali, Shridar Ganesan, Jon H. Chung
Financial support: Brian Alexander
Administrative support: Brian Alexander
Provision of study material or patients: Hossein Khiabanian, Primo N. Lara, Neeraj Agarwal, Andrea Necchi
Collection and assembly of data: Ethan S. Sokol, Dean Pavlick, Garrett M. Frampton, Jeffrey S. Ross, Jeffrey P. Gregg, Primo N. Lara, Neeraj Agarwal, Jon H. Chung
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Ethan S. Sokol
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche
Dean Pavlick
Stock and Other Ownership Interests: Roche
Garrett M. Frampton
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche
Jeffrey S. Ross
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Research Funding: Foundation Medicine
Jeffrey P. Gregg
Consulting or Advisory Role: AstraZeneca, Bristol-Myers Squibb, Roche, Foundation Medicine, Luminex
Speakers’ Bureau: AstraZeneca, Foundation Medicine, Bristol-Myers Squibb
Primo N. Lara
Consulting or Advisory Role: Janssen Pharmaceuticals, Merck, CellMax Life, Pfizer
Research Funding: Aragon Pharmaceuticals (Inst), Janssen Biotech (Inst), TRACON Pharma (Inst), Merck (Inst), Pharmacyclics (Inst), Incyte (Inst), Taiho Pharmaceutical (Inst)
Steffi Oesterreich
Stock and Other Ownership Interests: Ocean Genomics (I)
Research Funding: GE, Blueprint Medicine, H3 Biomedicine, AstraZeneca, Illumina
Neeraj Agarwal
Consulting or Advisory Role: Pfizer, Medivation, Astellas Pharma, Bristol-Myers Squibb, AstraZeneca, Nektar, Eli Lilly, Bayer AG, Foundation Medicine, Pharmacyclics, Exelixis, Janssen Oncology, Merck, Novartis
Research Funding: Bayer AG (Inst), Bristol-Myers Squibb (Inst), GlaxoSmithKline (Inst), Takeda Pharmaceuticals (Inst), Novartis (Inst), Pfizer (Inst), BN ImmunoTherapeutics (Inst), Exelixis (Inst), TRACON Pharma (Inst), Rexahn Pharmaceuticals (Inst), Amgen (Inst), AstraZeneca (Inst), Active Biotech (Inst), Bavarian Nordic (Inst), Calithera Biosciences (Inst), Celldex (Inst),Eisai (Inst), Genentech (Inst), Immunomedics (Inst), Janssen Pharmaceuticals (Inst), Merck (Inst), NewLink Genetics (Inst), Prometheus (Inst), Sanofi (Inst)
Andrea Necchi
Employment: Bayer AG (I)
Stock and Other Ownership Interests: Bayer AG (I)
Honoraria: Roche, Merck, AstraZeneca, Janssen Pharmaceuticals, Foundation Medicine, Bristol-Myers Squibb
Consulting or Advisory Role: Merck Sharp & Dohme, Roche, Bayer AG, AstraZeneca, Clovis Oncology, Janssen Pharmaceuticals, Incyte, Seattle Genetics, Astellas Pharma, Bristol-Myers Squibb, Rainier Therapeutics, GlaxoSmithKline, Ferring
Research Funding: Merck Sharp & Dohme (Inst), AstraZeneca (Inst), Ipsen (Inst)
Travel, Accommodations, Expenses: Roche, Merck Sharp & Dohme, AstraZeneca, Janssen Pharmaceuticals, Rainier Therapeutics
Other Relationship: Bayer AG (I)
Vincent A. Miller
Employment: Foundation Medicine
Leadership: Foundation Medicine, Revolution Medicines
Stock and Other Ownership Interests: Foundation Medicine, Mirati Therapeutics, Revolution Medicines
Patents, Royalties, Other Intellectual Property: Periodic royalties related to T790M patent awarded to Memorial Sloan Kettering Cancer Center
Brian Alexander
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Roche
Research Funding: Eli Lilly (Inst), Puma (Inst), Celgene (Inst)
Open Payments Link: https://openpaymentsdata.cms.gov/physician/854258/summary
Siraj M. Ali
Employment: Foundation Medicine, EQRX, EQRX (I)
Leadership: Incysus
Stock and Other Ownership Interests: Exelixis, Blueprint Medicines, Agios, Genocea Biosciences
Consulting or Advisory Role: Revolution Medicines, Azitra (I), Princepx Tx (I)
Patents, Royalties, Other Intellectual Property: Patents through Foundation Medicine, patents through Seres Health on microbiome stuff in non-neoplastic disease (I)
Shridar Ganesan
Employment: Merck (I)
Stock and Other Ownership Interests: Ibris, Inspirata, Merck (I)
Consulting or Advisory Role: Inspirata, Novartis, Roche, Foghorn Therapeutics, Foundation Medicine
Patents, Royalties, Other Intellectual Property: Two patents for digital imaging that may be licensed to Ibris and Inspirata
Travel, Accommodations, Expenses: Inspirata
Jon H. Chung
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
No other potential conflicts of interest were reported.
REFERENCES
- 1. Mateo J, Lord CJ, Serra V, et al: A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 30:1437-1447, 2019. [DOI] [PMC free article] [PubMed]
- 2.Pilarski R. The role of BRCA testing in hereditary pancreatic and prostate cancer families. Am Soc Clin Oncol Educ Book. 2019;39:79–86. doi: 10.1200/EDBK_238977. [DOI] [PubMed] [Google Scholar]
- 3.Mavaddat N, Peock S, Frost D, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105:812–822. doi: 10.1093/jnci/djt095. [DOI] [PubMed] [Google Scholar]
- 4. doi: 10.1038/s41586-019-1382-1. Jonsson P, Bandlamudi C, Cheng ML, et al: Tumour lineage shapes BRCA-mediated phenotypes. Nature 571:576-579, 2019 [Erratum: Nature 577:E1, 2020] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golan T, Hammel P, Reni M, et al: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 381:317-327, 2019. [DOI] [PMC free article] [PubMed]
- 6. doi: 10.1038/nature17676. Nik-Zainal S, Davies H, Staaf J, et al: Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534:47-54, 2016 [Erratum: Nature 566:E1, 2019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoppe MM, Sundar R, Tan DSP, et al. Biomarkers for homologous recombination deficiency in cancer. J Natl Cancer Inst. 2018;110:704–713. doi: 10.1093/jnci/djy085. [DOI] [PubMed] [Google Scholar]
- 9.Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. doi: 10.1016/S1470-2045(16)30559-9. [DOI] [PubMed] [Google Scholar]
- 10.Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–1961. doi: 10.1016/S0140-6736(17)32440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abida W, Bryce AH, Vogelzang NJ, et al. Preliminary results from TRITON2: A phase II study of rucaparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) associated with homologous recombination repair (HRR) gene alterations. Ann Oncol. 2018;29(suppl 8):i272. [Google Scholar]
- 12.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith MR, Sandhu SK, Kelly WK, et al: Phase II study of niraparib in patients with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD): Preliminary results of GALAHAD. J Clin Oncol 37, 2019 (suppl; abstr 202)
- 14.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 16.Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–892. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 17.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun JX, He Y, Sanford E, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLOS Comput Biol. 2018;14:e1005965. doi: 10.1371/journal.pcbi.1005965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. doi: 10.1200/JCO.18.00328. Slavin TP, Banks KC, Chudova D, et al: Identification of incidental germline mutations in patients with advanced solid tumors who underwent cell-free circulating tumor DNA sequencing. J Clin Oncol 36:3459-3465, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore KN, Secord AA, Geller MA, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20:636–648. doi: 10.1016/S1470-2045(19)30029-4. [DOI] [PubMed] [Google Scholar]
- 21.Necchi A, Raggi D, Giannatempo P, et al. Exceptional response to olaparib in BRCA2-altered urothelial carcinoma after PD-L1 inhibitor and chemotherapy failure. Eur J Cancer. 2018;96:128–130. doi: 10.1016/j.ejca.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 22. doi: 10.1200/PO.18.00264. Sweis RF, Heiss B, Segal J, et al: Clinical activity of olaparib in urothelial bladder cancer with DNA damage response gene mutations. JCO Precis Oncol 10.1200/PO.18.00264. [DOI] [PubMed] [Google Scholar]
- 23.Seligson ND, Kautto EA, Passen EN, et al. BRCA1/2 Functional loss defines a targetable subset in leiomyosarcoma. Oncologist. 2019;24:973–979. doi: 10.1634/theoncologist.2018-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pathania S, Bade S, Le Guillou M, et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat Commun. 2014;5:5496. doi: 10.1038/ncomms6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maxwell KN, Wubbenhorst B, Wenz BM, et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat Commun. 2017;8:319. doi: 10.1038/s41467-017-00388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaelin WG., Jr Synthetic lethality: A framework for the development of wiser cancer therapeutics. Genome Med. 2009;1:99. doi: 10.1186/gm99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hierro C, Matos I, Martin-Liberal J, et al. Agnostic-histology approval of new drugs in oncology: Are we already there? Clin Cancer Res. 2019;25:3210–3219. doi: 10.1158/1078-0432.CCR-18-3694. [DOI] [PubMed] [Google Scholar]
- 28. doi: 10.1038/nature12477. Alexandrov LB, Nik-Zainal S, Wedge DC, et al: Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [Erratum: Nature 502:258, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16:475. doi: 10.1186/s13058-014-0475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popova T, Manié E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 32.Birkbak NJ, Wang ZC, Kim J-Y, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2:366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Telli ML, Timms KM, Reid J, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22:3764–3773. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gulhan DC, Lee JJ-K, Melloni GEM, et al. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51:912–919. doi: 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 35.Riaz N, Blecua P, Lim RS, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8:857. doi: 10.1038/s41467-017-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshall CH, Sokolova AO, McNatty AL, et al. differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. 2019;76:452–458. doi: 10.1016/j.eururo.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]