

Abstract
Purpose
Dabrafenib and trametinib are approved for the management of advanced non–small-cell lung cancers (NSCLCs) that harbor BRAF V600E mutations. Small series and pan-cancer analyses have identified non-V600 alterations as therapeutic targets. We sought to examine a large genomic data set to comprehensively characterize non-V600 BRAF alterations in lung cancer.
Patients and Methods
A total of 23,396 patients with lung cancer provided data to assay with comprehensive genomic profiling. Data were reviewed for predicted pathogenic BRAF base substitutions, short insertions and deletions, copy number changes, and rearrangements.
Results
Adenocarcinomas represented 65% of the occurrences; NSCLC not otherwise specified (NOS), 15%; squamous cell carcinoma, 12%; and small-cell lung carcinoma, 5%. BRAF was altered in 4.5% (1,048 of 23,396) of all tumors; 37.4% (n = 397) were BRAF V600E, 38% were BRAF non-V600E activating mutations, and 18% were BRAF inactivating. Rearrangements were observed at a frequency of 4.3% and consisted of N-terminal deletions (NTDs; 0.75%), kinase domain duplications (KDDs; 0.75%), and BRAF fusions (2.8%). The fusions involved three recurrent fusion partners: ARMC10, DOCK4, and TRIM24. BRAF V600E was associated with co-occurrence of SETD2 alterations, but other BRAF alterations were not and were instead associated with CDKN2A, TP53, and STK11 alterations (P < .05). Potential mechanisms of acquired resistance to BRAF V600E inhibition are demonstrated.
Conclusion
This series characterized the frequent occurrence (4.4%) of BRAF alterations in lung cancers. Recurrent BRAF alterations in NSCLC adenocarcinoma are comparable to the frequency of other NSCLC oncogenic drivers, such as ALK, and exceed that of ROS1 or RET. This work supports a broad profiling approach in lung cancers and suggests that non-V600E BRAF alterations represent a subgroup of lung cancers in which targeted therapy should be considered.
INTRODUCTION
The rapid development of genotype-directed management of metastatic non–small-cell lung carcinoma (NSCLC) has emerged as the paradigm for precision oncology. This model is exemplified by the improved outcomes in patients with NSCLC that harbor EGFR mutations, ALK fusions, or ROS1 fusions who receive matched tyrosine kinase inhibitors (TKIs).1-3 For NSCLC that harbors BRAF V600E, the combination of dabrafenib and trametinib was approved recently on the basis of an overall response rate of 66% compared with 33% with dabrafenib monotherapy.4-7 In addition, a basket trial showed that BRAF V600E could be targeted successfully in solid tumors other than melanoma or NSCLC.8
Within lung cancers, small series have described other oncogenic BRAF point mutations in exons 11 and 15.9-12 However, because of the small sample size of prior studies and the focused sequencing methodologies that can miss important classes of genomic alterations, such as rearrangements, a complete landscape of BRAF alterations in lung cancers is lacking.13-15 Given the therapeutic action ability of diverse BRAF alterations, we hypothesized that analysis by comprehensive genomic profiling (CGP) would refine the BRAF landscape and identify additional subsets of patients who may be candidates for targeted therapy. To our knowledge, this is the largest series to examine BRAF alterations in lung cancer and identify recurrent BRAF kinase–impaired point mutations; kinase-activating mutations, including V600E, oncogenic small insertions and deletions; and rearrangements/fusions of BRAF.
PATIENTS AND METHODS
We reviewed 23,396 consecutive patients with lung cancer who underwent CGP during clinical care. The hybrid capture next-generation sequencing–based assay used identifies genomic alterations in 186, 236, or 315 genes: base substitutions, short insertions/deletions, copy number alterations, and gene fusions via intron baiting for 14, 19, and 31 genes, as previously described.16 DNA was extracted from 40-micron scrolls of formalin-fixed paraffin-embedded tissue, and CGP was performed on hybridization-captured, adaptor ligation–based libraries to a mean coverage depth of greater than ×500. Age, sex, and histology were abstracted from the accompanying pathology report submitted by the treating physician. Before sequencing, all patient cases were reviewed by a board-certified pathologist to establish adequacy of submitted material but not to confirm or overturn the submitted histologic diagnosis. Testing was performed in a Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited reference laboratory (Foundation Medicine, Cambridge, MA).
Ordinal relationships were examined with the Mann-Whitney U test; categoric relationships were examined with the Pearson χ2 test, and the Yates continuity correction was applied when applicable. Approval for this study, which included a waiver of informed consent and a Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). Literature review defined kinase-activating base substitutions in BRAF as follows: G464A, G464E, G464V, G466A, F468C, G469A, G469R, G469S, G469V, V471F, N581S, E586K, F595L, L597Q, L597R, L597S, L597V, and K601E. Base substitutions that inactivate BRAF kinase activity were defined as follows: G466E, G466R, G466V, G469E, D594A, D594E, D594G, D594H, D594N, D594V, D594Y, G596R, T599I.
RESULTS
The histologic breakdown and basic clinicopathologic features are listed in Table 1. In total, 1,061 individual BRAF alterations, which included base substitutions, small insertions/deletions, and rearrangements in BRAF, were identified within 1,048 patient cases (4.4% overall). Focal amplifications of BRAF were not included because of a lack of preclinical evidence to support an oncogenic role (Table 2). There were differences among histologic subtypes: 5.5% of adenocarcinomas and 1% (42 of 3,948) of squamous and small-cell tumors harbored BRAF alterations (Table 2). Among all lung adenocarcinoma and NSCLC NOS, 40% and 29% of BRAF alterations, respectively, were BRAF V600E (Table 2). Of the 32 SCCs with BRAF mutations, five were BRAF V600E, and the remainder were divided between kinase-activating and kinase-inactivating mutations. Of the 10 SCLCs with BRAF alterations, nine were kinase activating, one was V600E, and two were G469V (Fig 1A).
Table 1.
Clinicopathologic Features of 23,396 Patients With Lung Cancers Subjected to Comprehensive Genomic Profiling to Examine BRAF Alterations

Table 2.
Distribution of BRAF Alterations Across 1,048 Individual BRAF-Altered Lung Cancers

Fig 1.
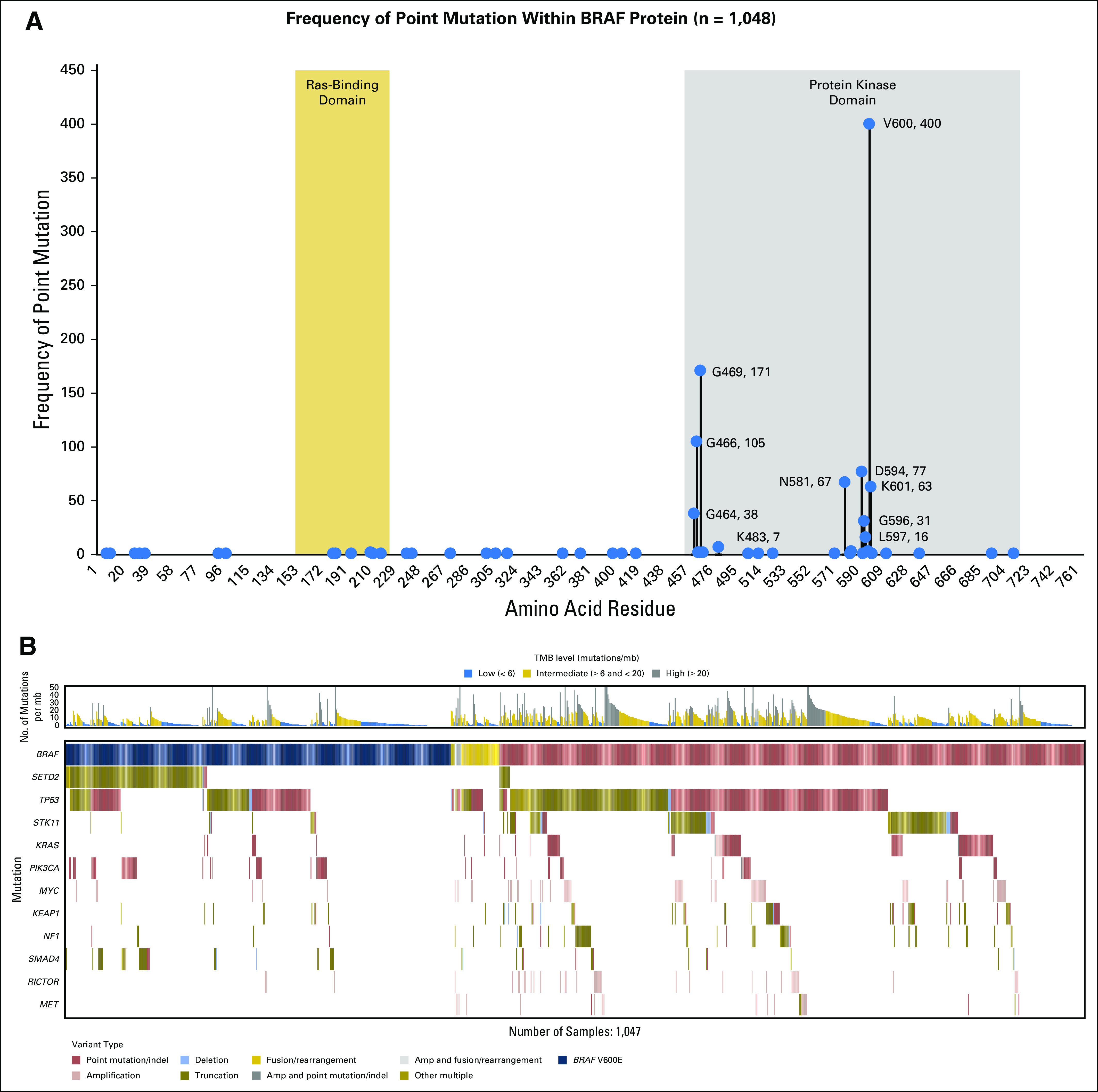
Intragenic distribution of BRAF point mutations and co-occurrence of BRAF alterations with alterations of other cancer-related genes within BRAF in a series of 23,396 patient cases with lung cancer. (A) Lollipop plot shows the distribution of point mutations in BRAF across lung cancers (n = 1,048). (B) Tile plot shows that BRAF V600E co-occur with SETD2 and that non-V600E BRAF mutations do not.
Overall, non-V600E activating mutations accounted for 37.9% (402 of 1,061) of all BRAF alterations. Within non-V600E mutations (n = 402), codon 469 (G469) alterations represented 34% (135 of 402), and G469A was the most common alteration (23%; 94 of 402; Table 2). Other recurrent G469 mutations included G469R (4%; 16 of 402), G469S (3.2%; 13 of 402), and G469V (10.1%; 41 of 402). Mutations at G464 accounted for 10% (38 of 402) of activating mutations; G464A (1.2%; five of 402), G464E (0.7%; three of 402), and G464V (7.5%; 30 of 402) were most common (Fig 1A). G466A occurred in 3.7% (15 of 402), and V471F occurred in 0.25% (one occurrence). In addition to mutations in the GxGxxG motif of the BRAF P-loop, we observed mutations at L597 (4%; 16 of 402), which consisted of L597Q (1.5%; six of 402), L597R (1.5%; six of 402), L597S (0.25%; one of 402), and L597V (0.75%; three of 402). Recurrent K601E represented 11.7% (47 of 402) of activating point mutations, and N581S represented 6.7% (27 of 402; Fig 1A).
Base substitutions that yielded kinase inactivation represented 18.2% (193 of 1,061) of all BRAF alterations in this series (Table 2). Mutations at the D594 position at the start of the DFG motif made up 40% (77 of 193) of these inactivating alterations, as follows: D594G (19.7%; 38 of 193), D594A (1%; two of 193), D594E (1%; two of 193), D594H (2%; four of 193), D594N (14%; 27 of 193), D594V (0.5%; one of 193), and D594Y (1%; two of 193). G596R was found in 13.5% (26 of 193) of tumors. BRAF F595L and G596D were found once each and may have affected kinase activity.17 Additional kinase-inactivating mutations included changes at position G466 in the P-loop in 54% (104 of 193), as follows: G466V (35.2%; 68 of 193), G466E (5.7%; 11 of 193), and G466R (3.1%; six of 193). Alterations of SETD2 were enriched in tumors that harbored BRAF V600E but not other BRAF alterations (P < .001; Fig 1B). Alterations of SMAD4 and PIK3CA also co-segregated with BRAF V600E relative to other BRAF alterations (P < .01). Conversely, alterations of KEAP1, NF1, MET, RICTOR, KRAS, MYC, STK11, and TP53 occurred more frequently in non-V600E BRAF–altered tumors (P < .05). Nearly half (44%) of SETD2 alterations in the BRAF V600E occurrences were loss of heterozygosity (LOH), which was more frequent than the occurrence of LOH (22%) across a large set of SETD2-altered NSCLC tumors (data not shown). KRAS was enriched in the BRAF non-V600E tumors with an odds ratio of 0.103 and an FDR-adjusted P value of 1.91E−09. This was assessed with χ2 to compare V600E (odds ratio > 1 indicates enrichment) and non-V600E (odds ratio < 1 indicates enrichment). This assessment included any known/likely KRAS variant.
Small activating insertions and deletions of BRAF were rare (2%; 23 of 1,061 tumors) and were predominantly in adenocarcinoma (91%; 21 of 23 tumors). Such deletions were L485_N486>F, L485_N486>Y (n = 2), N486_P490del (n = 3), V487_P492>A (n = 2), T488_P492del, and A489_Q493del (n = 3), which are all adjacent or within the alpha C-helix, as previously described.14,15 Other oncogenic BRAF deletions were G593_A598del, T599_V600>M, V600_W604>R, and V600_W604>E (n = 1 each). K483_M484>EI was mutated without a net change in length. Short insertions were G503_V504insVLR and A598_T599insT (n = 2), and two longer insertions, A598_T599insIFLHEDLTVKIGDFGLA and T599_V600insRVGDFGLAT, also were identified. Oncogenic BRAF deletions and insertions were largely mutually exclusive from other National Comprehensive Cancer Network–designated NSCLC oncogenic alterations except for one tumor that harbored both T599_V600insT and ERBB2 amplification, quantitatively estimated as seven copies (Table 2).
Rearrangements that consisted of N-terminal deletions (NTDs), exonic deletions, kinase domain duplications (KDDs), and fusions of BRAF were identified at a frequency of 4.3% (46 of 1,061 BRAF alterations; Fig 2). NTDs were identified in 0.76% (eight of 1,048 of tumors), and two tumors also harbored base substitutions in BRAF. One tumor harbored deletions of exons 3 through 8 and G464A, and the other, deletion of exons 4 through 8 and D594G. No other known oncogenic drivers were identified in either tumor. One tumor with an NTD harbored deletion of exons 2 through 9 as well as KRAS G12C. Tumors with limited exonic deletions of BRAF included one with exon 8 deleted and one with exon 7 deleted (Fig 2). KDD events occurred in 0.76% (eight of 1,048 tumors) and appended exons that coded for the full kinase domain of BRAF to the 3-prime end of the wild-type gene. One tumor had a breakpoint in intron 7, appended to exons 7 through 18; two tumors had a breakpoint in intron 8; four tumors had breakpoints in intron 9; and one tumor had a breakpoint in intron 10 (Fig 2). All tumors were otherwise wild type for RAS/RAF/MAPK family member alterations and NCCN-designated NSCLC driver alterations. Predicted fusions of BRAF were found in 2.9% (30 of 1,048) of tumors, which provided an overall frequency of 2.8% (30 of 1,061) of all BRAF alterations. Twenty-six tumors had identifiable fusion partners of BRAF, and the following recurrent fusions occurred twice each: ARMC10, DOCK4, and TRIM24; three tumors harbored SND1-BRAF. Fusions that involved AGAP3, AGK, AP3B1, BTFL34, EPS15, EYS, GHR, GRM8, LMO7, MKRN1, NUP214, PARP12, PTPN13, STAT3, TRIM4, TRIO, and ZC3HAV4 occurred once each (Fig 2).
Fig 2.

The spectrum of BRAF rearrangements in lung cancer. N-terminal deletions, kinase domain duplications, and BRAF fusions. Breakpoints in BRAF were largely conserved between introns 8 and 11.
Paired samples were available for a small subset of tumors (n = 16). Among seven BRAF V600E adenocarcinomas, five had new mutations in RAS family members, including four mutations in KRAS, and one in NRAS (Table 3). A patient with BRAF V600E NSCLC experienced disease response to vemurafenib for 7 months, and a progression sample demonstrated a BRAF rearrangement as well as the original BRAF V600E.
Table 3.
Mutations Associated With Progressive Disease in BRAF V600E Mutant Patient Cases

DISCUSSION
Here, we present, to our knowledge, the largest assessment of BRAF alterations and expand upon the understanding of activating genomic BRAF aberrations across lung cancers. Pathologic activation of the RAS/RAF/MEK/ERK (MAPK) pathway is observed across multiple tumor types, and BRAF alterations in lung cancer can be targeted by MEK inhibitors or pan-RAF inhibitors. Preclinical data suggest that MAPK pathway activation that results from BRAF activating (including non-V600E) alterations may be sensitive to targeting downstream signaling nodes MEK and ERK.18 Our data suggest that non-V600E BRAF alterations are recurrent in NSCLC and warrant additional clinical exploration.
RAF proteins (including BRAF) have similar structures, which contain three conserved regions (CR1, CR2, and CR3).19 CR1 contains RAS-binding and cysteine-rich domains (called RBD and CRD, respectively), that bind RAS. CR2 is a serine-threonine–rich domain, which functions as an inhibitory domain upon binding of the 14-3-3 regulatory protein. CR3 encompasses the kinase domain, which includes sites for binding of ATP (the P-loop) and BRAF substrates MEK1 and MEK2. This is also the site at which BRAF inhibitors bind. RAF proteins function as homo- and heterodimers, which is necessary to exert kinase activity. It is likely that differential sensitivity to inhibitors by type of BRAF alteration reflects varied activation mechanisms, elicited by different mutations. The canonical BRAF V600E kinase domain mutation was observed in 397 tumors. We observed BRAF G469A and G469V in 135 tumors, and this codon in the kinase P-loop retains the ability to form heterodimers with C-RAF. In this large patient subset, use of the multikinase inhibitors sorafenib, and the closely structurally related compound regorafenib, which have activity against C-RAF, an obligate physiologic heterodimerization partner for BRAF, may be a more rational approach. Indeed, sorafenib activity in in two NSCLC tumors with activating mutations, G469A and G469V, was demonstrated recently.17,20 Orthogonal support from translational studies demonstrated decreased signaling activity, with a dimerization-impaired form of BRAF G469A (R509H) compared with wild-type BRAF G469A, in contrast with only a slight (7%) reduction for the analogous dimerization-impaired form of BRAF V600E (R509H), which is consistent with the operation of the R509H form as a promoter.21 We hypothesize that response to single-agent BRAF inhibitors in G469 alterations would be limited by paradoxical activation of RAF/MEK/ERK signaling caused by the current approved BRAF inhibitors (vemurafenib, dabrafenib), and we expect that newer pan-RAF inhibitors, such as PLX8394, may have broader utility against both V600 and non-V600 mutant forms of BRAF in NSCLC.22
Across cancers, the mutagenic processes most frequently observed in each anatomic tumor type exhibit some well-described variation.23 In this series, we identified recurrent BRAF mutations at G464, G466, and particularly G469, which typically are not observed in melanoma.24 This observation hints at different underlying carcinogenic processes between melanoma (ultraviolet light–induced DNA damage) and lung cancer (often smoking-induced damage). Unfortunately, smoking histories were not available for this work. Similarly, deletions of the alpha C-helix in BRAF are found most frequently in KRAS wild-type pancreatic carcinoma and are analogous to activating EGFR exon 19 deletions in the C-helix of the epidermal growth factor receptor kinase domain.14,15 Although no NSCLC response to this class of deletion has been described yet, a patient with non-Langerhans histiocytic disease that harbored a BRAF C-helical deletion recently experienced disease response with trametinib.25 In this series, we observed for the first time in BRAF the replacement of residues L485_N486 at the end of the beta strand with the aromatic amino acid tyrosine (n = 2) or phenylalanine (n = 1). In preclinical studies of deletion in the alpha C-helix of BRAF, single to multiple amino acids deletions have been modeled with some gain in BRAF kinase activity—less than that of the 486 through 490 deletion, but this insertion of F/Y was not modeled. This change may mimic the poorly characterized L747P mutation in EGFR or the conserved exon 19 insertion, which also results in L747P mutation.26,27 It remains to be understood how L485_N486>F/Y activates BRAF and any associated sensitivity to a pan-RAF inhibitor. Larger series with treatment data will be needed to address a possible role for how tissue and/or the genomic context of a given BRAF alteration would affect clinical responsiveness. For example, in this data set, 0.7% of lung carcinoma tumors harbored focal BRAF amplification, which by itself is not known to serve as an oncogenic driver, but more than half of these co-occurred with BRAF non-V600E point mutations without other oncogenic driver alterations (data not shown). The co-occurrence of BRAF V600E and alterations of SETD2, SMAD4, and PIK3CA is novel and highly significant for SETD2 mutations (P < .001). An LOH assessment demonstrated an enrichment of SETD2 LOH in these tumors relative to all lung SCC tumors (44% v 25%; data not shown). Non-V600E BRAF–altered tumors were enriched for concurrent alterations of KEAP1, NF1, MET, RICTOR, KRAS, MYC, STK11, and TP53. STK11 alterations in particular may be functionally related to BRAF alterations, as was shown in melanoma cells in which BRAF V600E suppressed LKB1 function, which allowed activation of AMPK.28,29 Such interaction has not been described for non-V600E BRAF mutants, but it is conceivable that STK11 mutations on one allele, coupled with inactivation of residual wild-type STK11 protein by a mutated BRAF protein, may abrogate STK11 function.
We observed a diversity of BRAF rearrangements, including NTDs, KDDs, and BRAF fusions, in this series. Previously, variably sized deletions of exons 2 through 9 or less in BRAF (NTD-BRAF) were described only in preclinical models of vemurafenib-resistant melanoma and lung cancer.30,31 To our knowledge, this is the first report of NTD-BRAF in lung cancer samples (none, to our knowledge, with prior RAF-directed therapy), and it suggests that mechanisms other than splicing at the RNA level can underlie NTD. In addition to NTD-BRAF, we also report the selective deletion of exons 7 or 8, which had been unknown as activating BRAF (Fig 2). Moreover, we report recurrent BRAF KDDs in NSCLC, a genomic event first described in gliomas of the optic nerve.32 We previously reported a patient with acinic cell carcinoma that had BRAF KDDs who achieved a durable response to the pan-RAF inhibitor regorafenib.33,34 Preclinical work to demonstrate the sensitivity of BRAF KDDs that co-occur with BRAF V600E to a pan-RAF dimerization inhibitor suggests that the oncogenic activity of BRAF KDD is dimerization dependent.35 Additional investigation to determine the biology of KDD (ie, does BRAF KDD dimerize with wild-type CRAF, or auto- or homo-dimerize with the two kinase domains) interactions is needed. Although quite rare in this series, BRAF KDD responsiveness in other tumors highlights the importance of assessment of this alteration in NSCLC.
Fusions that involve the BRAF kinase domain occur in thyroid carcinoma, pediatric low-grade gliomas, melanoma, and other cancers.13,36-38 We expand on this understanding with the largest, to our knowledge, BRAF fusion series reported (n = 30) in lung cancers. Across the series, BRAF fusions lack the RAS-binding auto-inhibitory domain found in the N-terminal half of BRAF, akin to BRAF NTDs, and the N-terminal fusion partner often harbors a constitutive dimerization or oligomerization motif. Among melanomas that harbor BRAF fusions, response to trametinib is described, which indicates that NSCLC tumors that harbor BRAF fusions may also benefit from monotherapy with MEK inhibitors.13,39 In contrast, patients with pilocytic astrocytomas and presumed BRAF fusions experienced rapid progression with sorafenib treatment, which suggests that heterodimerization with CRAF is not needed for BRAF fusion activity, although some dimerization is required.40,41 Kinase fusions may emerge as resistance mechanisms to targeted therapy, such as epidermal growth factor receptor inhibitors.42,43 Limited clinical histories were available for patient cases in the series, but, for one tumor with EGFR exon 19 deletion (T790M negative), a TRIM24-BRAF fusion was observed with erlotinib resistance, which suggests that the BRAF fusion may drive resistance. Both the response to trametinib in BRAF fusion at diagnosis and the observed kinase fusions at resistance suggest BRAF fusions are a target that warrants exploration. Resistance to small molecule inhibitors is universal, and the landscape of BRAF-mutant lung cancers treated with BRAF inhibition is not known.44 Among a small subset of tumors with paired samples and clinical data, genomic alterations not present in the pretreatment specimen existed in the post-treatment specimens and may correlate with acquired resistance (Table 3). We observed a BRAF fusion upon resistance to vemurafenib, which mimicked a recent description of a fusion-based resistance to vemurafenib in melanoma.45 In a second tumor, loss of MAP2K4 and biallelic inactivation of SMARCA4 was seen at progression. Several tumors also had activating KRAS/NRAS mutations upon progression.
Overall, we report the largest series, to our knowledge, to examine all classes of BRAF alterations in lung cancers. Although limited by disease heterogeneity, incomplete clinical annotation, and no independent confirmation of histology, the series identifies multiple non-V600 aberrations that tend to be mutually exclusive with other oncogenic drivers in lung cancer. Whether non-V600 identifies a good prognostic group, as in colorectal cancer, is of interest but is not answerable from our data.46 Likewise, it is unclear whether non-V600E alterations are responsive to existing therapies; clinical trials are needed. The series provides a platform to investigate multiple hypotheses to refine the therapy for BRAF-altered lung cancers and may have treatment implications when clinical trials are not available for rare genomically defined subsets.
Footnotes
Presented in part at the Meeting of the European Society of Medical Oncology, Vienna, Austria, September 25-29, 2015.
AUTHOR CONTRIBUTIONS
Conception and design: Dean Pavlick, Samuel J. Klempner, Vamsidhar Velcheti, Nir Peled, Sherri Z. Millis, Venkataprasanth P. Reddy, Julia A. Elvin, Afshin Dowlati, Jeffrey S. Ross, Vincent A. Miller, Alexa B. Schrock, Sai-Hong Ignatius Ou, Siraj M. Ali
Collection and assembly of data: Dean Pavlick, Samuel J. Klempner, Vamsidhar Velcheti, Garrett M. Frampton, Nir Peled, Molly Murray, Young Kwang Chae, Laurie Gay, James H. Suh, Venkataprasanth P. Reddy, Afshin Dowlati, Jeffrey S. Ross, Sai-Hong Ignatius Ou, Siraj M. Ali
Provision of study material or patients: Samuel J. Klempner, Vamsidhar Velcheti, Afshin Dowlati, Sai-Hong Ignatius Ou
Data analysis and interpretation: Yuri Sheikine, Dean Pavlick, Samuel J. Klempner, Sally E. Trabucco, Jon H. Chung, Mark Rosenzweig, Kai Wang, Garrett M. Frampton, Nir Peled, Young Kwang Chae, Lee A. Albacker, Hatim Husain, Venkataprasanth P. Reddy, Afshin Dowlati, Ryan J. Hartmaier, Phil Stephens, Jeffrey S. Ross, Trever G. Bivona, Alexa B. Schrock, Shridar Ganesan, Sai-Hong Ignatius Ou, Siraj M. Ali
Administrative support: Molly Murray, Afshin Dowlati
Manuscript writing: All authors
Final approval of manuscript: All authors
Agree to be accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Yuri Sheikine
Consulting or Advisory Role: Foundation Medicine
Dean Pavlick
No relationship to disclose
Samuel Klempner
Consulting or Advisory Role: Lilly, Boston Biomedical, Celgene, Astellas Pharma
Speakers' Bureau: Foundation Medicine
Research Funding: Leap Therapeutics (Inst)
Sally E. Trabucco
Employment: Foundation Medicine
Research Funding: Foundation Medicine
Travel, Accommodations, Expenses: Foundation Medicine
Jon Chung
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Mark Rosenzweig
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Kai Wang
No relationship to disclose
Vamsidhar Velcheti
Honoraria: Novartis, Foundation Medicine, Merck, Bristol-Myers Squibb, Genentech, Roche
Consulting or Advisory Role: Clovis Oncology, Genentech, Bristol-Myers Squibb, Merck, Celgene, Foundation Medicine, AstraZeneca, MedImmune, Genoptix, Amgen, Takeda
Research Funding: Genentech (Inst), Trovagene (Inst), Eisai (Inst), OncoPlex Diagnostics (Inst), ALkermes (Inst), NantOmics (Inst), GEnoptix (Inst), Altor BioScience (Inst), Merck (Inst), Bristol-Myers Squibb (Inst), Atreca (Inst), Heat Biologics (Inst), Leap Therapeutics (Inst)
Travel, Accommodations, Expenses: AstraZeneca/MedImmune, Eisai, Foundation Medicine, Merck
Garrett M. Frampton
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Nir Peled
Consulting or Advisory Role: AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Lilly, MSD, Novartis, Pfizer, Roche
Speakers' Bureau: AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Lilly, MSD, Novartis, Pfizer, Roche
Molly Murray
Employment: Foundation Medicine
Young Kwang Chae
Consulting or Advisory Role: Foundation Medicine, Boehringer Ingelheim, Biodesix, Counsyl, AstraZeneca, Guardant Health
Speakers' Bureau: Merck, Genentech, Roche
Travel, Accommodations, Expenses: Hanmi
Lee A. Albacker
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Laurie Gay
Employment: Foundation Medicine
Stock and Other Ownership Interests: Gilead Sciences, Foundation Medicine
Hatim Husain
Consulting or Advisory Role: AstraZeneca, Abbvie, Foundation Medicine
Speakers' Bureau: AstraZeneca, Merck, Bristol-Myers Squibb
Research Funding: Pfizer (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Merck, Bristol-Myers Squibb, Foundation Medicine, Abbvie
James Suh
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Sherri Millis
Employment: Foundation Medicine
Venkataprasanth Reddy
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Travel, Accommodations, Expenses: Foundation Medicine
Julia Elvin
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Ryan J. Hartmaier
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Patents, Royalties, Other Intellectual Property: Nfe2L2 exon 2 and/or exon 3 loss from work conducted at Foundation Medicine (provisional patent filed) (Inst)
Afshin Dowlati
Consulting or Advisory Role: Abbvie, Stemcentrx, Ariad
Research Funding: Lilly (Inst), ImClone (Inst), Amgen (Inst), Bristol-Myers Squibb (Inst), OncoMed (Inst), EMD Serono (Inst), MedImmune (Inst)
Phil Stephens
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Jeffrey Ross
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Honoraria: Pfizer, EMD Merck Serono
Research Funding: Foundation Medicine
Trever Bivona
Stock and Other Ownership Interests: Driver
Honoraria: Novartis, AstraZeneca, Array BioPharma, Revolution Medicine
Consulting or Advisory Role: Revolution Medicine, Array BioPharma, Novartis, AstraZeneca
Speakers' Bureau: Novartis
Research Funding: Ignyta, Revolution Medicine
Vincent Miller
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Patents, Royalties, Other Intellectual Property: Receive periodic royalties related to T790M patent awarded to Memorial Sloan Kettering Cancer Center
Shridar Ganesan
Employment: Merck (I)
Stock and Other Ownership Interests: Ibris, Inspirata, Merck (I)
Consulting or Advisory Role: Inspirata, Novartis
Patents, Royalties, Other Intellectual Property: I hold two patents for digital imaging that may be licensed to Ibris and Inspirata
Travel, Accommodations, Expenses: Inspirata
Alexa Schrock
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Sai-Hong Ou
Honoraria: Pfizer, Roche Pharma AG, Genentech, Roche, ARIAD, Takeda, Novartis, AstraZeneca, Foundation Medicine
Consulting or Advisory Role: Pfizer, Roche, Genentech, Novartis, AstraZeneca, Takeda, ARIAD, Foundation Medicine
Speakers' Bureau: Genentech, AstraZeneca, Takeda, ARIAD
Research Funding: Pfizer (Inst), Roche Pharma AG (Inst), AstraZeneca (Inst), MedImmune (Inst), Clovis Oncology (Inst), ARIAD (Inst), Ignyta (Inst), Peregrine Pharmaceuticals (Inst), GlaxoSmithKline (Inst), Astellas Pharma (Inst), Chugai Pharma (Inst)
Siraj Ali
Employment: Foundation Medicine
Stock and Other Ownership Interests: Exelixis, Blueprint Medicines, Agios
Patents, Royalties, Other Intellectual Property: Patents via Foundation Medicine; patents via Seres Health on microbiomes in non-neoplastic disease (I)
REFERENCES
- 1.Rosell R, Carcereny E, Gervais R, et al. : Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation–positive non–small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13:239-246, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Solomon BJ, Mok T, Kim DW, et al. : First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371:2167-2177, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Shaw AT, Solomon BJ: Crizotinib in ROS1-rearranged non–small-cell lung cancer. N Engl J Med 372:683-684, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Planchard D, Besse B, Groen HJM, et al. : Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non–small-cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol 17:984-993, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Planchard D, Kim TM, Mazieres J, et al. : Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 17:642-650, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies H, Bignell GR, Cox C, et al. : Mutations of the BRAF gene in human cancer. Nature 417:949-954, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Kim G, McKee AE, Ning YM, et al. : FDA approval summary: Vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res 20:4994-5000, 2014 [DOI] [PubMed] [Google Scholar]
- 8.Hyman DM, Puzanov I, Subbiah V, et al. : Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373:726-736, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gautschi O, Peters S, Zoete V, et al. : Lung adenocarcinoma with BRAF G469L mutation refractory to vemurafenib. Lung Cancer 82:365-367, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Litvak AM, Paik PK, Woo KM, et al. : Clinical characteristics and course of 63 patients with BRAF-mutant lung cancers. J Thorac Oncol 9:1669-1674, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tissot C, Couraud S, Tanguy R, et al. : Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer 91:23-28, 2016 [DOI] [PubMed] [Google Scholar]
- 12.Villaruz LC, Socinski MA, Abberbock S, et al. : Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 121:448-456, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross JS, Wang K, Chmielecki J, et al. : The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 138:881-890, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster SA, Whalen DM, Özen A, et al. : Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell 29:477-493, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Chen SH, Zhang Y, Van Horn RD, et al. : Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov 6:300-315, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kordes M, Röring M, Heining C, et al. : Cooperation of BRAF(F595L) and mutant HRAS in histiocytic sarcoma provides new insights into oncogenic BRAF signaling. Leukemia 30:937-946, 2016 [DOI] [PubMed] [Google Scholar]
- 18.Chalmers ZR, Connelly CF, Fabrizio D, et al. : Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9:34, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roskoski R, Jr: RAF protein-serine/threonine kinases: Structure and regulation. Biochem Biophys Res Commun 399:313-317, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Pervere LM, Rakshit S, Schrock AB, et al. : Durable response to combination of dabrafenib and trametinib in BRAF V600E–mutated non–small-cell lung cancer. Clin Lung Cancer 18:e211-e213, 2017 [DOI] [PubMed] [Google Scholar]
- 21.Röring M, Herr R, Fiala GJ, et al. : Distinct requirement for an intact dimer interface in wild-type, V600E, and kinase-dead B-Raf signaling. EMBO J 31:2629-2647, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okimoto RA, Lin L, Olivas V, et al. : Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc Natl Acad Sci USA 113:13456-13461, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holderfield M, Deuker MM, McCormick F, et al. : Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14:455-467, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee LH, Gasilina A, Roychoudhury J, et al. : Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2:e89473, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang YT, Ning WW, Li J, et al. : Exon 19 L747P mutation presented as a primary resistance to EGFR-TKI: A case report. J Thorac Dis 8:E542-E546, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He M, Capelletti M, Nafa K, et al. : EGFR exon 19 insertions: A new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res 18:1790-1797, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. : Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548:234-238, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng B, Jeong JH, Asara JM, et al. : Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 33:237-247, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poulikakos PI, Persaud Y, Janakiraman M, et al. : RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480:387-390, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin L, Asthana S, Chan E, et al. : Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci USA 111:E748-E757, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, et al. : BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol 71:789-794, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klempner SJ, Bordoni R, Gowen K, et al. : Identification of BRAF kinase domain duplications across multiple tumor types and response to RAF inhibitor therapy. JAMA Oncol 2:272-274, 2016 [DOI] [PubMed] [Google Scholar]
- 34.Trojan J, Waidmann O: Role of regorafenib as second-line therapy and landscape of investigational treatment options in advanced hepatocellular carcinoma. J Hepatocell Carcinoma 3:31-36, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kemper K, Krijgsman O, Kong X, et al. : BRAF(V600E) kinase domain duplication identified in therapy-refractory melanoma patient-derived xenografts. Cell Reports 16:263-277, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciampi R, Knauf JA, Kerler R, et al. : Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest 115:94-101, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones DT, Kocialkowski S, Liu L, et al. : Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673-8677, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutchinson KE, Lipson D, Stephens PJ, et al. : BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 19:6696-6702, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menzies AM, Yeh I, Botton T, et al. : Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res 28:607-610, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sievert AJ, Lang SS, Boucher KL, et al. : Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA 110:5957-5962, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karajannis MA, Legault G, Fisher MJ, et al. : Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro-oncol 16:1408-1416, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allen JM, Schrock AB, Erlich RL, et al. : Genomic profiling of circulating tumor DNA in relapsed EGFR-mutated lung adenocarcinoma reveals an acquired FGFR3-TACC3 fusion. Clin Lung Cancer 18:e219-e222, 2017 [DOI] [PubMed] [Google Scholar]
- 43.Klempner SJ, Bazhenova LA, Braiteh FS, et al. : Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer 89:357-359, 2015 [DOI] [PubMed] [Google Scholar]
- 44.Cree IA, Charlton P: Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 17:10, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulkarni A, Al-Hraishawi H, Simhadri S, et al. : BRAF fusion as a novel mechanism of acquired resistance to vemurafenib in BRAF V600E mutant melanoma. Clin Cancer Res 23:5631-5638, 2017 [DOI] [PubMed] [Google Scholar]
- 46.Jones JC, Renfro LA, Al-Shamsi HO, et al. : Non-V600 BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol 35:2624-2630, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]