

Introduction
Genomic alterations in the Ewing sarcoma family of tumors (EFT) were discovered > 30 years ago with the identification of the reciprocal translocation, t(11;22)(q24;q12), otherwise known as EWS-FL1.1,2 In the time since, multiple other fusion partners with EWS have been identified that fit a similar Ewing sarcoma phenotype.3,4 When EWS fusions are not identified, tumors with histologic features of Ewing sarcoma have been labeled as primitive neuroectodermal tumors. In 2012, Pierron et al5 identified a subset of Ewing-like tumors harboring paracentric inversion on the short arm of chromosome X, resulting in the fusion of the BCOR and CCNB3 genes.5 Since that discovery, several small case series have further elucidated the clinical, morphologic, and genomic differences that make this diagnosis distinct from other round cell sarcomas, most notably Ewing sarcoma.6-8
Though distinct from Ewing sarcoma, most BCOR-CCNB3–fused sarcomas (BCS) are treated with upfront compressed chemotherapy with vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide plus local control with surgery and/or radiation. BCS shares similar event-free and overall survival rates with the standard EWS-FLI1–fused Ewing sarcoma using this treatment strategy.6-8 Despite the growing knowledge base related to BCS, little is known about potential drug targets related to this disease entity, especially with regard to treatment of disease recurrence. We highlight the treatment of a young patient who had multiply-relapsed disease with the US Food and Drug Administration–approved cyclin-dependent kinase 4/6 (CDK4/6) inhibitor palbociclib; the tumor harbored a BCOR-CCNB3 fusion and a germline variant in CDKN2B, and treatment resulted in a complete response and no evidence of disease 25 months into therapy.
Case History
Our male patient initially presented in 2010 at 1 year of age with a fixed mass on his back. Magnetic resonance imaging of the pelvis showed a large infiltrating presacral mass measuring 14 × 7.4 × 10.4 cm extending into the lower spinal canal, eroding the posterior right sacrum, and exerting a mass effect on both the rectum and bladder. A core needle biopsy was performed, which revealed a malignant, small, round, blue cell tumor along with small amounts of benign fibrofatty tissue and skeletal muscle. Tumor nuclei were round to oval with a fine-grained chromatin pattern and occasional small nucleoli or chromocenters. Immunohistochemical stains were positive for CD99, Fli1, and vimentin and were negative for NSE, synaptophysin, MYF4, GAF, CD45RB, and TdT—consistent with a primitive neuroectodermal tumor. No polymerase chain reaction–base fusion analysis or breakapart fluorescence in situ hybridization probe for EWSR1 was performed at the time. Three generations of family history were negative for malignancies on either side of the family, including melanoma or pancreatic cancer. A staging computed tomography scan of the chest and a bone scan showed no evidence of metastatic disease. The patient started chemotherapy per Children’s Oncology Group protocol AEWS0031, regimen B2, with ifosfamide, etoposide, vincristine, doxorubicin, and cyclophosphamide. Gross total resection was not feasible at the time per neurosurgery, and the patient received 57.6 Gy of proton beam radiation in October 2010. The patient remained in remission for > 2 years but then developed multiple local recurrences without metastases from 2013 to 2017 and underwent numerous surgeries, along with multiple different early-phase Children’s Oncology Group therapeutic studies, as outlined in the timeline in Figure 1A. After the most recent recurrence in October 2016, the patient was referred to our Pediatric Cancer Precision Genomics Program. Because of the findings outlined here in the Results, we chose to start palbociclib in February 2017. This patient has no evidence of disease on imaging 25 months into therapy (Fig 1B) and has had only hematologic toxicity that was grade 2 or less.
FIG 1.
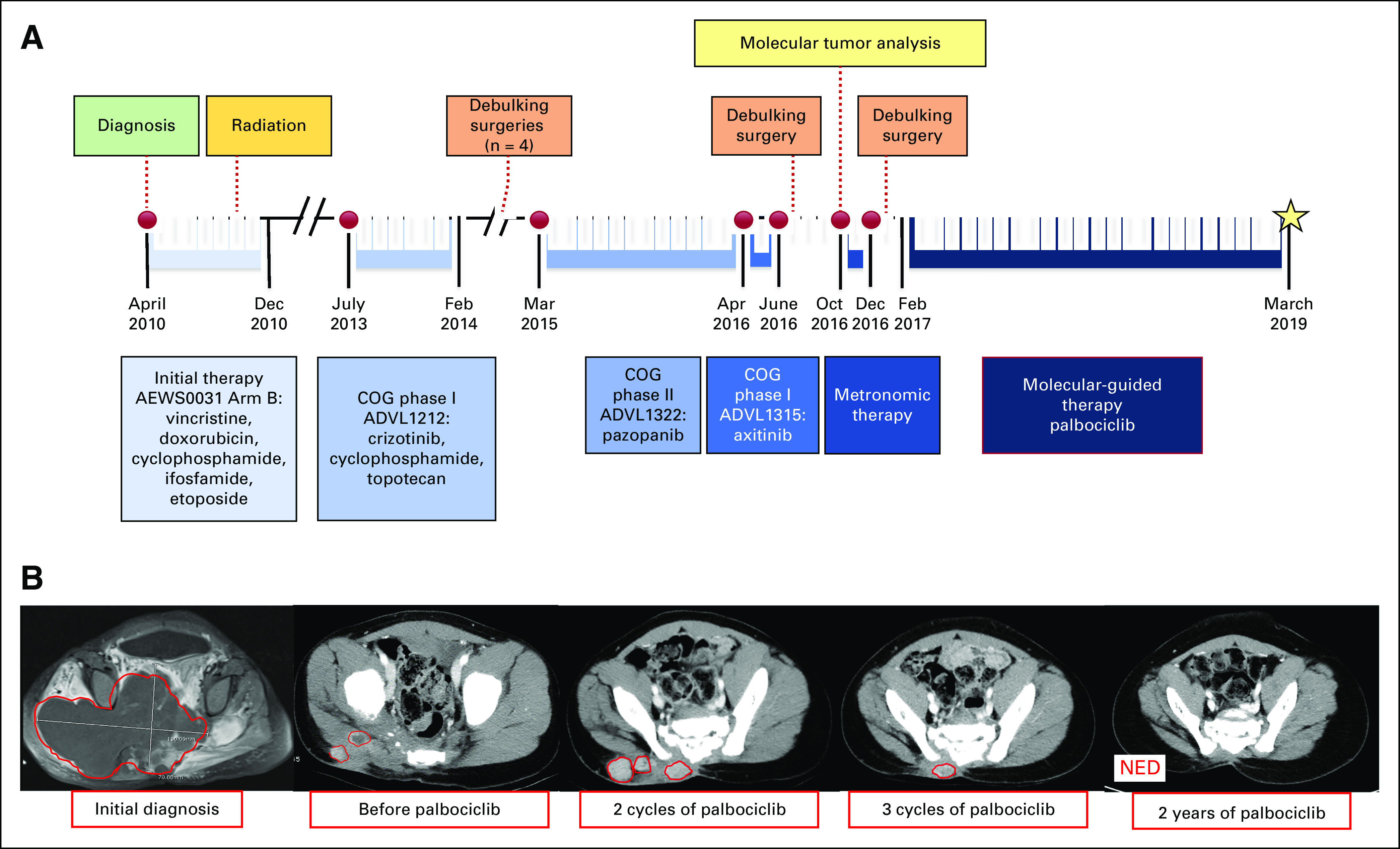
Patient timeline and response imaging. (A) The timeline of treatments and disease recurrences and progression in our patient. Red circles represent time of recurrence or progression. Hash marks represent prolonged period of time progressed through therapy (not to scale of remaining timeline). Patient is still continuing with palbociclib therapy after the most recent scans in March 2019. (B) Patient imaging displayed with initial diagnostic magnetic resonance imaging from 2010; subsequent relapse scans were done via computed tomography imaging. Red lines outline tumor boundaries. There was concern for progression after 2 cycles of palbociclib, but there was a 2-month lag between the “before palbociclib” scan and actually starting drug, so interval progression likely occurred in this timeframe. No new baseline scan was performed on the day palbociclib started. Each cycle of palbociclib was 28 days. NED, no evidence of disease.
Results
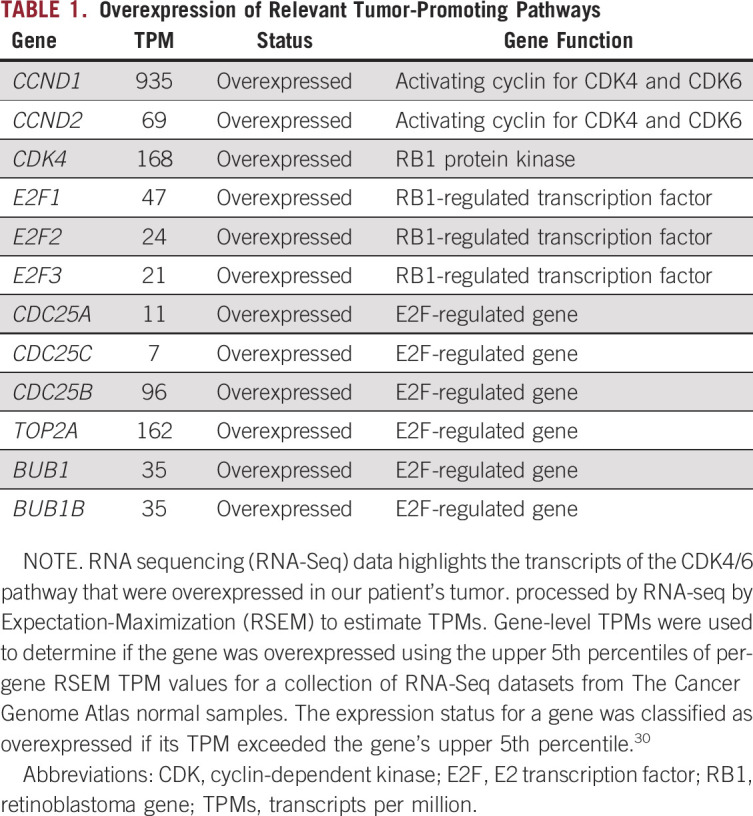
Whole-genome sequencing, RNA sequencing (RNA-Seq) analysis, germline exome sequencing, and protein evaluation were performed at the Clinical Laboratory Improvement Amendment (CLIA)–approved laboratory, NantOmics (Culver City, CA). Somatic DNA changes were determined by comparing the whole-genome DNA sequence from the tumor with the patient’s germline sequence at 33X coverage. The mutational burden of the tumor was relatively low at 75,046 somatic mutations, with only 88 somatic mutations mapping to protein coding regions (Circos plot in Fig 2A). The tumor harbored an in-frame fusion of the second base in the last codon of BCOR exon 15 (chrX:39,911,366) and the first base of CCNB3 exon 5 (chrX:50,051,505) (Fig 2B). Additionally, an undescribed somatic mutation in the SMO gene (SMO N476S) was identified in the tumor, and germline sequencing revealed a CDKN2B N41D missense variant, which was heterozygous in both the germline and tumor genomes of this patient. RNA-Seq was also performed by NantOmics, and mRNA transcripts were ranked by abundance, which could be associated with increased pathway activity and sensitivity to a targeted drug. Overexpression of relevant tumor-promoting pathways is displayed in Table 1 and Figure 2C.
FIG 2.

Transcriptome analysis reveals activation of the cyclin-dependent kinase 4/6 (CDK4/6)-RB pathway. (A) Circos plot of patient’s tumor genome. (B) Identification of BCOR-CCNB3 fusion. This graphic illustrates the fusion of BCOR exon 15 to CCNB3 exon 5. With the exception of the destruction box in CCNB3, all functional domains from each encoded protein remain intact in the BCOR-CCNB3 fusion protein. (C) The CDK4/6 pathway is a gene regulatory program controlled by multiple tiers of protein kinases and transcriptional regulators. Increases in cyclin-D or CDK4 or 6 protein can lead to phosphorylation of the RB1-E2F tumor suppressor complex. Upon phosphorylation of RB1, the E2F1-3 transcription factors are released from the complex and are able to bind to the promoters of target genes, driving activation of transcription. These target genes include E2F1-3 themselves, the CDC25 genes, TOP2A, BUB1, and BUB1B. WT, wild type.
TABLE 1.
Overexpression of Relevant Tumor-Promoting Pathways
Discussion
Germline and somatic whole-genome DNA sequencing combined with RNA sequencing was used with the goal of developing a treatment plan, and it surprisingly provided our team with a more defined diagnosis of a recurrent BCS. The specific intrachromosomal fusion between BCOR and CCNB3 in our patient’s tumor is identical to previously described cases.5,6 The BCOR gene itself can fuse to a number of 3′ partner genes in round cell sarcomas or additionally have internal tandem duplications, which have been reported to drive similar transcriptional patterns in a variety of sarcomas.5,8,9 Similar to previous studies of mRNA transcripts in BCS,5,8,9 both BCOR and CCNB3 transcripts were highly overexpressed in our patient’s tumor, as were the HOX-A, -B, and -C gene clusters (Table 2). Furthermore, analysis of our patient’s tumor (Table 1) matched the BCS-specific fingerprint of genes used in the Riggi_Ewing_sarcoma_progenitor signature, which can be used to distinguish between BCS and EWS.5,10 Most notable for this patient was the observation that multiple genes in the CDK4/6–RB pathway (Table 1; Fig 2C) were overexpressed, which made palbociclib, which has known pediatric dosing information, an attractive drug to use in this case.
TABLE 2.
Comparison of Patient Transcriptome With Published BCS Reference Publications

Curiously, our patient was diagnosed at a very young age compared with the literature on BCS. In the several case series describing BCS, the median age of diagnosis is in the teenage years, with the youngest patient recorded at age 2.5-8,11 Again, because of the age of presentation, one could be concerned about an inherited cancer syndrome. Alfaro-Cervello et al12 published a case report of a congenital undifferentiated sarcoma with BCOR-CCNB3 fusion possibly similar to our patient case. This congenital tumor also harbored a SMARCB1/INI1 gene deletion common to malignant rhabdoid tumor, epithelioid sarcomas, and epithelioid malignant peripheral nerve sheath tumor that also, when found germline, is known to cause rhabdoid tumor predisposition syndrome.12-16 In the case reported by Alfaro-Cervello et al,12 INI1 germline analysis was not performed. Our patient’s tumor had functionally intact INI1, which precludes an effective comparison. Our patient also harbored a germline heterozygous missense variant, CDKN2B N41D. It is unclear what role this germline CDKN2B N41D variant could play in sarcomagenesis, as cancer risks associated with CDKN2A/B gene variants include melanoma, pancreatic cancer, and astrocytomas.17,18 There is a recent short report from Jouenne et al19 that found an increased risk of soft tissue sarcoma development with germline loss of CDKN2A, though no data exist confirming this risk with CDKN2B variants. Additionally, little is known about this actual variant in CDKN2B. Sunita et al20 showed that the specific CDKN2B N41D variant, which encodes p15(INK4B), is unable to bind to the CDK6 protein, leading to loss of function of CDKN2B, which could lead to dysregulated control of S-phase entry. Though this variant’s contribution to tumorigenesis is intriguing, CKDN2B was normally expressed in our patient’s tumor, and there are no data suggesting that this impaired binding to CDK6 leads to mRNA overexpression along multiple levels of the CDK4/6 pathway.
Despite discovering alterations of several key regulators of the CDK4/6 pathway in this tumor, none have been proven to serve as clinical biomarker for sensitivity to CDK4/6 inhibitors.21 In a preclinical Ewing sarcoma orthotopic xenograft model with CDKN2A deletion, palbociclib was able to greatly suppress growth despite doxorubicin resistance of this model.22 In other sarcoma subtypes, palbociclib reduced tumor burden in murine preclinical models.23-25 Clinically, there is phase II evidence of palbociclib’s efficacy in adults with liposarcoma26,27 and leiomyosarcoma.28 Despite growing evidence in these sarcomas, there are no published data testing CDK4/6 inhibitors in BCS. Additionally, though phase I/II trials are underway, the only published response data for palbociclib in pediatrics is a case report of growing teratoma syndrome,29 thus making our use of this drug in a child novel.
To summarize, 3 independent observations supported consideration of therapeutic inhibition of the CDK4/6-RB1 pathway for this patient: (1) the presence of the BCOR-CCNB3 gene fusion believed to drive entry into the cell cycle, (2) direct detection of an active CDK4/6-RB1 pathway, and (3) the presence of a germline CDKN2B variant. Using this information, our Precision Genomics team chose to place our patient with multiply-relapsed disease on palbociclib; the patient has now benefited from > 2 years of disease remission. The sustained complete response with palbociclib in our patient makes this case a novel and interesting application of palbociclib use and argues for additional research using CDK4/6 inhibitors in BCS.
ACKNOWLEDGMENT
We thank Grzegorz J. Nalepa, Department of Pediatrics, Indiana University School of Medicine, the physician of this patient and who laid the clinical and scientific foundation for this work, which came to fruition after his passing.
SUPPORT
Supported by Grant No. U54HD16014 (to J.L.R. on behalf of the Precision Genomics Team at Indiana University School of Medicine) with the Indiana University Center for Pediatric Pharmacology and Precision Medicine.
AUTHOR CONTRIBUTIONS
Conception and design: Timothy F. Tramontana, Mark S. Marshall, Morgan R. Schmitt, Jacquelyn L. Carter, Jamie L. Renbarger, Michael J. Ferguson
Collection and assembly of data: Timothy F. Tramontana, Mark S. Marshall, Amy E. Helvie, Jennifer Ivanovich, Michael J. Ferguson
Data analysis and interpretation: Mark S. Marshall, Amy E. Helvie, Jennifer Ivanovich, Jamie L. Renbarger, Michael J. Ferguson
Provision of study material or patients: Amy E. Helvie
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Michael J. Ferguson
Consulting or Advisory Role: Bayer
No other potential conflicts of interest were reported.
REFERENCES
- 1.Whang-Peng J, Triche TJ, Knutsen T, et al. Cytogenetic characterization of selected small round cell tumors of childhood. Cancer Genet Cytogenet. 1986;21:185–208. doi: 10.1016/0165-4608(86)90001-4. [DOI] [PubMed] [Google Scholar]
- 2.Turc-Carel C, Aurias A, Mugneret F, et al. Chromosomes in Ewing’s sarcoma: I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12) Cancer Genet Cytogenet. 1988;32:229–238. doi: 10.1016/0165-4608(88)90285-3. [DOI] [PubMed] [Google Scholar]
- 3.Ginsberg JP, de Alava E, Ladanyi M, et al. EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s sarcoma. J Clin Oncol. 1999;17:1809–1814. doi: 10.1200/JCO.1999.17.6.1809. [DOI] [PubMed] [Google Scholar]
- 4.Shing DC, McMullan DJ, Roberts P, et al. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res. 2003;63:4568–4576. [PubMed] [Google Scholar]
- 5.Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–466. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- 6.Peters TL, Kumar V, Polikepahad S, et al. BCOR-CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod Pathol. 2015;28:575–586. doi: 10.1038/modpathol.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puls F, Niblett A, Marland G, et al. BCOR-CCNB3 (Ewing-like) sarcoma: A clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am J Surg Pathol. 2014;38:1307–1318. doi: 10.1097/PAS.0000000000000223. [DOI] [PubMed] [Google Scholar]
- 8.Kao YC, Owosho AA, Sung YS, et al. : BCOR-CCNB3 fusion–positive sarcomas: A clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol, 42:604-615, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson S, Perrin V, Guillemot D, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018;245:29–40. doi: 10.1002/path.5053. [DOI] [PubMed] [Google Scholar]
- 10.Riggi N, Suvà ML, Suvà D, et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008;68:2176–2185. doi: 10.1158/0008-5472.CAN-07-1761. [DOI] [PubMed] [Google Scholar]
- 11.Matsuyama A, Shiba E, Umekita Y, et al. Clinicopathologic diversity of undifferentiated sarcoma with BCOR-CCNB3 fusion: Analysis of 11 cases with a reappraisal of the utility of immunohistochemistry for BCOR and CCNB3. Am J Surg Pathol. 2017;41:1713–1721. doi: 10.1097/PAS.0000000000000934. [DOI] [PubMed] [Google Scholar]
- 12.Alfaro-Cervello C, Andrade-Gamarra V, Nieto G, et al. Congenital undifferentiated sarcoma associated to BCOR-CCNB3 gene fusion. Pathol Res Pract. 2017;213:1435–1439. doi: 10.1016/j.prp.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 13.Judkins AR, Mauger J, Ht A, et al. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol. 2004;28:644–650. doi: 10.1097/00000478-200405000-00013. [DOI] [PubMed] [Google Scholar]
- 14.Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012–4019. doi: 10.1158/0008-5472.CAN-04-3050. [DOI] [PubMed] [Google Scholar]
- 15.Hornick JL, Dal Cin P, Fletcher CDM. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542–550. doi: 10.1097/PAS.0b013e3181882c54. [DOI] [PubMed] [Google Scholar]
- 16.Sredni ST, Tomita T. Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol. 2015;18:49–58. doi: 10.2350/14-07-1531-MISC.1. [DOI] [PubMed] [Google Scholar]
- 17.Chan AK, Han SJ, Choy W, et al. Familial melanoma-astrocytoma syndrome: Synchronous diffuse astrocytoma and pleomorphic xanthoastrocytoma in a patient with germline CDKN2A/B deletion and a significant family history. Clin Neuropathol. 2017;36:213–221. doi: 10.5414/NP301022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campa D, Pastore M, Gentiluomo M, et al. Functional single nucleotide polymorphisms within the cyclin-dependent kinase inhibitor 2A/2B region affect pancreatic cancer risk. Oncotarget. 2016;7:57011–57020. doi: 10.18632/oncotarget.10935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jouenne F, Chauvot de Beauchene I, Bollaert E, et al. Germline CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma. J Med Genet. 2017;54:607–612. doi: 10.1136/jmedgenet-2016-104402. [DOI] [PubMed] [Google Scholar]
- 20.Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94:1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: Mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3:39–55. doi: 10.1016/j.trecan.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami T, Singh AS, Kiyuna T, et al. Effective molecular targeting of CDK4/6 and IGF-1R in a rare FUS-ERG fusion CDKN2A-deletion doxorubicin-resistant Ewing’s sarcoma patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget. 2016;7:47556–47564. doi: 10.18632/oncotarget.9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez M, Muñoz-Galván S, Jiménez-García MP, et al. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget. 2015;6:40557–40574. doi: 10.18632/oncotarget.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vlenterie M, Hillebrandt-Roeffen MH, Schaars EW, et al. Targeting cyclin-dependent kinases in synovial sarcoma: Palbociclib as a potential treatment for synovial sarcoma patients. Ann Surg Oncol. 2016;23:2745–2752. doi: 10.1245/s10434-016-5341-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Böhm MJ, Marienfeld R, Jäger D, et al. Analysis of the CDK4/6 cell cycle pathway in leiomyosarcomas as a potential target for inhibition by palbociclib. Sarcoma. 2019;2019:3914232. doi: 10.1155/2019/3914232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson MA, Tap WD, Keohan ML, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013;31:2024–2028. doi: 10.1200/JCO.2012.46.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: A phase 2 clinical trial. JAMA Oncol. 2016;2:937–940. doi: 10.1001/jamaoncol.2016.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elvin JA, Gay LM, Ort R, et al. Clinical benefit in response to palbociclib treatment in refractory uterine leiomyosarcomas with a common CDKN2A alteration. Oncologist. 2017;22:416–421. doi: 10.1634/theoncologist.2016-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schultz KA, Petronio J, Bendel A, et al. : PD0332991 (palbociclib) for treatment of pediatric intracranial growing teratoma syndrome. Pediatr Blood Cancer 62:1072-1074, 2015 [DOI] [PubMed] [Google Scholar]
- 30.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]