

Abstract
PURPOSE
Circulating cell-free tumor DNA (ctDNA) reflects the heterogeneous spectrum of tumor-specific mutations, especially in systemic disease. We validated plasma-based assays that allow the dynamic quantitative detection of ctDNA as a prognostic biomarker for tumor load and prediction of therapy response in melanoma.
MATERIALS and METHODS
We analyzed plasma-derived ctDNA from a large training cohort (n = 96) of patients with advanced-stage melanoma, with assays for the BRAFV600E and NRASQ61 driver mutations as well as TERTC250T and TERTC228T promoter mutations. An independent patient cohort (n = 35) was used to validate the utility of ctDNA monitoring under mitogen-activated protein kinase–targeted or immune checkpoint therapies.
RESULTS
Elevated plasma ctDNA level at baseline was an independent prognostic factor of disease progression when compared with serum S100 and lactate dehydrogenase levels in multivariable analyses (hazard ratio [HR], 7.43; 95% CI, 1.01 to 55.19; P = .05). The change in ctDNA levels during therapy correlated with treatment response, where increasing ctDNA was predictive for shorter progression-free survival (eg, for BRAFV600E ctDNA, HR, 3.70; 95% CI, 1.86 to 7.34; P < .001). Increasing ctDNA levels predicted disease progression significantly earlier than did routine radiologic scans (P < .05), with a mean lead time of 3.5 months. NRAS-mutant ctDNA was detected in a significant proportion of patients with BRAF-mutant tumors under therapy, but unexpectedly also at baseline. In vitro sensitivity studies suggested that this represents higher-than-expected intratumoral heterogeneity. The detection of NRASQ61 ctDNA in baseline samples of patients with BRAFV600E mutation who were treated with mitogen-activated protein kinase inhibitors significantly correlated with shorter progression-free survival (HR, 3.18; 95% CI, 1.31 to 7.68; P = .03) and shorter overall survival (HR, 4.08; 95% CI, 1.57 to 10.58; P = .01).
CONCLUSION
Our results show the potential role of ctDNA measurement as a sensitive monitoring and prediction tool for the early assessment of disease progression and therapeutic response in patients with metastatic melanoma.
INTRODUCTION
Circulating cell-free tumor DNA (ctDNA) has emerged as a potential noninvasive blood biomarker for monitoring tumor load and detecting clinically actionable driver and resistance mutations in patients with melanoma who are receiving therapy.115 Most studies published on ctDNA in melanoma have been, in essence, proof-of-principle reports investigating technical feasibility or focusing on ctDNA courses of individual patients (mostly BRAFV600E-positive patients) based on retrospective sample and data collections. Furthermore, the mutational status of melanoma tissue biopsy specimens was mostly used as the gold standard or reference for ctDNA analyses. However, this does not account for the divergence of mutational states in blood versus tissue, because of intra- and intertumor heterogeneity or the different detection sensitivities of blood- versus tissue-based assays. As more reports are published on the reliability of ctDNA analysis as a monitoring tool for tumor burden, the question emerges as to whether blood, rather than tissue, should be considered the primary source of genetic information to inform precision oncology.
Here, we analyzed a large training cohort and an independent validation cohort of patients with metastatic melanoma monitored for the most frequently occurring mutations in BRAF, NRAS, and the promoter region of the TERT gene—estimated to cover 80% of patients.1 We established ctDNA thresholds for disease monitoring and examined the relationship between ctDNA responses to therapies, including mitogen-activated protein kinase (MAPK) and immune checkpoint inhibition, and correlated the dynamic changes of ctDNA with clinical benefit. Furthermore, we tested the concordance of plasma ctDNA with the presence of mutations in matched tumor tissues, highlighting the importance of sensitive mutation detection at therapy baseline.
MATERIALS AND METHODS
Patient Training Cohort and Healthy Control Subjects
A total of 560 plasma samples from 96 patients with advanced stage III or IV melanoma were obtained from the Department of Dermatology, University Hospital Essen (Fig 1A; Data Supplement). Plasma samples were collected as part of standard care or within clinical trials. Patients were treated with MAPK signaling-targeted drugs (n = 58), immune checkpoint inhibitors (n = 33), or with signaling targeted therapy in combination with chemotherapy (n = 5; Data Supplement). Patients were eligible for plasma monitoring on the basis of histologically confirmed stage III or IV melanoma and positive detection status for BRAFV600E, NRASQ61, TERTC228T, or TERTC250T mutations in a previous tumor biopsy specimen. Patients were included in ctDNA monitoring when they had available baseline and a minimum of three consecutive plasma samples during systemic therapy collected at approximately 4- to 6-week follow-up intervals. Baseline plasma samples were collected an average of 15 days (95% CI, 9 to 21 days) before therapy start. Tumor tissue biopsy specimens were acquired an average of 114 days (95% CI, 74 to 155 days) before therapy start. Computed tomography (CT), magnetic resonance imaging (MRI), or ultrasound imaging (US) was carried out at baseline and at every 6 to 12 weeks (± 2 weeks of plasma sampling time points). Radiologic tumor response of the training cohort was evaluated in radiology departments or radiologists’ offices under real-life circumstances (ie, comparing cross-sectional images with the immediate preceding examination or baseline examination for decision-making). Based on free-text reports, tumor responses were retrospectively classified by the study investigators as complete response, partial response, stable disease, and progressive disease.
FIG 1.
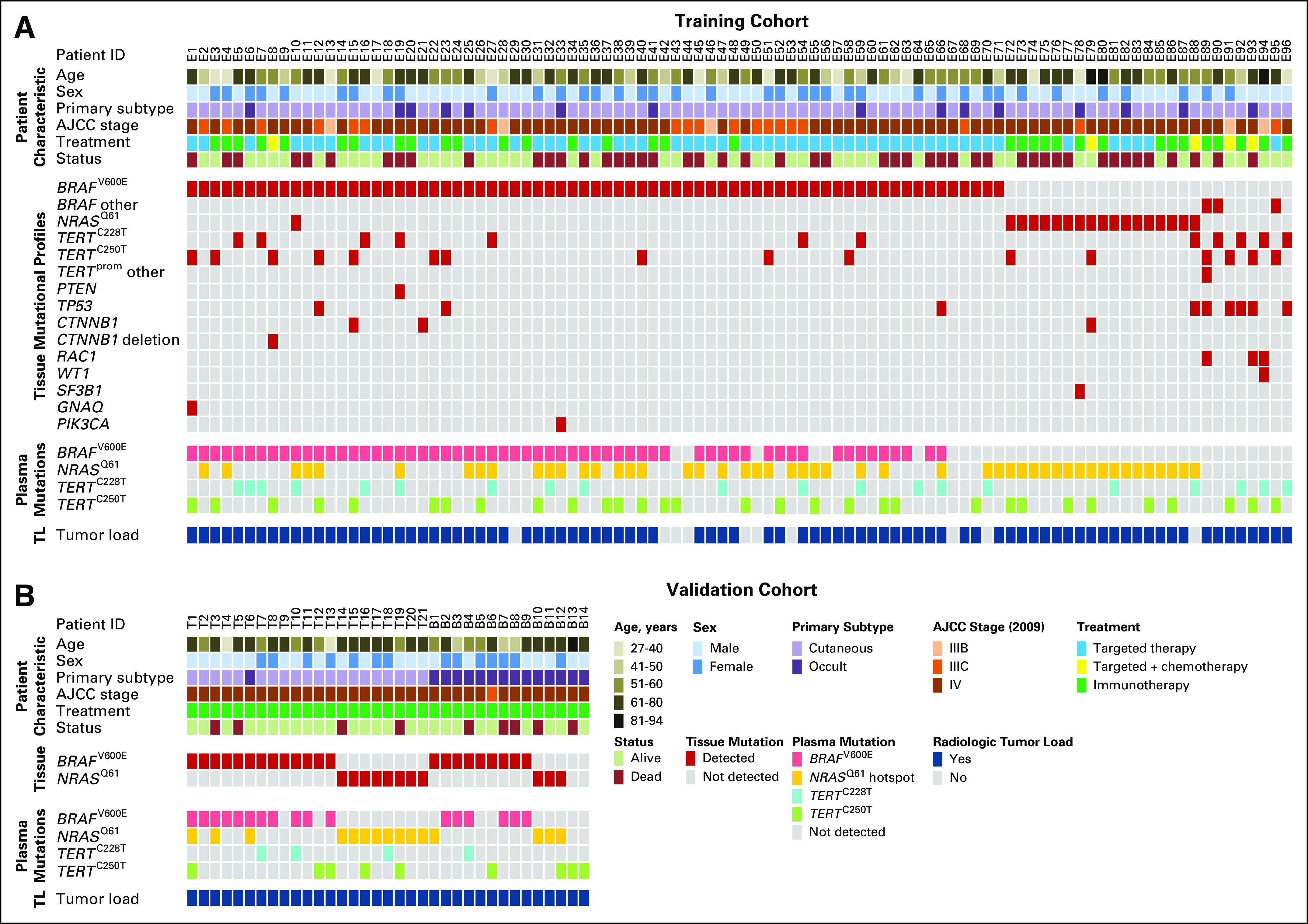
Basic characteristics of patients with melanoma who have BRAFV600E, NRASQ61, and TERTprom mutations. Overview of (A) the training cohort (n = 96 patients with stage III and IV disease) and (B) the validation cohort (n = 35 patients with stage III and IV disease). In the upper panels, the demographic and tumor characteristics are represented. The middle panels show mutations detected by DNA sequencing in respective tumor samples. The lower panels correspond to respective plasma samples analyzed with droplet digital polymerase chain reaction. Radiologic tumor-load information at baseline is represented at the bottom of the panels. See also the Data Supplement. AJCC, American Joint Committee on Cancer; ID, identification; TL, tumor load.
Aliquots from 96 standard plasma preparations from random, healthy blood donors were provided by the Institute of Transfusion Medicine of the Essen University Hospital. According to routine presampling questionnaires, plasma donors were negative for malignant diseases in their history. The experiments were approved by the Ethics Commission of the Medical Faculty of the University Duisburg-Essen (approval No. 16-7132-BO). Technical assay details and statistical data processing are described in the Data Supplement.
Patient Validation Cohort
In total, 104 plasma samples from 35 patients with advanced stage III or IV disease were prospectively collected either within pharmaceutical clinical trials, early access programs, or biobanking protocols at the Department of Dermatology of the Tübingen University Medical Center and the Department of Medical Oncology of the Universitair Ziekenhuis in Brussels (Fig 1B; Data Supplement). Informed consent procedures were followed (approval Nos. 316/2018BO2 and BUN 143201421920, respectively). Patients from Tübingen were treated with pembrolizumab (n = 17) or nivolumab plus ipilimumab (n = 4) at the Department of Dermatology of the Tübingen University Medical Center. The patients at Universitair Ziekenhuis in Brussels were treated with pembrolizumab (n = 14). The eligibility criteria were the same as for the training cohort. Patients with available baseline and a minimum of two consecutive plasma samples during systemic therapy were included in ctDNA monitoring. Baseline plasma samples were collected an average of 5 days (95% CI, 1 to 11 days) before therapy start. Radiologic tumor response evaluation of the validation cohort was performed according to immune-related response criteria (Brussels) or Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 (Tübingen).
RESULTS
ctDNA Level Correlated With Melanoma Tumor Burden and Was a Risk Factor for Disease Progression at Baseline
Previous reports suggested a correlation between ctDNA levels and tumor burden in patients with melanoma.2-7 For this study, candidate mutations for ctDNA monitoring were selected on the basis of the most frequently occurring genomic changes in melanoma (estimated to cover 80% of patients; Data Supplement). After technical validation (Data Supplement), we assessed plasma ctDNA levels of our training cohort at baseline by droplet digital polymerase chain reaction (ddPCR). As expected, the baseline ctDNA level showed a significant correlation with increasing tumor stage (Welch’s t test P < .05; Figs 2A-2D). The routinely used tumor monitoring marker lactate dehydrogenase (LDH) was weakly associated with tumor stage, and serum S100 level had no association with tumor stage (Fig 2E). The rank correlation between ctDNA and serum LDH levels (ρ = 0.43, 0.27, and 0.18 in the BRAFV600E, NRASQ61, and TERTprom data sets, respectively; Figs 2F, 2G, and 2H) and the correlation with serum S100 levels (ρ = 0.38, 0.27, and 0.12; Figs 2I, 2J, and 2K) was also weak.
FIG 2.
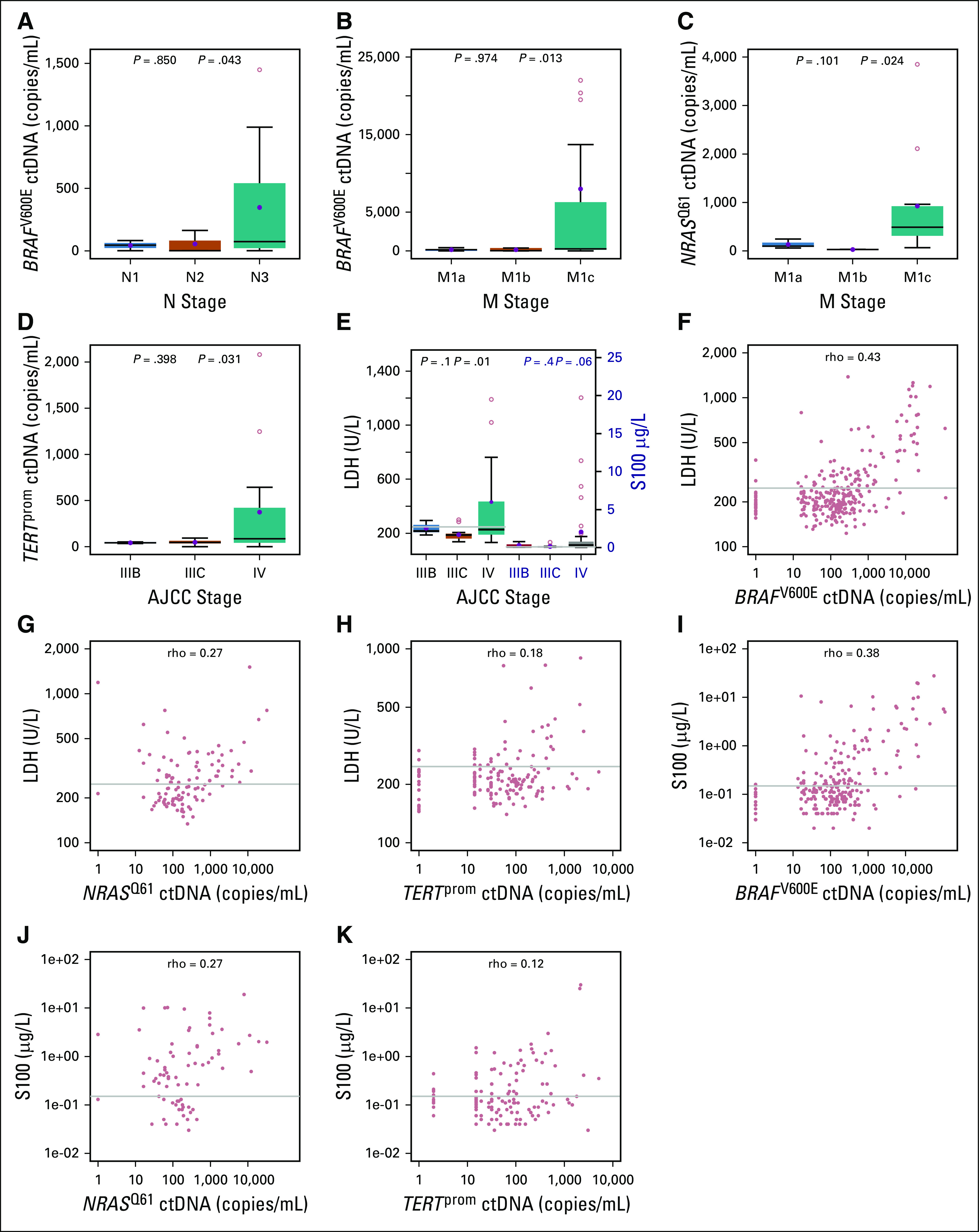
Correlation of baseline circulating cell-free tumor DNA (ctDNA) levels with metastatic stage and routine serum markers in the training cohort. Correlation of baseline BRAFV600E ctDNA levels with increasing metastatic tumor load in (A) lymph nodes (N1, n = 3; N2, n = 3; and N3, n = 14) and (B) organs (M1a, n = 5; M1b, n = 5; M1c, n = 41). (C) Correlation of NRASQ61 ctDNA levels with organ metastasis (M1a, n = 3; M1b, n = 2; M1c, n = 11). (D) Correlation of mutant TERTprom ctDNA levels with metastatic progression from locoregional (stage IIIB, n = 2; or IIIC, n = 6) to systemic disease (stage IV; n = 14) in patients with TERTprom-mutated melanomas. (E) Correlation of baseline serum lactate dehydrogenase (LDH) and S100 levels with metastatic progression in BRAFV600E-positive patients (gray line, upper limit of normal). Box-and-whisker plots represent median values and interquartile range. Blue dots represent mean values. Welch t test was used to calculate statistical significance. (F-K) Correlation of serum LDH and S100 levels with BRAFV600E (n = 274 and n = 216, respectively), NRASQ61 (n = 97 and n = 67, respectively), and mutant TERTprom (n = 152 and n = 124, respectively) ctDNA levels in the training cohort. Sample pairs were analyzed for the Spearman correlation coefficient (ρ). The upper limit of normal (gray line) for LDH is 247 IU/L and for S100 is 0.15 µg/L. AJCC, American Joint Committee on Cancer.
To determine the physiologic background noise of the selected ctDNAs, we acquired plasma samples of healthy donors with no known history of malignant disease (Data Supplement). We found that a ctDNA level above the physiologic thresholds identified by subsequent receiver operating characteristic (ROC) analyses (Data Supplement) was a significant risk factor in univariate analysis (hazard ratio [HR], 8.48; 95% CI, 1.16 to 62.05; log-rank test P = .03) for disease progression of patients with melanoma. Elevated levels of LDH (HR, 1.79; 95% CI, 1.08 to 2.98; P = .02) but not S100 (HR, 1.30; 95% CI, 0.78 to 2.18; log-rank test P = .31) represented a significant predictor of progression in univariate analysis. However, in multivariable analysis, only the elevated ctDNA level significantly correlated with disease progression (HR, 7.43; 95% CI, 1.01 to 55.19; log-rank test P = .05; Data Supplement).
ctDNA Profiles of Patients With Melanoma Correlated With Radiologic Response and Tumor Progression Under Therapy
In addition, receiver operating characteristic (ROC) curves were computed for longitudinally assessed BRAFV600E, NRASQ61, and TERTprom ctDNA, as well as serum LDH and S100 levels, on the basis of timely matching CT/MRI results from patients with melanoma. The areas under the curve of all ctDNA assays were superior to LDH and S100. For example, the difference in area under the curve between BRAFV600E ctDNA and LDH was 23.6% (Delong’s test P < .001) and between BRAFV600E and S100 was 20.7% (P < .001; Data Supplement). Furthermore, the ROC analysis allowed determination of ctDNA thresholds that we could use subsequently to evaluate the predictive impact of ctDNAs during therapy (Data Supplement).
To assess ctDNA levels under targeted and immune checkpoint inhibition, 540 plasma samples from 76 patients were analyzed by ddPCR and compared with radiologic staging. Patients with radiologic evidence of response had statistically significant decreasing ctDNA levels irrespective of the mutation or drug type. Nonresponders had rising ctDNA levels (Figs 3A and 3B, and Figs 4A, 4B, 4C, and 4D). Targeted therapies seemed to decrease ctDNA levels faster than immune checkpoint inhibitors (mean decrease of 31% v 15% at week 4; P < .001; combined analysis of data from Figs 3A and 3B, and Figs 4A, 4B, 4C, and 4D, differences remained significant: unpaired t test P < .001 for all time points). The detected changes in ctDNA level correlated with the duration of progression-free survival (PFS) under therapy. For example, in the BRAFV600E data set, the dynamic increase in ctDNA (at the second sampling time point [ie, 10 weeks after receiving therapy]) relative to the baseline was significantly associated with decreased PFS irrespective of the treatment (immunotherapy: HR, 3.98, 95% CI, 1.32 to 11.97, log-rank test P = .05, Fig 3C; targeted therapy: HR, 3.49, 95% CI, 1.45 to 8.40, P = .015, Fig 3D).
FIG 3.

Changes in the BRAFV600E circulating cell-free tumor DNA (ctDNA) levels correlate with therapy response and progression-free survival (PFS). Changes in mean BRAFV600E ctDNA levels after therapy initiation relative to baseline (BL) in patients who received (A) immune checkpoint inhibition therapy (n = 18 patients) and (B) signaling targeted therapy (n = 33). Follow-up (FU) sampling was performed every 4 to 6 weeks. (*) P < .01, (†) P < .001, and (‡) P < .001 from unpaired t test. The data represent mean ± SEM. Kaplan-Meier plots are for PFS of the same patients with melanoma as assessed by routine radiologic scans. (C) Immune checkpoint inhibition (patients with ctDNA decrease [n = 6] v increase [n = 12]) and (D) signaling targeted therapy (patients with ctDNA decrease [n = 12] v increase [n = 21]). Categorization into decrease versus increase was based on the ctDNA change at the second sampling time point relative to BL. The hazard ration (HR) is indicated for ctDNA increase. The P value was determined by the log-rank test. (E, F) Scatter dot plots of BRAFV600E ctDNA levels of responders versus nonresponders grouped according to (E) radiologic response 10 weeks after receiving any therapy or (F) radiologic PFS at 6 months of therapy (Mann-Whitney U test). Points represent individual patients; median with interquartile range is indicated for each plot. Gray lines indicate ctDNA thresholds as determined by receiver operating characteristic analyses (Data Supplement).
FIG 4.

Changes of NRASQ61 and TERTprom circulating cell-free tumor DNA (ctDNA) levels correlated with therapy response and progression-free survival (PFS). Changes in mean NRASQ61 and TERTprom ctDNA levels after therapy initiation relative to baseline (BL) in patients who received (A, C) immune checkpoint inhibition therapy, (n = 10 and n = 12, respectively) and (B, D) signaling targeted therapy, (n = 7 and n = 10, respectively). Follow-up (FU) sampling was performed every 4 to 6 weeks. (*) P < .05, (†) P < .01, (‡) P < .001, and (§) P < .001 from unpaired t test. The data are reported as mean ± SEM. Scatter dot plots of (E) NRASQ61 and (F) TERTprom ctDNA levels of patients assessed at week 10 after receiving any therapy, grouped according to radiologic PFS at 6 months (Mann-Whitney U test). Points represent individual patients; median with interquartile range is indicated for each plot. Gray lines indicate ctDNA thresholds as determined by receiver operating characteristic analyses (Data Supplement).
Regarding absolute copy numbers of BRAFV600E ctDNA across all patients, the median value of nonresponders was 209.5 copies/mL at 10 weeks of treatment (ie, significantly higher than in responders [16 copies/mL; Mann-Whitney U test P < .001]; Fig 3E) and above the threshold defined by ROC analysis (35.85 copies/mL; Data Supplement). Accordingly, the median was 494.5 copies/mL in patients with a PFS shorter than 6 months (ie, significantly higher than in patients with a PFS longer than 6 months [Mann-Whitney U test P < .001]; Fig 3F). Fisher’s exact test confirmed the statistically significant association of the ROC-determined ctDNA threshold and treatment response (P < .001) or 6-month PFS (P = .05).
Similarly, for NRASQ61 and TERTprom ctDNA, patients with a PFS longer than 6 months had significantly fewer copies when compared with patients with shorter PFS (Mann-Whitney U test P < .001 and P = .01 respectively; Figs 4E and 4F). Also, the ROC-determined thresholds (NRASQ61: 51.5 copies/mL; TERTprom: 32.83 copies/mL) significantly discriminated between a PFS longer than 6 months or shorter than 6 months (Fisher’s exact test P = .03 for both). Patients in the NRASQ61 and TERTprom data sets who responded to treatment also had lower absolute ctDNA copy numbers than did nonresponders (Mann-Whitney U test P = .06 and .008, respectively; Data Supplement). Again, the ctDNA threshold significantly discriminated between responders and nonresponders (Fisher’s exact test P = .02 and .005, respectively).
To validate our findings, 35 patients were included who received immune checkpoint inhibition therapy at the University Medical Center Tübingen and at Universitair Ziekenhuis in Brussels. Patients with radiologic therapy response had statistically significant decreasing ctDNA levels irrespective of the mutation or drug type. Nonresponders had increasing ctDNA levels (pooled data of all 35 patients with BRAFV600E, NRASQ61, and TERTprom mutations are shown in Figure 5A). The increase in ctDNA level (at the second sampling time point [ie, 12 weeks after receiving therapy]) relative to the baseline measurement was significantly associated with decreased PFS (HR, 8.53; 95% CI, 2.47 to 29.40; log-rank test P < .001; Fig 5B). Regarding absolute copy numbers of ctDNA across all patients treated, the median ctDNA value of nonresponders was 144 copies/mL at 12 weeks of treatment, which was significantly higher than in responders (12 copies/mL; Mann-Whitney U test P < .001; Fig 5C) and above the threshold defined by ROC analysis in the training cohort (Data Supplement). The median was 127 copies/mL in patients with a PFS shorter than 6 months and, thus, significantly higher than in patients with a PFS longer than 6 months (14 copies/mL; Mann-Whitney U test P = .003; Fig 5D). Fisher’s exact test confirmed a significant association of the ROC-determined ctDNA threshold (Data Supplement) with treatment response (P < .001) or 6-month PFS (P = .002).
FIG 5.
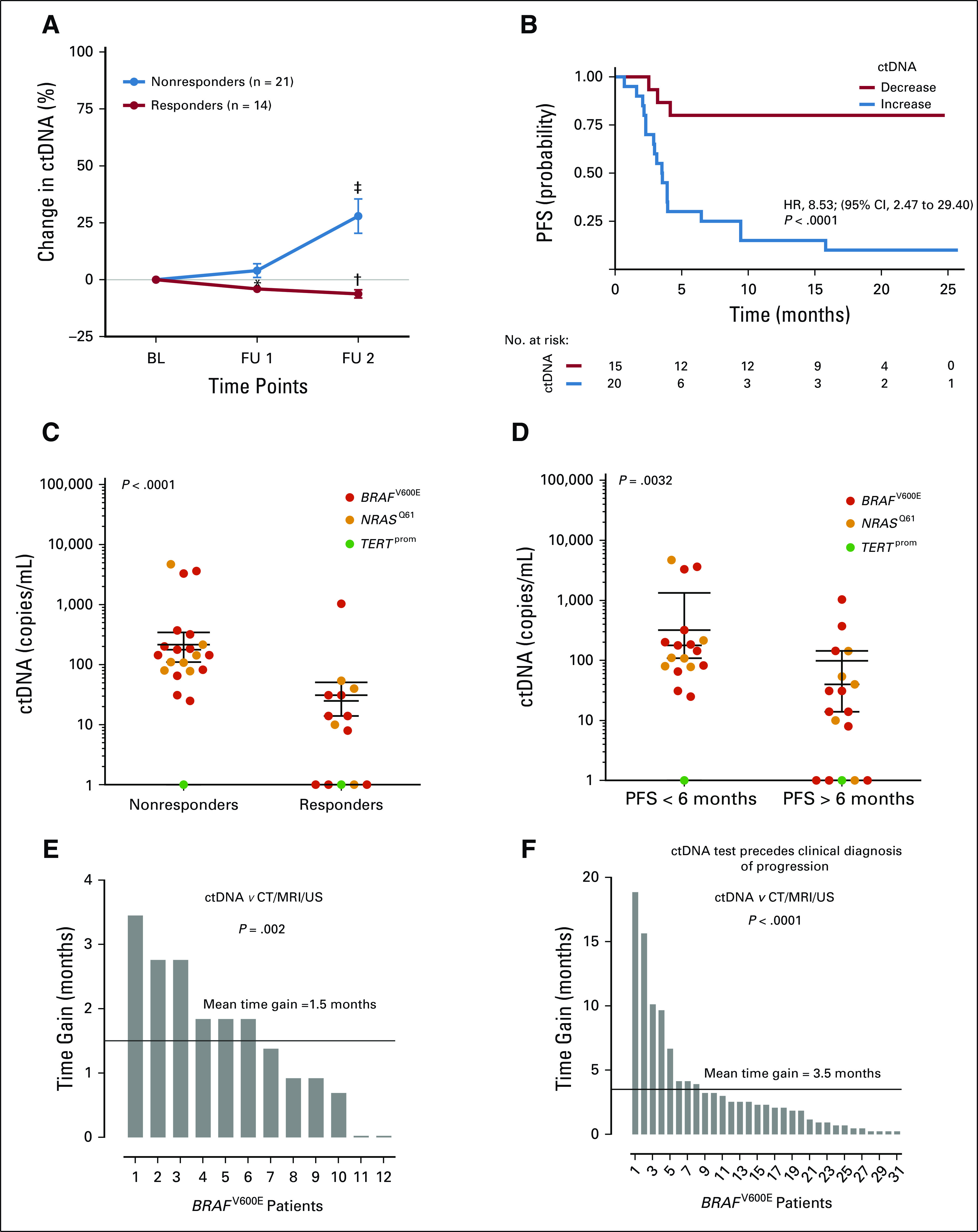
Validation cohort and time gain in assessment of disease progression. (A-D) circulating cell-free tumor DNA (ctDNA) dynamics correlated with clinical outcome in the validation cohort. (A) Changes in mean ctDNA levels after therapy initiation relative to baseline (BL) from patients who received immune checkpoint inhibition therapy (n = 35 patients). Follow-up (FU) sampling was performed every 6 weeks. (*) P < .05, (†) P < .01, and (‡) P < .001 from unpaired t test. The data are reported as mean ± SEM. (B) Kaplan-Meier plot for progression-free survival (PFS) of the same patients with melanoma who received immune checkpoint inhibition therapy who were assessed by routine radiologic scans (patients with ctDNA decrease [n = 15] v increase [n = 20]). Categorization into decrease versus increase was based on the ctDNA change at the second sampling time point relative to BL. Hazard ratio (HR) is indicated for ctDNA increase. The P value was determined by the log-rank test. (C-D) Scatter dot plots of BRAFV600E, NRASQ61, and TERTprom ctDNA levels of responders versus nonresponders, grouped according to (C) radiologic response 12 weeks after receiving any therapy or (D) radiologic PFS at 6 months of therapy (Mann-Whitney U test). Points represent individual patients; median with interquartile range is indicated for each data set. ctDNA as an early predictive parameter for therapy response and failure (data from training cohort). (E) Decreasing BRAFV600E ctDNA levels (as compared with the last sampling time point) preceded radiologic detection of response in 12 of 15 responders with an average lead-time window of 1.5 months (range, 0.023 to 3.45 months). Wilcoxon signed-rank test P values are reported. (F) Increasing BRAFV600E ctDNA levels preceded radiologic progression in 31 of 36 nonresponders, with an average lead-time window of 3.5 months (range, 0.23 to 18.86 months). CT, computed tomography; MRI, magnetic resonance imaging; US, ultrasound.
Changes in ctDNA Profile Indicated Therapy Response and Failure Earlier Than Did Radiologic Scans
A decrease in BRAFV600E ctDNA levels compared with the previous sampling time point preceded radiologic detection of response in 12 (80%) of 15 responders in the training cohort, with an average lead-time window of 1.5 months (range, 0.023 to 3.45 months; Wilcoxon signed-rank test P = .003; Fig 5E). In 31 (86%) of 36 nonresponders, an increase in ctDNA level preceded radiologic progression, with an average lead-time window of 3.5 months (range, 0.23 to 18.86 months; Wilcoxon signed-rank test P < .001; Fig 5F). Similarly, an increase in NRASQ61 ctDNA preceded radiologic progression in 14 (87.5%) of 16 patients and an increase in mutant TERTprom promoter ctDNA in 14 (82%) of 17 patients, with average lead-time windows of 3 months (range, 0.03 to 16.63 months; Wilcoxon signed-rank test P = .001) and 4 months (range, 0.49 to 13.80 months; Wilcoxon signed-rank test P = .001; Data Supplement).
Baseline NRASQ61 ctDNA Was an Independent Predictor of Progressive Disease and Overall Survival in Patients Treated With MAPK Inhibitors
Mutations in NRASQ61 have been reported in patients with resistance to BRAF inhibitors.8,9 Thus, we tested the presence of NRASQ61 ctDNA in longitudinal plasma samples from patients with positive BRAFV600E tumor status under MAPK-targeted therapy (n = 33). Unexpectedly, we detected NRASQ61 ctDNA in 21 of the 33 patients at baseline before treatment. In contrast to the dogma of mutual exclusivity, NRASQ61 ctDNA clearly co-occurred with BRAFV600E ctDNA, however, in the majority of the cases, at lower copy levels (16 of 21 cases). Moreover, the presence of baseline NRASQ61 ctDNA significantly correlated with shorter PFS (HR, 3.18; 95% CI, 1.31 to 7.68; log-rank test P = .03; Fig 6A) and shorter OS (HR, 4.08; 95% CI, 1.57 to 10.58; log-rank test P = .01; Fig 6B) under MAPK inhibitor (MAPKi) treatment. The detection of NRASQ61 ctDNA at baseline of patients with BRAFV600E who were treated with MAPKi (n = 53) was significantly associated with disease progression in univariate analysis (HR, 2.95; 95% CI, 1.35 to 6.44; log-rank test P = .006). Elevated LDH levels (HR, 2.31; 95% CI, 1.12 to 4.75; P = .02) but not S100 levels (HR, 2.04; 95% CI, 0.99 to 4.22; log-rank test P = .51) represented a significant risk factor for progression in univariate analysis, too. However, in multivariable analysis, only elevated NRASQ61 ctDNA levels significantly predicted disease progression (HR, 2.69; 95% CI, 1.17 to 6.16; log-rank test P = .02).
FIG 6.
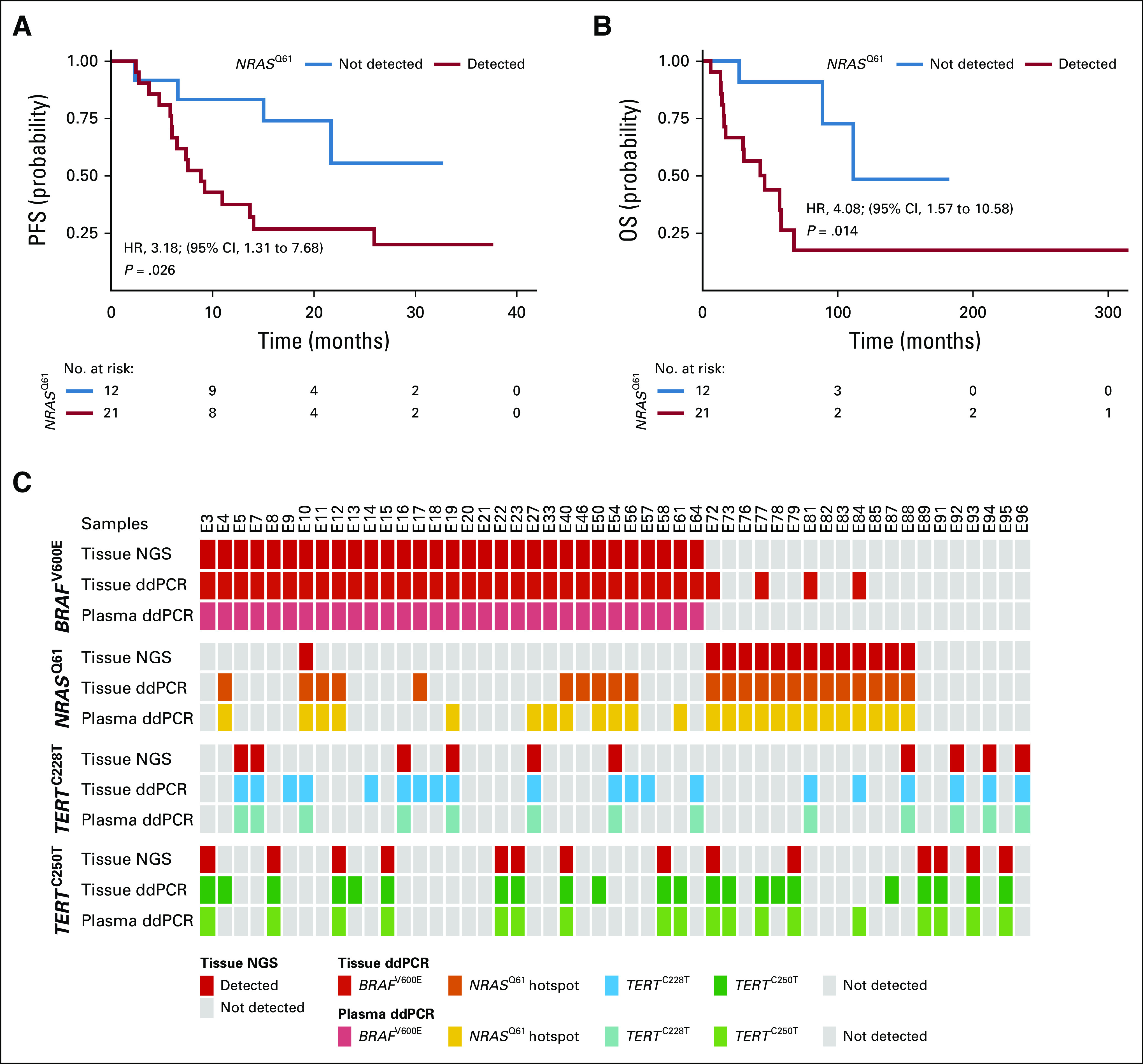
Detection of NRASQ61 at baseline (BL) is a predictor of worse clinical outcome in patients treated with mitogen-activated protein kinase (MAPK) inhibitors. Kaplan-Meier plots for (A) radiologic progression-free survival (PFS) and (B) overall survival (OS) of patients with BRAFV600E-positive tumors who received MAPK signaling targeted therapy (n = 21 with positive NRASQ61 detection in BL plasma v n = 12 without detection). Hazard ratio (HR) is reported for circulating cell-free tumor DNA (ctDNA) detected. P values were determined by the log-rank test. (C) Overview of 51 BRAFV600E-, NRASQ61-, and TERTprom-matched tumor tissue and plasma samples of patients who had available tumor biopsy specimen at therapy BL. Tumor samples were routinely analyzed for BRAFV600E, NRASQ61, and TERTprom mutations with amplicon-based next-generation sequencing (NGS; Fig. 1) and afterward reanalyzed by ddPCR. Plasma was sampled at BL (ie, before systemic therapy initiation) and analyzed for ctDNAs by droplet digital polymerase chain reaction (ddPCR).
To test if potential differences in the assay sensitivities of next-generation sequencing (NGS) versus ddPCR accounted for the observed discrepancies of the patients’ NRASQ61 mutational states, we obtained genomic DNA isolated from 51 melanoma tissue samples that previously underwent amplicon-targeted NGS and reanalyzed them on the ddPCR platform. Indeed, of 12 patients with BRAFV600E mutation whose plasma samples were positive for NRASQ61 ctDNA at baseline, but with negative tumor tissue according to NGS, eight were also positive in tissue samples when tested with the ddPCR assay (Fig 6C; Data Supplement). However, in another four cases, only plasma ddPCR could detect the positive mutational NRASQ61 state, whereas the tumor sample remained negative by ddPCR. The opposite scenario (ie, positive NRASQ61 state in tissue and negative in plasma) was found in two cases.
DISCUSSION
To make precision oncology a reality, highly predictive, noninvasive monitoring technologies must be identified. The application of ctDNA to metastatic melanoma has been reported to be a promising tool in terms of detecting the relevant tumor-derived mutant DNA over time.2-6,10-19
Here, we also considered the clinical significance of background mutations in the ctDNA of healthy individuals for statistical normalization at baseline and then later under therapy. This consideration is critical because mutant ctDNA, derived from aging cells or from undiscovered cancer cells, also can occur throughout the lifetime of individuals without a known diagnosis of cancer.20 Regarding differences between healthy plasma donors and patients with melanoma with radiologically confirmed active tumor mass, we intended to establish a molecular cutoff value with true clinical relevance using ROC analysis. This reproducibly identified high-risk patients for disease progression at baseline in the training and validation cohort. We conclude that concrete ctDNA thresholds are prognostic for outcome and represent a reliable practical tool for routine diagnostics even in the context of background mutant ctDNA.
Another goal of this study was to statistically validate a therapeutic predictive threshold on the basis of ROC analyses of longitudinally assessed plasma samples from patients with melanoma with timely matching CT/MRI scans. Accordingly, patients whose ctDNA levels were above a threshold of 35.85 copies/mL BRAFV600E, 51.5 copies/mL NRASQ61, and 32.83 copies/mL TERTprom ctDNA over 6 to 8 weeks despite therapy had significantly shorter PFS as compared with patients whose ctDNA levels remained below these thresholds. All the ctDNA assays applied here were significantly better indicators of measurable disease than were LDH or S100 levels.
Despite the practicability of molecular thresholds, our study indicates that the mean change in the ctDNA level over consecutive time points compared with the baseline represents an additional significant indicator of treatment effectiveness. Our study also showed that ctDNA changes relative to the previous sampling time point or even the rate of ctDNA increase per week (Data Supplement) can serve as an early hint of progression in patients receiving therapy.
Tumor tissue–based mutational testing (eg, by NGS with a sensitivity of approximately 1%21) is currently used as clinical state-of-the-art guidance for therapy selection for patients with metastatic melanoma. It does not account for the issue of intertumor and intrapatient heterogeneity. Instead, liquid biopsies should better assess the full genetic heterogeneity of solid cancers at the systemic disease level. Here, we analyzed baseline plasma samples with highly sensitive ddPCR and compared the results with existing NGS-based tissue profiles. Most importantly, we found NRASQ61 ctDNA copies in the baseline plasma of 21 of 33 longitudinally monitored patients who had NRASQ61-negative tumor tissue according to NGS. This may indicate that the amplicon-targeted NGS protocol, which was routinely applied to assess the mutational tumor status in our cohorts, may not be sensitive enough to detect low-frequency NRAS mutations within bulk tumors (eg, when originating only from minor cell subpopulations). To test this assumption, we obtained genomic DNA isolated from 51 melanoma tissue samples that previously underwent amplicon-targeted NGS and reanalyzed them on the ddPCR platform. Indeed, of 12 patients with BRAFV600E mutation whose plasma samples were positive for NRASQ61 ctDNA at baseline, while having a negative NGS status for NRASQ61, tumor tissue of eight also was positive when tested with the ddPCR assay. In contrast, enhancement of the NGS coverage from 103 to 177 and reduction of the mutant discovery threshold from 5% to 1% (mutant allele frequency) did not increase the NRASQ61 detection rate in a control experiment with selected tissue samples (Data Supplement). Importantly, in another four cases, only plasma ddPCR could detect the positive mutational NRASQ61 state. This may indicate that NRASQ61 clones could already pre-exist somewhere in the body before initiation of any MAPK pathway–targeted therapy. The presence of baseline NRASQ61 ctDNA in patients with BRAFV600E mutation before initiation of MAPKi therapy significantly correlated with shorter PFS and OS. Previous data suggest that NRASQ61 ctDNA emerges upon MAPKi therapy.3,16 Accordingly, we observed increasing levels of NRASQ61 during MAPKi therapy; however, the absolute NRASQ61 copy numbers remained below the BRAFV600E copy level.3,16
In summary, our study findings support the importance of ctDNA analysis as a valid clinical tool for the provision of real-time information on current treatment response and prediction of future response probability. In addition, the comparison of plasma-based ctDNA analysis with tumor tissue–based, amplicon-targeted NGS indicated considerable differences in diagnostic sensitivity, particularly with regard to intrapatient tumor heterogeneity. Studies that are powered to measure the difference between tissue-based and plasma-based diagnostics are needed to test if liquid biopsies represent a complementary tool or even outperform tissue-based genetic testing for primary therapeutic decision-making.
ACKNOWLEDGMENT
We thank the patients and their families for participating in this research. We acknowledge the staff at the medical centers of Tübingen, Brussels, and Essen. We thank M. Herlyn for providing the WM3734 and 451-LuBR cell lines.
Footnotes
Supported by the European Union's Horizon 2020 research and innovation program under Marie Sklodowska-Curie Grant No. 641458 (R.V., J.M.S.D.) and the Kom Op Tegen Kanker organization (B.N.).
AUTHOR CONTRIBUTIONS
Conception and design: Renáta Váraljai, Batool Shannan, Heike Chauvistré , Felix C.E. Vogel, Susanne Horn, Dirk Schadendorf, Alexander Roesch
Financial support: Julia Newton-Bishop, Dirk Schadendorf
Administrative support: Klaus Griewank, Dirk Schadendorf, Alexander Roesch
Provision of study materials or patients: Teofila Seremet, Peter A. Horn, Bart Neyns, Dirk Schadendorf, Alexander Roesch
Collection and assembly of data: Renáta Váraljai, Kilian Wistuba-Hamprecht, Teofila Seremet, Antje Sucker, Jan-Malte Placke, Peter A. Horn, Nils von Neuhoff, Susanne Horn, Jürgen C. Becker, Bart Neyns, Benjamin Weide, Dirk Schadendorf, Alexander Roesch
Data analysis and interpretation: Renáta Váraljai, Teofila Seremet, Joey Mark S. Diaz, Jérémie Nsengimana, Klaus Griewank, Julia Newton-Bishop, Susanne Horn, Jürgen C. Becker, Andreas Stang, Bart Neyns, Dirk Schadendorf, Alexander Roesch
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Teofila Seremet
Employment: Janssen (I), GlaxoSmithKline (I)
Honoraria: Novartis, Janssen
Consulting or Advisory Role: Novartis
Speakers' Bureau: Novartis
Travel, Accommodations, Expenses: Novartis, MSD Oncology, Janssen, LEO Pharma
Klaus Griewank
Patents, Royalties, Other Intellectual Property: Exon 4 mutations in GNAQ and GNA11, a rare gene mutation in melanoma. I receive annual payments.
Travel, Accommodations, Expenses: Roche
Jan-Malte Placke
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Peter A. Horn
Stock and Other Ownership Interests: share certificates (Aktien) of different companies
Patents, Royalties, Other Intellectual Property: Patent applications
Jürgen Becker
Consulting or Advisory Role: Merck Serono, Amgen, eTheRNA, Sanofi/Regeneron
Speakers' Bureau: Amgen, Merck Serono, Novartis, Sanofi/Regeneron, Merck Serono (Inst), Alcedis (Inst), IQvia (Inst), Amgen (Inst), Bristol-Myers Squibb (Inst)
Travel, Accommodations, Expenses: 4SC, Merck Serono
Julia Newton-Bishop
Travel, Accommodations, Expenses: Unknown mixed
Bart Neyns
Honoraria: Bristol-Myers Squibb, Novartis, Roche, Merck Sharp & Dohme
Consulting or Advisory Role: Bristol-Myers Squibb, Novartis, Roche, Merck Sharp & Dohme, Amgen
Speakers' Bureau: Novartis
Research Funding: Pfizer (Inst), Novartis (Inst), Merck KGaA (Inst)
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Novartis, Roche, Merck Sharp & Dohme, Amgen
Benjamin Weide
Honoraria: Roche, MSD, Bristol-Myers Squibb
Consulting or Advisory Role: CureVac, Philogen
Research Funding: Bristol-Myers Squibb (Inst), MSD (Inst), Philogen (Inst)
Dirk Schadendorf
Honoraria: Roche, Novartis, Amgen, Bristol-Myers Squibb, Merck Sharp & Dohme, Sysmex, Immunocore, Grünenthal Group, Merck Serono, Agenus, Array BioPharma, AstraZeneca, LEO Pharma, Incyte, Pfizer, Pierre Fabre, Philogen, Regeneron, 4SC, Mologen
Consulting or Advisory Role: Roche, Novartis, Bristol-Myers Squibb, Merck Sharp & Dohme, Merck Serono, Amgen, Immunocore, Incyte, 4SC, Pierre Fabre, Mologen, Sanofi/Regeneron
Speakers' Bureau: Roche, Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, Amgen, Incyte, Pierre Fabre
Research Funding: Bristol-Myers Squibb (Inst), Novartis (Inst)
Travel, Accommodations, Expenses: Roche, Bristol-Myers Squibb, Amgen, Merck, Merck Serono, Novartis
Alexander Roesch
Honoraria: Novartis
Consulting or Advisory Role: Novartis
Research Funding: Bristol-Myers Squibb, Novartis (Inst), Adtec (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.de Unamuno Bustos B, Murria Estal R, Pérez Simó G, et al. Towards personalized medicine in melanoma: Implementation of a clinical next-generation sequencing panel. Sci Rep. 2017;7:495. doi: 10.1038/s41598-017-00606-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong SQ, Raleigh JM, Callahan J, et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precis Oncol. 2017;1:1–14. doi: 10.1200/PO.16.00009. [DOI] [PubMed] [Google Scholar]
- 3.Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015;6:42008–42018. doi: 10.18632/oncotarget.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipson EJ, Velculescu VE, Pritchard TS, et al. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother Cancer. 2014;2:42. doi: 10.1186/s40425-014-0042-0. http://www.ncbi.nlm.nih.gov/pubmed/25516806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanmamed MF, Fernández-Landázuri S, Rodríguez C, et al. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin Chem. 2015;61:297–304. doi: 10.1373/clinchem.2014.230235. http://www.ncbi.nlm.nih.gov/pubmed/25411185 [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Long GV, Boyd S, et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol. 2017;28:1130–1136. doi: 10.1093/annonc/mdx026. [DOI] [PubMed] [Google Scholar]
- 7.Gangadhar TC, Savitch SL, Yee SS, et al. Feasibility of monitoring advanced melanoma patients using cell-free DNA from plasma. Pigment Cell Melanoma Res. 2018;31:73–81. doi: 10.1111/pcmr.12623. http://doi.wiley.com/10.1111/pcmr.12623%0Ahttp://www.ncbi.nlm.nih.gov/pubmed/28786531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haselmann V, Gebhardt C, Brechtel I, et al. Liquid profiling of circulating tumor DNA in plasma of melanoma patients for companion diagnostics and monitoring of BRAF inhibitor therapy. Clin Chem. 2018;64:830–842. doi: 10.1373/clinchem.2017.281543. [DOI] [PubMed] [Google Scholar]
- 11.Santiago-Walker A, Gagnon R, Mazumdar J, et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin Cancer Res. 2016;22:567–574. doi: 10.1158/1078-0432.CCR-15-0321. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Cao M, Mayo-de-Las-Casas C, Molina-Vila MA, et al. BRAF mutation analysis in circulating free tumor DNA of melanoma patients treated with BRAF inhibitors. Melanoma Res. 2015;25:486–495. doi: 10.1097/CMR.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 13.Knol AC, Vallée A, Herbreteau G, et al. Clinical significance of BRAF mutation status in circulating tumor DNA of metastatic melanoma patients at baseline. 25. Exp Dermatol. 2016:783–788. doi: 10.1111/exd.13065. [DOI] [PubMed] [Google Scholar]
- 14.Chang GA, Tadepalli JS, Shao Y, et al. Sensitivity of plasma BRAFmutant and NRASmutant cell-free DNA assays to detect metastatic melanoma in patients with low RECIST scores and non-RECIST disease progression. Mol Oncol. 2016;10:157–165. doi: 10.1016/j.molonc.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreuer M, Meersseman G, Van Den Herrewegen S, et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J Transl Med. 2016;14:95. doi: 10.1186/s12967-016-0852-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girotti MR, Gremel G, Lee R, et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2016;6:286–299. doi: 10.1158/2159-8290.CD-15-1336. [DOI] [PubMed] [Google Scholar]
- 17.Lee JH, Long GV, Menzies AM, et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti–programmed cell death 1 antibodies. JAMA Oncol. 2018;4:717–721. doi: 10.1001/jamaoncol.2017.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xi L, Pham TH-T, Payabyab EC, et al. Circulating tumor DNA as an early indicator of response to T-cell transfer immunotherapy in metastatic melanoma. Clin Cancer Res. 2016;22:5480–5486. doi: 10.1158/1078-0432.CCR-16-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee RJ, Gremel G, Marshall A, et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann Oncol. 2018;29:490–426. doi: 10.1093/annonc/mdx717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pantel K. Blood-based analysis of circulating cell-free DNA and tumor cells for early cancer detection. PLoS Med. 2016;13:e1002205. doi: 10.1371/journal.pmed.1002205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rapisuwon S, Vietsch EE, Wellstein A. Circulating biomarkers to monitor cancer progression and treatment. Comput Struct Biotechnol J. 2016;14:211–222. doi: 10.1016/j.csbj.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]