

INTRODUCTION
Dyskeratosis congenita, the original telomere syndrome, was clinically described more than 100 years ago on the basis of individuals who presented with a distinct rash, abnormal nails, and whitening of the tongue.1 From this rare syndrome, our clinical and molecular understanding of telomere syndromes has evolved and has now expanded to include aplastic anemia, myelodysplastic syndrome, and pulmonary fibrosis.2-5 The identification of patients with telomere syndromes is of significant clinical importance because these patients are exquisitely sensitive to alkylating chemotherapeutic agents and ionizing radiation.6-8
Precision medicine efforts that deploy tumor-normal sequencing have used various molecular assays and platforms to interrogate and annotate the cancer census genes.9,10 TERT, a gene that encodes a key protein involved in telomere maintenance, has been analyzed predominantly to identify genomic alterations that occur in tumors.11,12 Although constitutional telomere syndromes are recognized, they are rarely considered in the oncologist’s differential diagnosis unless a diagnosis of a telomere syndrome occurred before the patient’s cancer diagnosis.13
From January 2016 to May 2019, unselected patients with advanced solid tumors were presented with the option to participate and consent to a Memorial Sloan Kettering Cancer Center institutional review board–approved protocol (#12-245; ClinicalTrials.gov identifier: NCT01775072) of tumor and germline DNA sequencing. Patients viewed a standard pretest educational video on germline genetic testing. All patients with pathogenic or likely pathogenic variants were offered genetic counseling. Variants of uncertain significance were not reported. Electronic medical records were reviewed for demographic and clinical variables, including family history.
Here, we describe the frequency of individuals with germline TERT mutations and their associated clinical characteristics in the first 11,096 individuals who underwent agnostic germline molecular testing, and for four (57.1%) of seven individuals in this group with germline TERT mutations, telomere length assessment was possible (Table 1).
TABLE 1.
Clinical Characteristics of Patients With Pathogenic Germline TERT Mutations

MOLECULAR METHODS
Sequencing, Variant Calling, and Results Reporting
Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a 468-gene targeted capture panel, was used for tumor sequencing, while germline analysis included initially a 76-gene and then an 88-gene hereditary predisposition panel9,14,15 (Data Supplement). All variants with less than 1% population frequency in the Exome Aggregation Consortium (ExAC) database were interpreted. A clinical molecular geneticist or molecular pathologist interpreted variants per American College of Medical Genetics criteria.16 Mutations were classified as high (relative risk [RR], > 4), moderate (RR, 2 to 4), or low (RR, < 2) penetrance or as recessive.
Comparison of Germline Data to Public Databases
To assess associations of specific variants and tumor phenotype, population allele frequencies (AFs) in cases were compared with AFs in noncancer cases obtained from the ExAC17 public database minus cases obtained from The Cancer Genome Atlas.14 Comparisons of AFs in Ashkenazi Jewish cases were restricted to Ashkenazi Jewish individuals in the genome aggregation database release 2.01.18 AFs were compared by Fisher’s exact test in R version 3.3.2 using RStudio version 1.0.136 (RStudio, Boston, MA) to compute the odds ratios, CIs, and P values. Clinical variables in subsets defined by mutation status were compared by analysis of variance using GraphPad Prism version 7.01 software (GraphPad Software, San Diego, CA).19-21
TERT Sequencing
The average coverage of TERT was a minimum of more than 150× coverage to comply with quality control standards. The Memorial Sloan Kettering Cancer Center germline pipeline does not call TERT promoter variants as does the somatic pipeline, but it does call exons with ± 20 base pairs.9
Telomere Lengths
To assess telomere length, peripheral blood lymphocytes and granulocytes were sent to RepeatDx (North Vancouver, British Columbia, Canada) and measured through cytometric fluorescence in situ hybridization.22,23
CASE SERIES
Of 11,096 individuals who underwent MSK-IMPACT testing from January 2016 to May 2019, seven were found to harbor a germline pathogenic variant in TERT. Cancer types in our cohort included four female patients with breast cancer (ages at diagnosis, 28, 37, 46, and 58 years), one male patient with gall bladder adenocarcinoma (age 33 years), one male patient with pancreatic ampullary adenocarcinoma (age 45 years), and one male patient with two malignancies: lymphoma at age 58 years and prostate cancer at age 66 years. In the six patients with a solid tumor as their first malignancy, all were younger than the median age of diagnosis in the general population for their type of cancer24-27 (Data Supplement).
Clinical histories of three (42.8%) of the seven patients were suggestive of telomere syndromes. Individual i, diagnosed with breast cancer, endured therapy-induced radiation pneumonitis and refractory thrombocytopenia; individual iv, diagnosed with ampulla of Vater cancer, experienced premature graying and thrombocytopenia that preceded a cancer diagnosis at age 19 years; and individual vii revealed a history of bone marrow failure in a family member with a segregating TERT mutation and showed abnormal pulmonary function testing (Table 1).
For two individuals, telomere length testing through cytometric fluorescence in situ hybridization identified telomere lengths in the 1st percentile or less; in two additional individuals, telomere lengths were in the 10th percentile or less; and in one additional individual, telomere length testing failed because of low blood counts that did not recover after chemotherapy (Fig 1). For two individuals, it was not possible to obtain telomere lengths. Somatic mutation analysis of all five individuals with germline TERT mutations and tumor specimens available for analysis showed somatic TP53 driver mutations, which are generally associated with poor prognosis28-30 (Fig 2).
FIG 1.
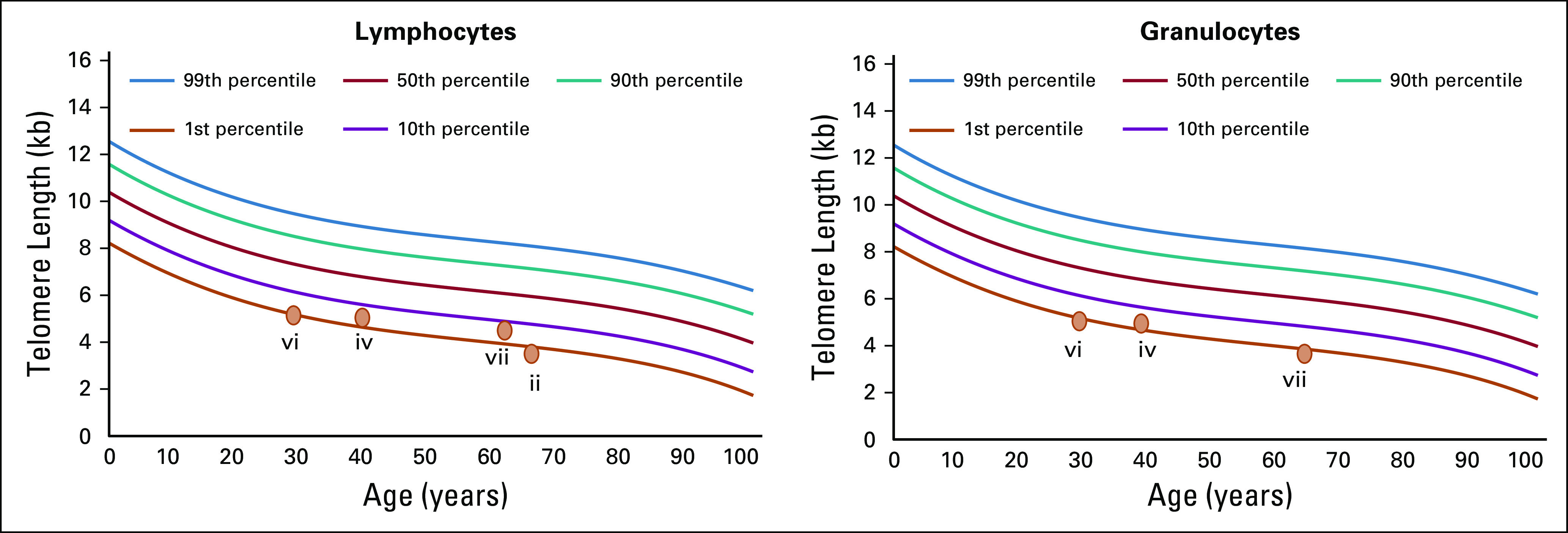
Present cytometric fluorescence in situ hybridization lymphocyte results for individuals ii, iv, vi, and vii and granulocyte results for individuals iv, vi, and vii.
FIG 2.
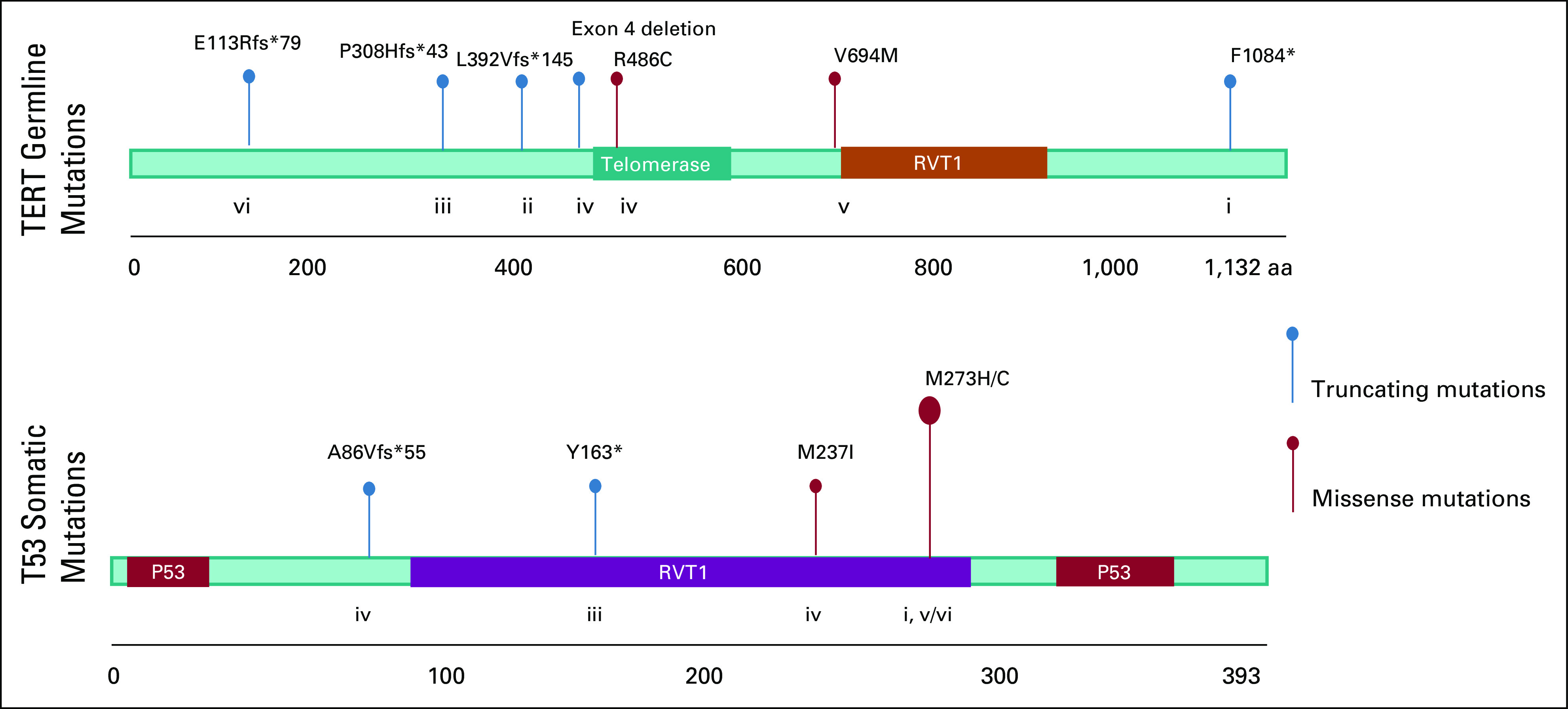
Germline TERT mutational lollipop illustration (n = 7) and somatic TP53 mutational lollipop illustration (n = 5). Individuals ii and vii were without tumor specimens available for sequencing, and individual vi was found to have two somatic TP53 mutations. aa, amino acid.
DISCUSSION
An understanding of the genetic basis that contributes to therapeutic sensitivities is important for oncology patients who receive chemotherapy, radiation, and other biologic therapeutics. The identification of patients with known therapeutic vulnerabilities is important for patient care. Patients with telomere syndromes may manifest overt or subtle clinical findings.5 Moreover, for patients with both obvious and subtle telomere syndromes, exquisite treatment sensitivities have been reported.6,7 Although only one in 1,571 individuals in this series of patients with advanced cancer showed a germline TERT mutation, this could translate to more than 1,000 patients diagnosed with malignancies a year in the United States who may have increased therapeutic sensitivities. Because telomere syndrome disorders are complex and exhibit anticipation, incomplete penetrance, multiple genes that underpin disease, recognized modifiers, and both autosomal dominant and autosomal recessive patterns of inheritance, this estimate will be refined with time. However, until a clearer picture is possible, it seems reasonable for individuals with constitutional germline TERT mutations to be considered for monitoring given the potential long-term sequalae from chemotherapy and radiation as well as increased organ-specific damage.7,8,31
Integration of germline, somatic, and clinical data for patients in our cohort was also notable for somatic TP53 mutations and a younger age at cancer diagnosis compared with the general population. Moreover, multiple TERT single nucleotide polymorphisms have been shown to be associated with telomere length and breast and ovarian cancer risk.31 All this information together with telomere lengths may provide insights for stratifying patients with regard to age at presentation, outcome, and tumor evolution.
Future studies that interrogate for germline mutations in other genes implicated in telomere biology (ie, CTC1, DKC1, NHP2, NOP10, TERC, TINF2, RTEL1) in the setting of cancer may reveal additional individuals with potential therapeutic sensitivities. In addition, studies that attempt to elucidate a clearer role in oncogenesis are needed. In the context of Li Fraumeni syndrome (ie, individuals with germline TP53 mutations), short telomeres are associated with earlier onset of cancer, which likely results from genomic instability.32 Somatic data from this study also supports the interplay between TP53 and telomere regulatory genes.
Footnotes
Supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics and members of the Molecular Diagnostics Service in the Department of Pathology and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. M.F.W. also receives grant support from the V Foundation, Corning Fund, and Crawford Fund for Pediatric Genomics. All authors at Memorial Sloan Kettering are supported by the Memorial Sloan Kettering Cancer Center Support Grant/Core Grant through National Cancer Institute Grant No. P30 CA008748.
AUTHOR CONTRIBUTIONS
Conception and design: Michael F. Walsh, Zsofia K. Stadler, Kenneth Offit
Financial support: Michael F. Walsh, Kenneth Offit
Administrative support: Temima Wildman, Kimberly Amoroso, Kenneth Offit
Provision of study material or patients: Michael F. Walsh, Kimberly Amoroso, Liying Zhang
Collection and assembly of data: Rosalba Sacca, Temima Wildman, Kimberly Amoroso, Jennifer Kennedy, Ozge Birsoy, Zoe Steinsnyder, Alicia Latham, Maria I. Carlo, Zsofia K. Stadler
Data analysis and interpretation: Rosalba Sacca, Temima Wildman, Liying Zhang, Ozge Birsoy, Diana Mandelker, Maria I. Carlo, Karen Cadoo, Yelena Kemel, Mark Robson, Zoe Steinsnyder, Zsofia K. Stadler
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Liying Zhang
Employment: Shanghai Genome Center (I)
Leadership: Shanghai Genome Center (I)
Stock and Other Ownership Interests: Shanghai Genome Center (I)
Honoraria: Future Technology Research, BGI Group, Illumina, Roche Diagnostics Asia Pacific
Travel, Accommodations, Expenses: Shanghai Genome Center (I), Roche Diagnostics Asia Pacific
Alicia Latham
Other Relationship: Conquer Cancer Foundation
Maria I. Carlo
Consulting or Advisory Role: Pfizer
Other Relationship: Prostate Cancer Foundation, Robert Wood Johnson Foundation
Karen Cadoo
Research Funding: AstraZeneca (Inst), Syndax (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Mark Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca
Research Funding: AstraZeneca (Inst), Myriad Genetics (Inst), InVitae (Inst), AbbVie (Inst), Tesaro (Inst), Medivation (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Pfizer
Other Relationship: Research to Practice, Clinical Care Options, Physician Education Resource
Zsofia K. Stadler
Consulting or Advisory Role: Allergan (I), Genentech (I), Roche (I), Regeneron Pharmaceuticals (Inst), Optos (I), Adverum (I), Biomarin (I), Alimera Sciences (I), Novartis (I), Spark Therapeutics (I), Fortress Biotech (I), REGENXBIO (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Zinsser F. Atrophia cutis reticularis cum pigmentatione, dystrophia unguium et leukoplakia oris. Ikonger Dermatol. 5:219–223. doi: 10.1007/BF00476707. [DOI] [PubMed] [Google Scholar]
- 2.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alder JK, Guo N, Kembou F, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med. 2011;184:904–912. doi: 10.1164/rccm.201103-0520OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alder JK, Parry EM, Yegnasubramanian S, et al. Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat. 2013;34:1481–1485. doi: 10.1002/humu.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [Erratum: Nat Rev Genet 14:235, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agrusa JE, Bertuch AA, DiNardo CD, et al. Severe therapy-related toxicities after treatment for Hodgkin lymphoma due to a pathogenic TERT variant and shortened telomeres. Pediatr Blood Cancer. 2019;66:e27779. doi: 10.1002/pbc.27779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Fuente J, Dokal I. Dyskeratosis congenita: Advances in the understanding of the telomerase defect and the role of stem cell transplantation. Pediatr Transplant. 2007;11:584–594. doi: 10.1111/j.1399-3046.2007.00721.x. [DOI] [PubMed] [Google Scholar]
- 8.Dietz AC, Orchard PJ, Baker KS, et al. Disease-specific hematopoietic cell transplantation: Nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46:98–104. doi: 10.1038/bmt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rusch M, Nakitandwe J, Shurtleff S, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun. 2018;9:3962. doi: 10.1038/s41467-018-06485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang FW, Hodis E, Xu MJ, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valentijn LJ, Koster J, Zwijnenburg DA, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. 2015;47:1411–1414. doi: 10.1038/ng.3438. [DOI] [PubMed] [Google Scholar]
- 13.Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [Erratum: Nat Med 23:1004, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broad Institute Frequently asked questions. http://exac.broadinstitute.org/faq.
- 18.Broad Institute Genome aggregation database. http://gnomad.broadinstitute.org.
- 19.Easton DF, Pharoah PD, Antoniou AC, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–2257. doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet Med. 2015;17:70–87. doi: 10.1038/gim.2014.147. [DOI] [PubMed] [Google Scholar]
- 21.Weitzel JN, Blazer KR, MacDonald DJ, et al. Genetics, genomics, and cancer risk assessment: State of the art and future directions in the era of personalized medicine. CA Cancer J Clin. 2011;61:327–359. doi: 10.3322/caac.20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poon SS, Lansdorp PM. Quantitative fluorescence in situ hybridization (Q-FISH). Curr Protoc Cell Biol 18:18.4, 2001. [DOI] [PubMed]
- 23.Baerlocher GM, Lansdorp PM. Telomere length measurements using fluorescence in situ hybridization and flow cytometry. Methods Cell Biol. 2004;75:719–750. doi: 10.1016/s0091-679x(04)75031-1. [DOI] [PubMed] [Google Scholar]
- 24.Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev. 2017;26:632–641. doi: 10.1158/1055-9965.EPI-16-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317:2402–2416. doi: 10.1001/jama.2017.7112. [DOI] [PubMed] [Google Scholar]
- 26.American Cancer Society Cancer facts and figures 2019. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf.
- 27.Rostain F, Hamza S, Drouillard A, et al. Trends in incidence and management of cancer of the ampulla of Vater. World J Gastroenterol. 2014;20:10144–10150. doi: 10.3748/wjg.v20.i29.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffith OL, Spies NC, Anurag M, et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat Commun. 2018;9:3476. doi: 10.1038/s41467-018-05914-x. [Erratum: Nat Commun 9:4850, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olivier M, Langerød A, Carrieri P, et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006;12:1157–1167. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 30.Pharoah PD, Day NE, Caldas C. Somatic mutations in the p53 gene and prognosis in breast cancer: A meta-analysis. Br J Cancer. 1999;80:1968–1973. doi: 10.1038/sj.bjc.6690628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bojesen SE, Pooley KA, Johnatty SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabori U, Nanda S, Druker H, et al. Younger age of cancer initiation is associated with shorter telomere length in Li-Fraumeni syndrome. Cancer Res. 2007;67:1415–1418. doi: 10.1158/0008-5472.CAN-06-3682. [DOI] [PubMed] [Google Scholar]