

INTRODUCTION
The overall survival of patients with high-grade serous ovarian cancer (HGSOC) has not improved during the past 20 years.1 Studies have revealed that genomic intratumor heterogeneity correlates with poor survival2,3 and specific patterns of malignant cell spread in HGSOC.4 The physical distribution of malignant clones across the peritoneal cavity may be nonrandom, because some sites harbor genetically diverse clones.5 These region-specific properties may modulate malignant cell invasion and expansion, thereby shaping evolutionary selection.3,6 Our understanding of genomic heterogeneity in HGSOC is limited to single biopsies; little is known about individual spatial and temporal variation across various tumor sites.2-5 Hence, developing imaging methods for guiding tissue sampling to physiologically and metabolically distinct tumor habitats is desirable. Such approaches will be vital for new clinical trials, especially those combining immunologic and genomically targeted therapies.
Here we use lesion-specific three-dimensional (3D) molds for phenotypic image-guided tumor sampling to ensure spatial colocation of imaging, histology, and genomic data, critical for understanding tumor biology. Phenotypic imaging maps of heterogeneity (ie, imaging habitats) of two HGSOC sites were obtained by combining perfusion, diffusion, and metabolic maps derived from multiparametric imaging. We evaluated if this phenotypic imaging-based heterogeneity reflects the underlying histologic and/or genetic heterogeneity of the tumor.
CASE REPORT
A 69-year-old woman diagnosed with stage IIIC HGSOC provided written informed consent to a prospective single-institution protocol approved by the institutional review board of Memorial Sloan Kettering Cancer Center (IRB No. 14-061). Four days before primary cytoreductive surgery the patient underwent same-day multiparametric magnetic resonance imaging (mpMRI; Discovery MR750 3T MRI system, GE Healthcare, Chicago, IL) of the abdomen and pelvis and 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET)/computed tomography (CT; Discovery PET/CT system, GE Healthcare). Details of image acquisition, analysis, and MR-PET/CT coregistration are described in the Appendix and in Appendix Table A1.
Cluster-Guided Specimen Sampling
Imaging habitats were identified using k-means clustering of the standardized uptake value (SUV), diffusion coefficient (D), perfusion fraction (f), and transfer constant (dynamic contrast-enhanced parameter Ktrans) voxels, with the number of clusters (k) being fixed to k = 3.7 Custom-made 3D molds of the lesions were printed using a 3D printer (MakerBot Replicator 2; MakerBot, Brooklyn, NY) on the basis of manual segmentation of the right ovarian tumor and metastatic implant on the axial T2-weighted MRI (Appendix). Tissue samples of each imaging cluster were obtained, cut in half, and formalin-fixed and paraffin-embedded for histopathologic analysis or snap-frozen for genomic analysis (Fig 1A).
FIG 1.
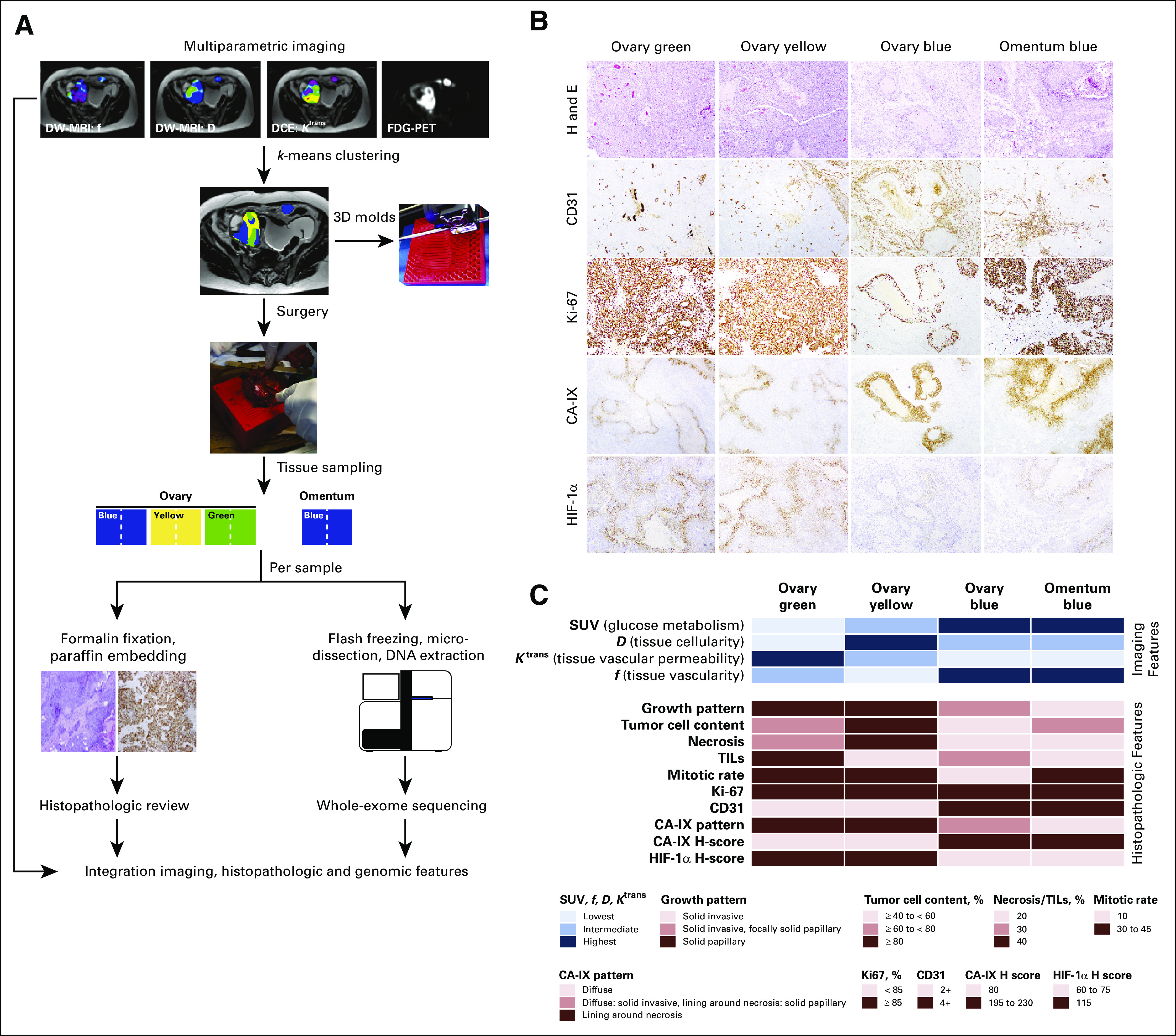
Study design and histopathologic review of the imaging-based high-grade serous ovarian cancer areas. (A) Phenotypically distinct areas of a high-grade serous ovarian cancer (HGSOC) were identified by k-means clustering of imaging features derived from magnetic resonance imaging (MRI) and positron emission tomography (PET)/computed tomography. The distinct imaging habitats (labeled blue, yellow, and green) were sampled from the surgically removed primary HGSOC and metastatic implant using a three-dimensional (3D) mold. Half of each imaging-based tissue area was formalin-fixed paraffin-embedded for histopathologic review and immunohistochemical analysis, and the other half was flash-frozen for whole-exome sequencing analysis. (B) Micrographs of representative hematoxylin and eosin (H and E)–stained sections of the imaging-based HGSOC areas (top row), and immunohistochemical analysis of CD31, Ki-67, CA-IX, and HIF-1α. (C) Imaging features associated with the distinct color areas as defined by k-means clustering of standardized uptake values (SUV), diffusion coefficient (D), dynamic contrast-enhanced DCE parameter (Ktrans), and water volume fraction flowing through microvessels (f) are plotted on top, and the histopathologic features of the distinct imaging-based tumor areas are shown at the bottom. Color-coding according to the legend. DW, diffusion-weighted; FDG, 18F-fluorodeoxyglucose; TIL, tumor-infiltrating lymphocyte.
Histologic Review and Immunohistochemistry
The three right ovarian mass HGSOC imaging-based habitats (labeled blue, yellow, or green) and the omental implant (blue) were reviewed by two pathologists blinded to the habitat assignment. CA-IX, CD31, HIF-1α, and Ki-67 immunohistochemistry was performed as described.8-10 Stromal tumor-infiltrating lymphocytes and Ki-67 were quantified according to the breast cancer recommendations.11,12 CD31 was assessed in the most active areas of neovascularization (microvessel counting, density grading 1+ to 4+).13 The expression of CA-IX (membranous) and HIF-1α (nuclear) was quantified using the H-score (range, 0 to 300).14
Whole-Exome Sequencing
Whole-exome sequencing (WES) of the microdissected tumor and matched normal DNA samples was performed as described.15,16 One hundred nine nonsynonymous somatic mutations (74%) not detected in all tumor areas by WES were independently validated using a custom hybrid-capture targeted massively parallel sequencing assay. Mutational signatures were defined using deconstructSigs.17 Large-scale transitions18 were derived from allele-specific segmented WES data.15,16 A maximum parsimony tree was built based on the nonsynonymous somatic mutations and gene copy number alterations, as described.19 Unsupervised hierarchical clustering was performed using Ward’s algorithm and Euclidean distance, as described,20 and the stability was assessed using pvclust.21
Image-Based Clustering of an HGSOC
Four parameters, SUV from FDG PET/CT (reflective of glucose metabolism), Ktrans from dynamic contrast-enhanced MRI (reflective of tissue vascular permeability), and D (reflective of tissue cellularity) and f (reflective of tissue vascularity) from diffusion-weighted MRI, were used to derive phenotypic imaging maps (clusters) of the right ovarian mass and metastatic implant. The clusters were labeled blue (lowest Ktrans, highest SUV, highest f), yellow (highest D, lowest f), or green (lowest D, highest Ktrans, lowest SUV). The right ovarian cancer contained all three distinct imaging-based clusters, whereas the omental metastatic implant was entirely composed of one cluster (blue; Fig 1A), suggestive of phenotypic imaging heterogeneity in the primary HGSOC. Tissue from the distinct imaging-based clusters for immunohistochemical and genetic analyses was obtained through a custom-made 3D mold (Fig 1A).
Imaging-Based Clusters Are Underpinned by Distinct Growth Patterns and Expression of Hypoxia-Related Markers
Phenotypic imaging-based clusters were associated with distinct histopathologic growth patterns. Although overall the ovarian mass displayed a solid growth pattern, the ovary green and ovary yellow areas showed an underlying papillary architecture (Fig 1B). In contrast, the ovary blue and omentum blue habitats displayed solid growth patterns and invaded into a reactive desmoplastic stroma; however, focal areas of underlying papillary architecture were observed in the ovary blue sample (Fig 1B). The expression levels and patterns of the hypoxia-marker HIF-1α and its downstream target CA-IX correlated with the distinct areas defined by multiparametric imaging (Fig 1C). Consistent with the imaging findings, which demonstrated that the ovary blue and omentum blue areas displayed the highest f, an MRI marker of tissue vascularity, CD31 immunohistochemical assessment revealed a higher density of tumor neovascularization (4+) in the ovary blue and omentum blue areas than in the ovary green and ovary yellow areas (2+; Fig 1C). Furthermore, distinct patterns and levels of CA-IX expression were observed among these areas, with ovary yellow and green having H-scores of 80, compared with 230 and 195 in ovary blue and omentum blue, respectively. For HIF-1α, a reverse pattern was found, with ovary yellow and ovary green areas displaying high levels of HIF-1α expression (H-scores, 115) compared with ovary blue and omentum blue (H-scores, 75 and 60, respectively; Figs 1B and 1C).
Imaging-Based Clusters Are Underpinned by Distinct Repertoires of Genetic Alterations
High-depth WES of microdissected tumor and normal samples revealed 50 nonsynonymous somatic mutations, including a TP53 p.P47Tfs*5 frameshift mutation, which were shared among all four imaging-based tumor areas (Fig 2). Twenty-eight nonsynonymous somatic mutations were shared exclusively between ovary blue and omentum blue, and 46 between the ovary green and ovary yellow components (Fig 2A). High-level amplifications shared exclusively between the ovary green and ovary yellow HGSOC components (eg, chromosomes 2q11.1-11.2, 7q11.21) or between the ovary blue and omentum blue areas (eg, chromosome 16p12.1-12.3; Fig 2B) were detected. Although all imaging habitats displayed genomics features of homologous recombination DNA repair deficiency (dominant mutational signature 3, high large-scale transition scores; Fig 2C), hierarchical clustering of the 146 nonsynonymous somatic mutations or the 718 gene copy number alterations not shared among all four imaging-based areas revealed that both at the mutational and gene copy number levels, ovary blue and omentum blue clustered together and were distinct from ovary yellow and ovary green (Fig 2D). A phylogenetic tree constructed based on the presence/absence of somatic mutations and gene copy number alterations (Fig 2E) revealed that the ovary green and ovary yellow tumor areas are likely derived from a clone distinct from the dominant clone found in ovary blue and omentum blue areas and that the omentum blue area likely originated from the ovarian blue area (Fig 2). Several somatic mutations found to be subclonal in ovary blue, including mutations affecting AUTS2, EIF2AK4, and TRIM41, became clonal in omentum blue (Fig 2A), and a subset of genes affected by copy number losses in ovary blue became homozygously deleted in omentum blue (eg, chromosome 18q23; Fig 2D).
FIG 2.

Genomics analysis of the imaging-based high-grade serous ovarian cancer areas. (A) Nonsynonymous somatic mutations identified in each of the imaging-based high-grade serous ovarian cancer (HGSOC) areas (bottom). Cancer cell fractions (CCFs) of the mutations identified (top). Color-coding according to the legend. Clonal mutations are depicted by an orange box, and loss of heterozygosity by a diagonal bar. (B) Copy number alterations of the distinct imaging-based HGSOC areas. In the genome plots, segmented log2 ratios (y-axis) are plotted according to their genomic positions (x-axis). Alternating blue and gray demarcate the chromosomes. (C) Mutational signatures (top) and large-scale state transitions (LSTs; bottom) of the imaging-based HGSOC areas. The cutoff of a high LST score of 15 or greater is marked by a dotted line. (D) Unsupervised hierarchical clustering of the somatic mutations (left) and of the gene copy number alterations (right) identified not to be shared by all four imaging-based HGSOC areas using Ward’s algorithm and Euclidean distance. Left, genes affected by mutations are shown in black; right, amplification, gain, loss, and deletion are shown in dark red, red, blue, and dark blue, respectively. The values on the edges of the clustering are derived from pvclust analysis to assess the stability of the clusters; both the approximately-unbiased and bootstrap-probability P values were 100%. (E) Phylogenetic tree depicting the evolution of the imaging-based HGSOC areas constructed using the maximum parsimony method. The colored branches represent each of the subclones identified, and selected somatic mutations and gene copy number alterations that define a given clone are illustrated along the branches. The length of the branches is representative of the number of somatic mutations and copy number alterations that distinguish a given clone from its ancestral clone. Amp, amplification; HomDel, homozygous deletion; indel, small insertion and deletion; SNV, single nucleotide variant.
DISCUSSION
Several studies have demonstrated that HGSOCs display spatial and temporal genetic heterogeneity,2-6,22 with tumors composed of genetically distinct clones and exhibiting distinct evolutionary trajectories and mechanisms of therapy resistance.4,23,24 Our observations support the contention that phenotypic and genetic analysis of single biopsy or single tumor samples may not provide a sufficiently accurate representation of HGSOC heterogeneity.4 This proof-of-principle study suggests that multiparametric imaging assessment may provide a noninvasive surrogate for intratumor genetic heterogeneity and may guide precise tissue sampling. Additional clinical validation of our findings is warranted. Given the evidence supporting the impact of intratumor genetic heterogeneity on tumors’ metastatic ability and resistance to therapeutic interventions,25 the results of this proof-of-principle observation provide the basis for studies to define the type of sampling required for the assessment of pre- and post-treatment HGSOCs. It should be noted, however, that multiparametric imaging for tumor heterogeneity and tumor evolution assessment requires not only manual segmentation of the imaging data but also standardization of the image acquisition protocols across different imaging platforms.
Targeted therapy and immunotherapy are being progressively applied earlier in the treatment of patients with cancer, and certain agents are anticipated to become part of front-line therapy. Germane to the successful adoption of these treatments as standard of care is the need for robust biomarkers to define the subset of patients who are likely to respond. Minimally invasive methods for disease monitoring in the form of circulating cell-free plasma DNA hold great promise in the assessment of genetic heterogeneity within a patient.26-28 Our imaging habitat–based method would constitute a noninvasive and complementary approach to such endeavors, given that it may allow for the sampling of selected image habitats without having to analyze every section of every tumor, an impossible task particularly in the metastatic and recurrent settings. More importantly, this may enable anticipation of resistance to targeted therapy or immunotherapy by unraveling known resistance mechanisms already present before therapy but restricted to specific anatomic sites within the tumor.
Appendix
Supplementary Methods
Magnetic resonance imaging protocol.
The examination was performed on a 3T magnetic resonance imaging (MRI) whole-body scanner unit (Discovery MR750; GE Healthcare, Chicago, IL) using a dedicated multichannel torso phased-array coil as the receive coil. The standard protocol included the following sequences acquired in an axial plane: two-dimensional T2-weighted fast spin-echo, T1-weighted gradient-echo. Single-shot intravoxel incoherent motion (IVIM)–based diffusion-weighted images were obtained using echo-planar imaging with a pair of motion-probing gradients along three orthogonal axes, using a chemical shift selective fat-saturation technique. Dynamic contrast-enhanced (DCE) images were acquired before and after the intravenous injection of gadopentetate dimeglumine (Magnevist; Berlex Laboratories, Montville, NJ; 0.1 mmol per kilogram of body weight at a rate of 2 mL/s) using an automatic injector (Medrad, Bayer, Pittsburgh, PA). The imaging volume was positioned so that the reconstructed slice positions matched the locations of the diffusion-weighted images. A summary of the MRI acquisition parameters is available in Appendix Table 1.
Positron emission tomography/computed tomography imaging protocol.
The patient was scanned on a dedicated positron emission tomography (PET)/computed tomography (CT) system (Discovery 690; GE Healthcare, Chicago, IL). A standard-of-care acquisition protocol was applied with an intravenous injection of approximately 400 to 455 MBq 18F-fluorodeoxyglucose after at least 6 hours of fasting and documentation of blood glucose less than 200 mg/dL followed by a 60-minute uptake period. Subsequently, a low-dose, attenuation-correction CT scan (120 to 140 kV, approximately 80 mA) was acquired with the patient in the supine position, followed by acquisition of PET emission images of the whole body (3 minutes per bed position, five to six bed positions).
Imaging analysis and MR-PET coregistration.
Volumetric regions of interest were outlined by the two study radiologists (E.S. and H.A.V.) on the axial fast spin-echo T2-weighted images and on the axial PET images, covering both the primary and metastatic lesions, using ImageJ software. Using anatomic landmarks available on axial T2-weighted MR images and on the axial PET and low-dose CT images, the tumor regions outlined on MRI were coregistered with PET. The image registration was performed in two steps. First, the registration was performed in the inferior-posterior z-direction. This was achieved by calculating the mean location (corresponding to the center of mass of the tumor) on the MRI image (T2W) and the corresponding mean location on the PET/CT image. Second, a rigid two-dimensional slice-by-slice registration was performed. Rigid registration was selected to ensure that the geometry (and the voxel count) of the region of interest (ROI) for each slice was maintained between MRI and PET. Using anatomic landmarks, a translational matrix was estimated between MRI and PET for each slice. The translational matrix was applied to the ROI for each slice to determine the displacement of the ROI.
The IVIM biexponential model proposed by Le Bihan et al (Le Bihan D, et al: Radiology 168:497-505, 1988; Le Bihan D, et al: Radiology 161:401-407, 1986; Le Bihan D, et al: Magn Reson Med 10:324-337, 1989) was used to estimate the diffusion parameters for each lesion outlined, including diffusion coefficient (D) and the volume fraction of the water flowing through the microvessels (f). The segmented analysis procedure (Callot V, et al: Magn Reson Med 50:531-540, 2003; Yao L, et al: Acad Radiol 7:27-32, 2000) was used to estimate IVIM parameters on a voxel-wise basis. Pharmacokinetic analysis of the DCE data were carried out using a one-compartment Tofts model (Tofts PS: J Magn Reson Imaging 7:91-101, 1997). For each outlined lesion, voxel-wise estimates of the volume transfer constant between the blood plasma and the extravascular extracellular space (Ktrans [min−1]) was calculated. Arterial input function was calculated using a biexponent model as previously described (Weinmann HJ, et al: Physiol Chem Phys Med NMR 16:167-172, 1984; Priest AN, et al: Magn Reson Med 63:1044-1049, 2010).
Gaussian smoothing was applied to all raw MR images before processing with variance of half a voxel, to smooth the parameters. Voxels were fitted on a voxel-wise basis for IVIM and DCE data. The quantitative diffusion parameters (D and f) and DCE parameter (Ktrans) were generated voxel-wise. For voxels that had estimated parameters that exceeded the bounds set by the fitting curve, the values were set to NaN (not-a-number). Once all voxels were fitted, the voxels with NaN value were replaced by an average of the surrounding voxels so that each voxel had an estimated parameter for the purpose of clustering. The standardized uptake values (SUV) of the voxels contained within each lesion on 18F-fluorodeoxyglucose PET was calculated based on the standard expression given by Kinahan et al (Kinahan PE, et al: Transl Oncol 2:223-230, 2009).
Three-dimensional printing methodology.
Custom made three-dimensional (3D) molds were printed based on manual segmentation of the ovarian primary tumor and metastatic implants on the preoperative axial T2-weighted MR images. The lesions were outlined on every axial slice where they were visible and automatically converted into a 3D object using open source software (MIPAV). The 3D model was then compared with the MR images and manually adjusted to resolve any discrepancies using MeshLab (MeshLab, Visual Computing Lab–ISTI-CNR), an extensible mesh processing system, aimed at editing and rendering unstructured 3D triangular meshes. The final 3D models of each lesion were imported into OpenSCAD (OpenSCAD, The OpenSCAD Developers), 3D CAD modeling software, which was used to create an internal cavity that exactly shaped each lesion according to the MRI shape and contour. The slits for slicing each lesion were designed into the molds at 5-mm intervals, which corresponded to the slice thickness and locations of the axial T2W fast relaxation fast spin echo MR images. In addition, the model was labeled with left, right, anterior, posterior, superior, and inferior markers to allow for proper orientation when collecting samples in the operating room. The molds were printed in two halves (to allow insertion of the specimen) using a 3D printer (MakerBot Replicator 2; MakerBot, Brooklyn, NY), which deposited consecutive 0.20-mm layers of polylactic acid plastic, producing the final lesion-specific molds.
TABLE A1.
Summary of MRI Imaging Acquisitions

Footnotes
Presented in part at the Radiology Society of North America Meeting, Chicago, IL, November 25-30, 2018, and the European Congress of Radiology, Vienna, Austria, February 27-March 3, 2019.
Supported in part by the Kaleidoscope of Hope Foundation; National Institutes of Health/National Cancer Institute Cancer Center Support Grant No. P30CA008748; Breast Cancer Research Foundation grant (J.S.R.-F.); and a Cycle for Survival grant (B.W.).
AUTHOR CONTRIBUTIONS
Conception and design: Britta Weigelt, Hebert Alberto Vargas, Evis Sala
Financial support: Evis Sala
Administrative support: Evis Sala
Provision of study material or patients: Dennis S. Chi, Evis Sala, Ginger J. Gardner
Collection and assembly of data: Britta Weigelt, Hebert Alberto Vargas, Felipe C. Geyer, Niamh Conlon, Lien N. Hoang, Achim A. Jungbluth, Anastasios D. Papanastasiou, Ramon E. Sosa, Robert A. Soslow, Dennis S. Chi, Ginger J. Gardner, Evis Sala
Data analysis and interpretation: Britta Weigelt, Hebert Alberto Vargas, Pier Selenica, Felipe C. Geyer, Yousef Mazaheri, Pedro Blecua, Alexandra Snyder, Charlotte K.Y. Ng, Robert A. Soslow, Ginger J. Gardner, Ronglai Shen, Jorge S. Reis-Filho, Evis Sala
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Britta Weigelt
Consulting or Advisory Role: Genentech (I), Invicro (I), Ventana Medical Systems (I), VolitionRx (I), Paige.AI (I), Goldman Sachs (I)
Felipe C. Geyer
Employment: Novartis Institutes for BioMedical Research
Niamh Conlon
Employment: AbbVie (I)
Stock and Other Ownership Interests: Merck (I)
Alexandra Snyder
Employment: Adaptive Biotechnologies, Merck
Stock and Other Ownership Interests: Merck
Consulting or Advisory Role: Driver Group
Research Funding: Bristol-Myers Squibb
Travel, Accommodations, Expenses: Genentech, Bristol-Myers Squibb
Robert A. Soslow
Speakers' Bureau: Ebix/Oakstone
Patents, Royalties, Other Intellectual Property: Methods for diagnosis and treatment of endometrial cancer. Assignee: The Regents of the University of California August 18, 2015 (Inst); Application: Compositions and methods for the diagnosis and treatment of ovarian cancers that are associated with reduced SMARCA4 gene expression or protein function (Inst); Royalties from published books: Cambridge University Press and Springer Publishing
Dennis S. Chi
Leadership: CSurgeries
Stock and Other Ownership Interests: Bovie Medical, Verthermia, Intuitive Surgical, TransEnterix
Consulting or Advisory Role: Bovie Medical, Verthemia
Ginger J. Gardner
Honoraria: BioAscent
Travel, Accommodations, Expenses: BioAscent
Ronglai Shen
Research Funding: GRAIL
Jorge S. Reis-Filho
Consulting or Advisory Role: Genentech, Invicro, Ventana Medical Systems, VolitionRx, Paige.AI, Goldman Sachs
Evis Sala
Honoraria: Siemens Healthineers
Speakers' Bureau: Siemens Healthineers
Travel, Accommodations, Expenses: Siemens Healthineers
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Schwarz RF, Ng CK, Cooke SL, et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015;12:e1001789. doi: 10.1371/journal.pmed.1001789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang AW, McPherson A, Milne K, et al. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell. 2018;173:1755–1769.e22. doi: 10.1016/j.cell.2018.03.073. [DOI] [PubMed] [Google Scholar]
- 4.Bashashati A, Ha G, Tone A, et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol. 2013;231:21–34. doi: 10.1002/path.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McPherson A, Roth A, Laks E, et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet. 2016;48:758–767. doi: 10.1038/ng.3573. [DOI] [PubMed] [Google Scholar]
- 6.Jiménez-Sánchez A, Memon D, Pourpe S, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170:927–938.e20. doi: 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carano RA, Ross AL, Ross J, et al. Quantification of tumor tissue populations by multispectral analysis. Magn Reson Med. 2004;51:542–551. doi: 10.1002/mrm.10731. [DOI] [PubMed] [Google Scholar]
- 8.Guerini-Rocco E, Hodi Z, Piscuoglio S, et al. The repertoire of somatic genetic alterations of acinic cell carcinomas of the breast: An exploratory, hypothesis-generating study. J Pathol. 2015;237:166–178. doi: 10.1002/path.4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Ahmadie HA, Alden D, Qin LX, et al. Carbonic anhydrase IX expression in clear cell renal cell carcinoma: An immunohistochemical study comparing 2 antibodies. Am J Surg Pathol. 2008;32:377–382. doi: 10.1097/PAS.0b013e3181570343. [DOI] [PubMed] [Google Scholar]
- 10.Schoolmeester JK, Greipp PT, Keeney GL, et al. Ovarian hemangiomas do not harbor EWSR1 rearrangements: Clinicopathologic characterization of 10 cases. Int J Gynecol Pathol. 2015;34:437–444. doi: 10.1097/PGP.0000000000000171. [DOI] [PubMed] [Google Scholar]
- 11.Salgado R, Denkert C, Demaria S, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann Oncol. 2015;26:259–271. doi: 10.1093/annonc/mdu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowsett M, Nielsen TO, A’Hern R, et al. Assessment of Ki67 in breast cancer: Recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. 2011;103:1656–1664. doi: 10.1093/jnci/djr393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weidner N, Semple JP, Welch WR, et al. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 14.Detre S, Saclani Jotti G, Dowsett M. A “quickscore” method for immunohistochemical semiquantitation: Validation for oestrogen receptor in breast carcinomas. J Clin Pathol. 1995;48:876–878. doi: 10.1136/jcp.48.9.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng CKY, Piscuoglio S, Geyer FC, et al. The landscape of somatic genetic alterations in metaplastic breast carcinomas. Clin Cancer Res. 2017;23:3859–3870. doi: 10.1158/1078-0432.CCR-16-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigelt B, Bi R, Kumar R, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst. 2018;110:1030–1034. doi: 10.1093/jnci/djy028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popova T, Manié E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 19.Schultheis AM, Ng CK, De Filippo MR, et al. Massively parallel sequencing-based clonality analysis of synchronous endometrioid endometrial and ovarian carcinomas. J Natl Cancer Inst. 2016;108:djv427. doi: 10.1093/jnci/djv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeLair DF, Burke KA, Selenica P, et al. The genetic landscape of endometrial clear cell carcinomas. J Pathol. 2017;243:230–241. doi: 10.1002/path.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki R, Shimodaira H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 22.Cooke SL, Ng CK, Melnyk N, et al. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010;29:4905–4913. doi: 10.1038/onc.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salomon-Perzyński A, Salomon-Perzyńska M, Michalski B, et al. High-grade serous ovarian cancer: The clone wars. Arch Gynecol Obstet. 2017;295:569–576. doi: 10.1007/s00404-017-4292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. Erratum: Nature 527:398, 2015. [DOI] [PubMed] [Google Scholar]
- 25.Caswell DR, Swanton C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017;15:133. doi: 10.1186/s12916-017-0900-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 27.Siravegna G, Marsoni S, Siena S, et al. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14:531–548. doi: 10.1038/nrclinonc.2017.14. [DOI] [PubMed] [Google Scholar]
- 28.Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet) Cancer Cell. 2017;31:172–179. doi: 10.1016/j.ccell.2017.01.002. [DOI] [PubMed] [Google Scholar]