

Abstract
PURPOSE
A subset of estrogen receptor–positive (ER-positive) breast cancer (BC) contains high levels of tumor-infiltrating lymphocytes (TILs), similar to triple-negative BC (TNBC). The majority of immuno-oncology trials target TNBCs because of the greater proportion of TIL-rich TNBCs. The extent to which the immune microenvironments of immune-rich ER-positive BC and TNBC differ is unknown.
PATIENTS AND METHODS
RNA sequencing data from The Cancer Genome Atlas (TCGA; n = 697 ER-positive BCs; n = 191 TNBCs) were used for discovery; microarray expression data from Molecular Taxonomy of Breast Cancer International Consortium (METABRIC; n = 1,186 ER-positive BCs; n = 297 TNBCs) was used for validation. Patients in the top 25th percentile of a previously published total TIL metagene score distribution were considered immune rich. We compared expression of immune cell markers, immune function metagenes, and immuno-oncology therapeutic targets among immune-rich subtypes.
RESULTS
Relative fractions of resting mast cells (TCGA Padj = .009; METABRIC Padj = 4.09E-15), CD8+ T cells (TCGA Padj = .015; METABRIC Padj = 0.390), and M2-like macrophages (TCGA Padj= 4.68E-05; METABRIC Padj = .435) were higher in immune-rich ER-positive BCs, but M0-like macrophages (TCGA Padj = 0.015; METABRIC Padj = .004) and M1-like macrophages (TCGA Padj = 9.39E-08; METABRIC Padj = 6.24E-11) were higher in immune-rich TNBCs. Ninety-one immune-related genes (eg, CXCL14, CSF3R, TGF-B3, LRRC32/GARP, TGFB-R2) and a transforming growth factor β (TGF-β) response metagene were significantly overexpressed in immune-rich ER-positive BCs, whereas 41 immune-related genes (eg, IFNG, PD-L1, CTLA4, MAGEA4) were overexpressed in immune-rich TNBCs in both discovery and validation data sets. TGF-β pathway member genes correlated negatively with expression of immune activation markers (IFNG, granzyme-B, perforin) and positively with M2-like macrophages (IL4, IL10, and MMP9) and regulatory T-cell (FOXP3) markers in both subtypes.
CONCLUSION
Different immunotherapy strategies may be optimal in immune-rich ER-positive BC and TNBC. Drugs targeting the TGF-β pathway and M2-like macrophages are promising strategies in immune-rich ER-positive BCs to augment antitumor immunity.
INTRODUCTION
The presence of tumor infiltrating lymphocytes (TILs), quantified either histologically or by immune-related gene expression, is associated with better survival in human epidermal growth factor receptor 2 (HER2)–positive breast cancers (BCs), triple-negative BCs (TNBCs), and highly proliferative estrogen receptor (ER)–positive BCs.1-5 In addition, high TIL counts are associated with increased sensitivity to chemotherapy in all subtypes, reflected by higher pathologic complete response rates to neoadjuvant chemotherapy.6,7 High TIL counts and expression of programmed death-ligand 1 (PD-L1) in the tumor microenvironment are highly correlated and both predict greater benefit from immune checkpoint inhibitor therapy in metastatic TNBC.6-9 High TIL counts (and PD-L1 expression) are more frequent among TNBCs than ER-positive BCs.4,6,10,11 Initial single-agent phase I/II trials with immune checkpoint inhibitors showed higher response and clinical benefit rates in TNBCs12,13 than in unselected patients with ER-positive BCs.14,15 Therefore, the majority of immunotherapy trials today target TNBCs and have demonstrated promising activity.16 The first immune checkpoint inhibitor, atezolizumab, was recently approved to treat PD-L1–positive, metastatic TNBC in combination with nab-paclitaxel.13
CONTEXT
Key Objective
Does the immune microenvironment of immune-rich estrogen receptor–positive breast cancer (ER-positive BC) differ from the immune microenvironment of immune-rich triple-negative BC (TNBC)?
Knowledge Generated
Relative fractions of mast cells and M2-like macrophages are higher in the immune microenvironment of immune-rich ER-positive BC, whereas fractions of M0-like and M1-like macrophages are higher in immune-rich TNBC. TGF-β pathway members and response genes are upregulated at the gene expression level in immune-rich ER-positive BC.
Relevance
Our results support the use of distinct immunotherapy strategies in immune-rich ER-positive BC versus TNBC, and drugs targeting M2-like macrophages or TGF-β signaling may be effective for immune-rich ER-positive BC.
However, ER-positive BCs account for the majority of newly diagnosed BCs and cause most BC-related deaths.17,18 A subset of ER-positive BCs has high TIL counts, similar to levels seen in TNBCs.4 If the immune microenvironment of immune-rich ER-positive BC is similar to that of immune-rich TNBC, one could assume that similar treatment strategies would work in both subtypes. Conversely, if immune-rich ER-positive BCs have different immune cell compositions than immune-rich TNBCs, these differences could inform the design of subtype-specific immunotherapy trials. The goal of our analysis was to compare differences in immune gene expression between immune-rich ER-positive BC and immune-rich TNBC and assess differential expression of current immunotherapy targets.
PATIENTS AND METHODS
Data Sources
Gene-level summarized RNA sequencing (RNA-seq) expression data from TCGA (n = 1,094) for discovery analyses and microarray expression data from METABRIC (n = 2,273) with ER and HER2 status annotation for validation analyses were obtained from public access portals (https://portal.gdc.cancer.gov; https://ega-archive.org/studies/EGAS00000000098). HER2-positive patients were excluded, and only ER-positive/HER2-negative (herein designated ER-positive; TCGA, n = 627; METABRIC, n = 1,186) and ER-negative/HER2-negative (herein designated TNBC; TCGA, n = 191; METABRIC, n = 279) patients were included in this analysis. We obtained formalin-fixed paraffin-embedded tissues from 63 TNBCs and 53 ER-positive, treatment-naïve, stage I to III BCs from Yale Pathology Tissue Archives, matched by distribution of histologic grade, for quantitative immunofluorescence (QIF) and automated quantitative analysis (AQUA).
Assessment of Immune Cell Content and Selection of Immune-Rich Patients
A gene expression signature score developed by Danaher et al19 was used to score each TCGA and METABRIC patient for total TILs. The total TIL gene signature score is the average of scores representing 11 immune cell subpopulations (CD45, macrophages, CD8+ T cells, T cells, cytotoxic cells, exhausted CD8+ cells, Th1 cells, B cells, neutrophils, natural killer [NK] cells, CD56dim NK cells). Log2+1-transformed expression was used to calculate total TIL gene signature scores from TCGA (RNA-seq) and METABRIC (microarray) data. The TIL metagene score distribution was plotted for the pooled ER-positive BC and TNBC patients, and patients with scores in the top 25th percentile of the distribution were defined as immune-rich.
For 156 basal-like cancers in TCGA (ER-positive BC, n = 21; TNBC, n = 135), histologic TIL counts were also available.20,21 For these patients, we assessed the correlation between histologic TIL count and TIL gene expression score. We also assessed correlations between TIL gene signature score and PAM50 subtype, tumor mutation burden (TMB), and Ki-67 expression level in the TCGA data when this information was available (ER-positive BC, n = 191; TNBC, n = 697). PAM50 subtypes and TMB categories were obtained from a previous publication, and Ki-67 categories were defined as greater than or less than the median log2+1-transformed Ki-67 expression across TCGA patients.22
Survival Analysis
Progression-free survival (PFS) and overall survival (OS) times were available for 807 TCGA patients (ER-positive BCs, n = 189; TNBCs, n = 618).23 PFS and OS were compared by ER subtype within immune-rich patients (ER-positive BCs, n = 114; TNBCs, n = 84) and within non-immune-rich patients (ER-positive BCs, n = 504; TNBCs, n = 105) using the “survival” and “survminer” R packages.24
Assessment of Immune Cell Subpopulations
CIBERSORT, an analytical tool that uses support vector regression, was used to estimate the relative proportions of 9 aggregated immune cell populations and 22 subpopulations in immune-rich ER-positive BCs and immune-rich TNBCs.25,26 The average relative fraction and standard error of the mean of each immune cell population were compared by subtype. For TCGA patients, previously calculated gene expression signature scores of overall macrophages, M1-like macrophages, M2-like macrophages, and the transforming growth factor β (TGF-β) signaling response were obtained from previous publications and were compared between patients with ER-positive BCs and those with TNBCs.27-29 Slopes of linear regression curves between total TILs and metagene expression signatures were compared by using a t test based on standard error in the linear regression models.30 Formalin-fixed paraffin-embedded tissue microarrays were stained for the nuclear marker 4′,6-diamidino-2-phenylindole and the pan-macrophage marker CD68, and multiplexed QIF and AQUA were used to quantify expression levels in Yale ER-positive BC and TNBC patients as previously described.31
Differential Gene Expression Analysis
The “DESeq2” (TCGA, RNA-seq data) and “limma” (METABRIC, microarray data) R packages were used for differential gene expression analysis of 779 immune-related genes (NanoString PanCancer IO 360 Panel, NanoString Technologies, Seattle, WA) and 137 currently available immuno-oncology drug target genes between immune-rich ER-positive BCs and immune-rich TNBCs.32-34 Availability of RNA expression data for each member gene in TCGA and METABRIC databases is summarized in the Data Supplement. Genes with false discovery rate Padj < .05 were considered significantly differentially expressed for a broad investigation of differentially regulated immune pathways.
Statistical Analysis
All statistical analyses were performed in R v 3.5.0. P values were adjusted for false discovery rate by the Benjamini-Hochberg method in the CIBERSORT and differential gene expression analyses. The t test was used for parametric analyses, Wilcoxon rank sum test was used for nonparametric analyses of non-normally distributed data, Fisher’s exact test was used to analyze contingency tables, and the log-rank test was used to compare survival curves. The “corrplot” R package was used to derive and plot Pearson’s correlation coefficients for the expression of immune genes and immuno-oncology drug target genes, and hierarchical clustering was used for visualization.35
RESULTS
TIL Metagene Score Distribution in Patients With ER-Positive BC or TNBC
By using the signature described by Danaher et al19 to quantify cell types and total TIL presence, we determined that macrophages and CD8+ T cells were the 2 most abundant immune cell types in both cancer subtypes in TCGA (Fig 1). We observed the same in METABRIC. In TCGA, the 75th percentile TIL metagene score was 5.92, and it categorized 119 ER-positive BCs (19%) and 89 TNBCs (45%) as being immune rich. In METABRIC, the 75th percentile score was 6.26, and it categorized 225 ER-positive BCs (19%) and 140 TNBCs (50%) as being immune rich. The total TIL gene signature scores correlated moderately but significantly with histologic TIL scores in TCGA (R = 0.44; P = 1.18E-08; Fig 2A) when these data were available. In TCGA, there was no statistically significant difference between the distribution of PAM50 subtypes or Ki-67 expression levels between immune-rich and non–immune-rich ER-positive BCs. Conversely, immune-rich TNBCs demonstrated higher Ki-67 expression than non–immune-rich TNBCs (Fig 2B-D). Patients with immune-rich ER-positive BCs had significantly higher TMB than patients with non–immune-rich ER-positive BCs; this difference was not seen among patients with TNBCs. For a subset of TCGA patients with available survival information (n = 804), PFS and OS were compared by subtype (Data Supplement). Immune-rich patients had similar PFS and OS regardless of ER status. In contrast, among the non–immune-rich cancers, OS was significantly longer in patients with ER-positive BCs than in patients with TNBCs (P = .043), as expected.
FIG 1.
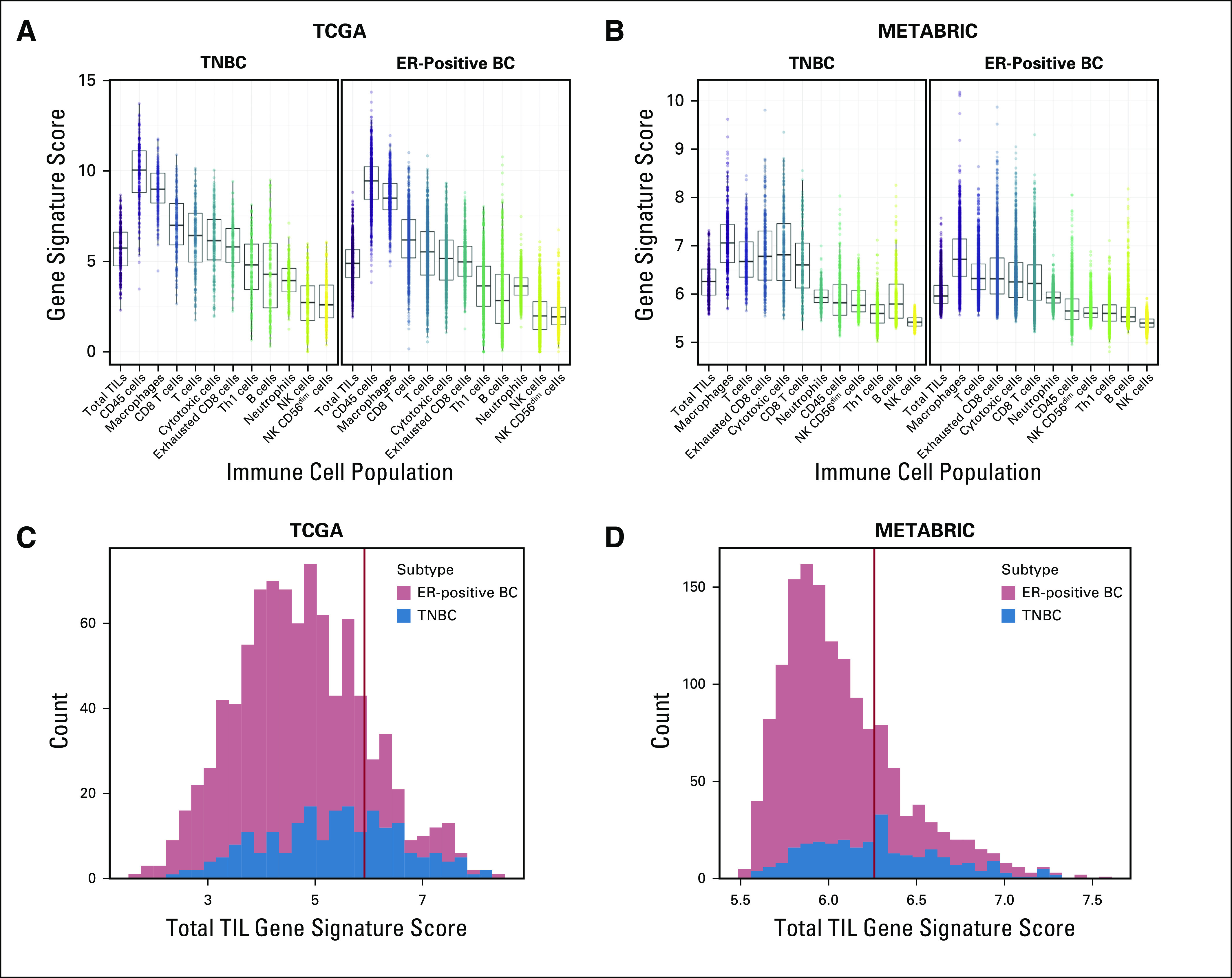
Identification of patients with immune-rich estrogen receptor–positive breast cancers (ER-positive BCs) and triple-negative BCs (TNBCs). (A-B) Gene signature scores were calculated for 11 immune cell subpopulations for patients with ER-positive BCs and TNBCs in (A) The Cancer Genome Atlas (TCGA) and (B) Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) databases, as described by Danaher et al.19 The x-axis indicates immune cell subpopulations plus the total tumor-infiltrating lymphocyte (TIL) population. The y-axis indicates the calculated gene signature scores for each subpopulation. For patients in (C) TCGA and (D) METABRIC, the distributions of total TIL gene signature scores were plotted on the x-axis, and patients in the top 25th percentile (designated by red vertical lines) were designated immune-rich (75th percentile score: TCGA, 5.92; METABRIC, 6.26). The TCGA cohort had a total of 627 patients with ER-positive BC and 191 patients with TNBC. The METABRIC cohort had a total of 1,186 patients with ER-positive BC and 279 with TNBC. NK, natural killer.
FIG 2.
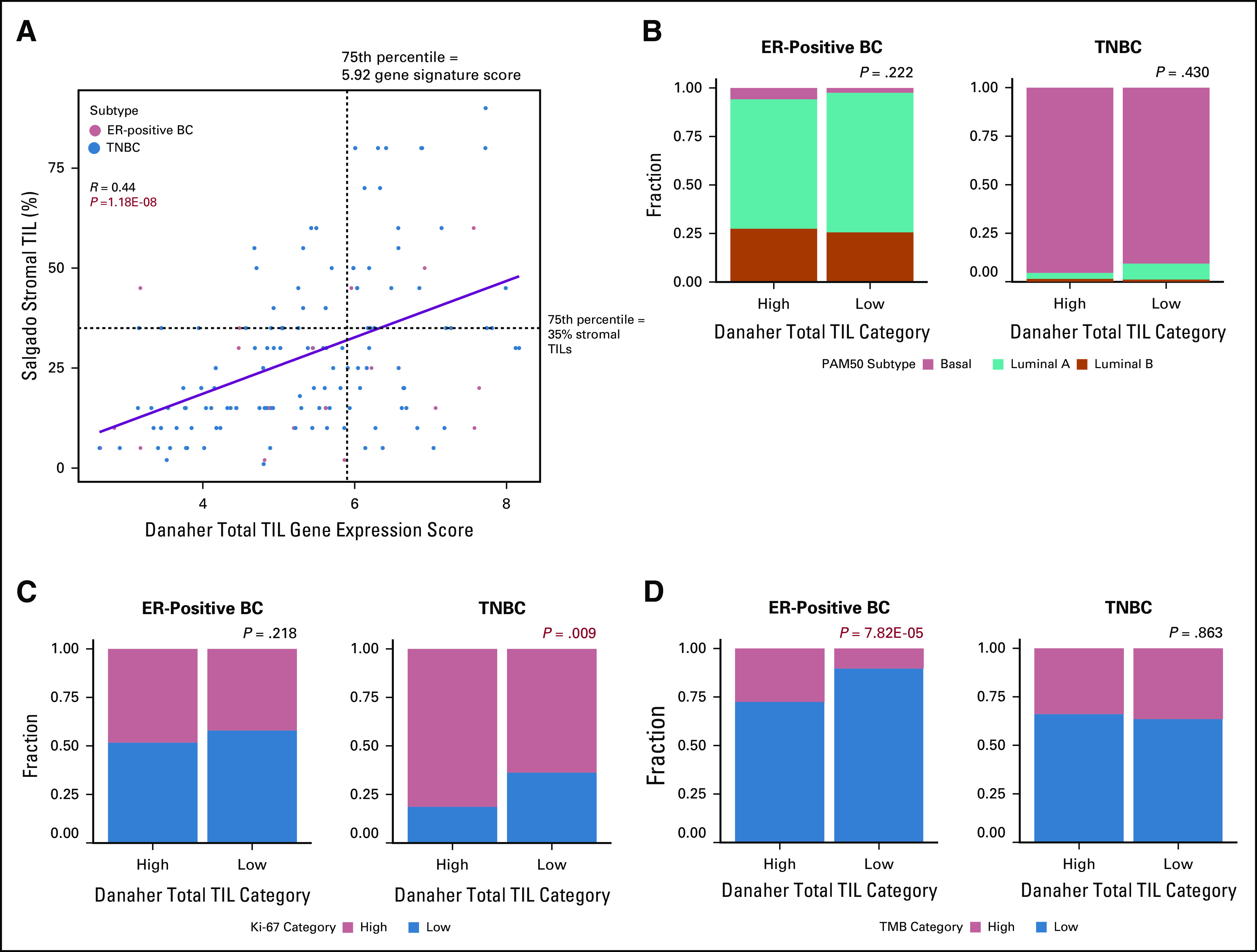
Validation of total tumor-infiltrating lymphocyte (TIL) gene signature scores. (A) For a subset of basal-like patients from The Cancer Genome Atlas (TCGA) with previously published histologic TIL scores (n = 156) according to Danaher et al,19 the total TIL gene signature scores (x-axis) were correlated with histologic stromal TIL counts (y-axis). The 75th percentile of histologic (35% stromal TILs) and gene signature TIL scores (5.92) are represented by dashed black lines. Tumor subtype is reflected by point color (estrogen receptor–positive breast cancers [ER-positive BCs], n = 21; triple-negative BCs ([TNBCs], n = 135). The linear regression line with 95% CIs is shown in blue. Pearson’s R = 0.44; P = 1.18E-08. (B-D) For all patients with ER-positive BCs (n = 697) and TNBCs (n = 191) in TCGA, patients were classified as immune cell “High” (Danaher et l19 total TIL gene signature score in the top 25th percentile) or “Low” (Danaher total TIL gene signature scores in the bottom 75th percentile), represented on the x-axis. Distributions of (B) PAM50 subtypes, (C) Ki-67 expression categories and (D) tumor mutation burden (TMB) categories were compared between patients with immune-rich high and low ER-positive BCs and TNBCs separately. PAM50 and TMB categories were obtained from previous publications, and Ki-67 categories were defined as greater than (high) or less than (low) the median Ki-67 expression level across all patients.
Immune Cell Subpopulations in the Tumor Microenvironment of Immune-Rich ER-Positive BCs and Immune-Rich TNBCs
By using CIBERSORT, we found higher relative fractions of T cells (Padj = .009) and mast cells (Padj = .0005) in patients with immune-rich ER-positive BCs compared with those who had immune-rich TNBCs (Fig 3A) in TCGA. In patients with immune-rich TNBCs, the relative fractions of macrophages (Padj = .026) and NK cells (Padj = .0007) were higher relative to those in patients with immune-rich ER-positive BCs. The higher relative fractions of T cells (Padj = .005) and mast cells (Padj = 1.73E-20) in immune-rich ER-positive BCs and the higher relative fraction of macrophages (Padj = 1.22E-06) in immune-rich TNBCs were also observed in METABRIC (Fig 3B). QIF also showed higher overall expression of the pan-macrophage marker CD68 in TNBC tissues relative to tissue from ER-positive BCs from Yale Pathology Archives (P = .011; Fig 3C).
FIG 3.
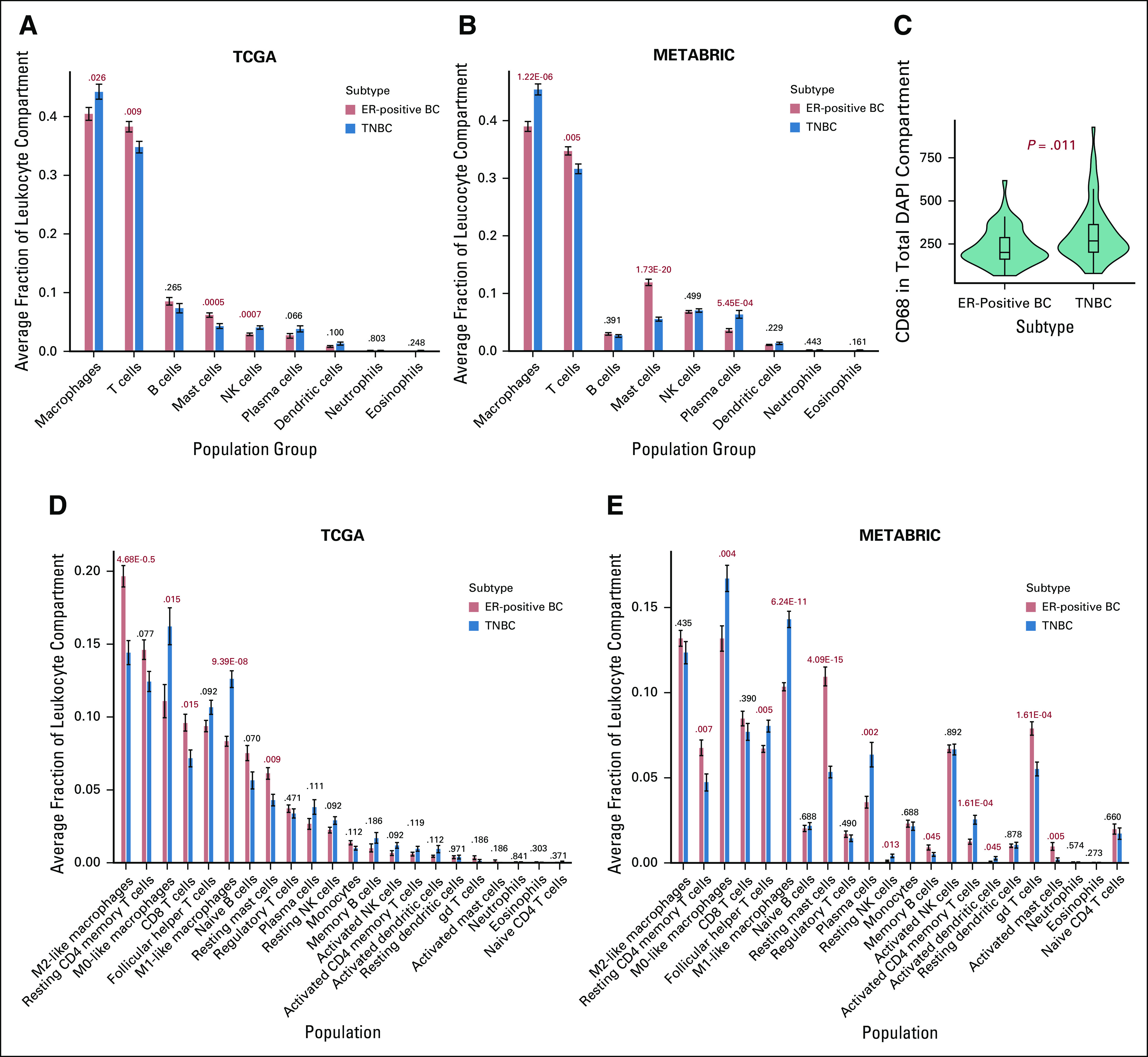
Quantification of immune cell populations in the tumor environment of immune-cell rich patients. (A-B) CIBERSORT deconvolution was used to quantify relative fractions of immune cell populations in the tumor leukocyte compartment of immune-rich patients. For patients from (A) The Cancer Genome Atlas (TCGA) and (B) Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) databases, 9 aggregate immune cell populations are listed on the x-axis, and the average fraction of each immune cell population for a certain subtype is quantified on the y-axis. Error bars represent standard error of the mean. Subtypes are designated by bar color. False discovery rate Padj values are provided above each subtype comparison. (C) Quantitative immunofluorescence (QIF) signal was compared between estrogen receptor–positive breast cancer (ER-positive BC;,n = 53) and triple-negative BC (TNBC; n = 63) formalin-fixed paraffin-embedded tumor microarray tissues. Subtype is shown on the x-axis, and CD68 QIF signal in the total 4′,6-diamidino-2-phenylindole (DAPI) compartment is quantified on the y-axis. For (D) TCGA and (E) METABRIC patients, 22 immune cell populations derived by CIBERSORT are listed on the x-axis, and the average fraction of each immune cell population for a certain subtype is quantified on the y-axis. Error bars represent standard error of the mean. Subtypes are designated by bar color. False discovery rate Padj values are provided for TCGA (ER-positive BCs, n = 119; TNBCs, n = 86) and METABRIC (ER-positive BCs, n = 225; TNBCs, n = 140) patients. NK, natural killer.
Next, we examined differences among 22 immune cell subpopulations. In immune-rich ER-positive tumors in TCGA, the relative fractions of CD8+ T cells (Padj = .015) and resting mast cells (TCGA Padj = .009) were higher relative to those in immune-rich TNBC tumors in both data sets (Fig 3D). In addition, despite lower overall macrophage content, the relative fraction of M2-like macrophages was also higher in immune-rich ER-positive BCs (Padj = 4.68E-05). In contrast, the relative fractions of M0-like macrophages (Padj = .015) and M1-like macrophages (Padj = 9.39E-08) were significantly higher in patients with immune-rich TNBCs compared with those with immune-rich ER-positive BCs. Differences in relative fractions of resting mast cells (Padj = 4.09E-15), M0-like macrophages (Padj = .0043), and M1-like macrophages (Padj = 6.24E-11) by immune-rich subtype were validated in METABRIC (Fig 3E).
We also examined the distribution of previously published overall macrophage, M1-like macrophage, M2-like macrophage, and TGF-β signaling response metagene scores that were available for the TCGA patients only.28,29 Consistent with the findings above, immune-rich TNBCs had higher overall macrophage (P = .041) and M1-like macrophage (P = 4.79E-09) metagene expression scores relative to immune-rich ER-positive BCs. Immune-rich ER-positive BCs had higher M2-like macrophage (P = .012) and TGF-β response (P = 0.035) metagene expression scores (Fig 4A). The subtype-specific differences in M1-like macrophage (P = 1.85E-04) and TGF-β response (P = .029) metagene scores significantly increased with higher total TIL gene signature scores, whereas the differences in overall macrophage (P = .786) and M2-like macrophage (P = .097) scores between cancer subtypes were not specific to immune-rich patients (Fig 4B). Slopes of the linear regression lines between total TILs and immune metagene expression scores, along with statistical comparisons of the slopes by subtype, are summarized in the Data Supplement.
FIG 4.
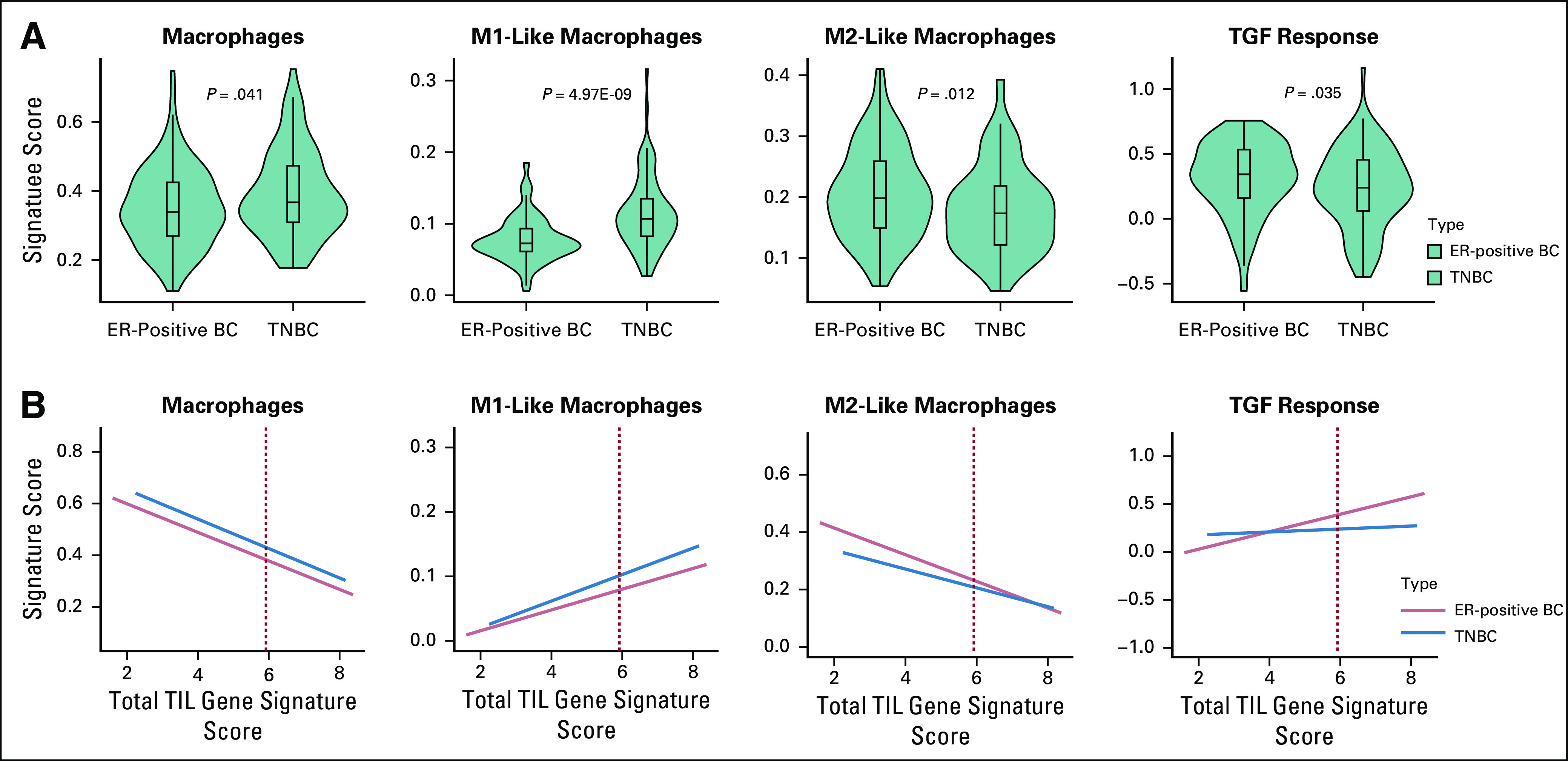
Comparison of macrophage population and transforming growth factor β (TGF-β) response metagene expression scores between patients with immune-rich estrogen receptor–positive breast cancers (ER-positive BCs) and those with triple-negative BCs (TNBCs) in The Cancer Genome Atlas (TCGA). (A) Metagene expression scores for overall macrophages, M1-like macrophages, M2-like macrophages, and TGF-β response were compared between immune-rich ER-positive BC (red, n = 119) and immune-rich TNBC (blue, n = 86) patients in TCGA. Subtype is listed on the x-axis, and previously calculated metagene scores are quantified on the y-axis. (B) For all patients with ER-positive BCs (n = 697) and TNBCs (n = 191) in TCGA, gene expression signature scores for overall macrophages, M1-like macrophages, M2-like macrophages, and TGF-β response (y-axis) were plotted against total TIL gene signature scores (x-axis). Red dashed lines indicate the 75th percentile scores of total TIL gene signature scores, above which patients were considered immune rich. Linear regression lines for each subtype with 95% CIs are shown in red (ER-positive BC) or blue (TNBC).
Differential Expression of Immune-Related Genes and Immuno-Oncology Drug Target Genes Between Immune-Rich ER-Positive BCs and Immune-Rich TNBCs
At the individual gene level, 132 of 754 immune-related genes were significantly overexpressed in TCGA immune-rich ER-positive BCs. Ninety-one of these immune-related genes, including mast cell genes MS4A2 and CPA3 and potential therapeutic targets CXCL14, CX3CR1, and CSF3R, were also upregulated in METABRIC patients with immune-rich ER-positive BCs (Data Supplement). In immune-rich TNBCs, 303 immune-related genes were overexpressed compared with immune-rich ER-positive BCs in TCGA, and 218 of these were validated in METABRIC. We also examined differences in the expression of immuno-oncology drug targets currently in clinical development. Sixteen of 136 available therapeutic target genes were overexpressed in TCGA patients with immune-rich ER-positive BCs, and 12 of these, including 3 members of the TGF-β signaling pathway (TGF-β3, LRRC32/GARP, and TGFβ-R2), were also overexpressed in METABRIC patients with immune-rich ER-positive BCs (Table 1; Data Supplement). Fifty-nine immuno-oncology drug target genes were overexpressed in immune-rich TNBCs in TCGA, and 41 of these, including IFNG, LAG3, CD274/PD-L1, CTLA4, and the cancer-testis antigen, MAGEA4, were also overexpressed in immune-rich TNBCs in METABRIC.
TABLE 1.
Highly Expressed Currently Available Immuno-Oncology Drug Targets in Immune-Rich ER-Positive BCs Relative to Immune-Rich TNBCs

Correlation Between TGF-β Pathway Expression and Other Immune-Related Genes
TGF-β is a pleotropic cytokine that can stimulate regulatory T cells, induce M2-like macrophage polarization, and suppress effector T cells, NK cells, and dendritic cells. Furthermore, TGF-β signaling has been linked to an immune-excluded phenotype in cancer. We therefore examined correlations between the differentially expressed TGF-β pathway members (TGF-β3, LRRC32, and TGF-βR2) and the expression levels of the 53 immuno-oncology drug target genes that were differentially expressed in both data sets (12 upregulated in immune-rich ER-positive BCs, 41 upregulated in immune-rich TNBCs) across immune-rich TNBCs and ER-positive BCs. In both TCGA and METABRIC, expression of the TGF-β pathway members correlated positively with each other and negatively with most immune activation markers (Fig 5A-B; Data Supplement). Similar correlation patterns were seen between TGF-β pathway members and immune activation marker expression when only immune-rich ER-positive BCs were analyzed (Fig 5C-D). Across all 754 available immune-related genes, TGF-β pathway members were negatively correlated with expression of IFNG (TGF-β3, r = –0.38; TGF-βR2, r = –0.33; LRRC32, r = –0.47), granzyme-B (TGF-β3, r = –0.42; TGF-βR2, r = –0.39; LRRC32, r = –0.46), perforin (TGF-β3, r = –0.24; TGF-βR2, r = –0.14; LRRC32, r = –0.33), T-cell markers, including CTLA4 (TGF-β3, r = –0.17; TGF-βR2, r = –0.21; LRRC32, r = –0.21), ICOS (TGF-β3, r = –0.17; TGF-βR2, r = –0.13; LRRC32, r = –0.23), and CD274 (TGF-β3, r = –0.18; TGF-βR2, r = 0.11; LRRC32, r = –0.24), as well as TNF pathway members TNF (TGF-β3, r = –0.29; TGF-βR2, r = –0.09; LRRC32, r = –0.27) and TNFRSF9 (TGF-β3, r = –0.18; TGF-βR2, r = –0.19; LRRC32, r = –0.29) in TCGA (Data Supplement). These negative correlations were all validated in METABRIC. We also noted a positive correlation between expression of TGF-β1 and the M2-like macrophage markers IL4 (r = 0.24), IL10 (r = 0.38), and MMP9 (r = 0.43) as well as the regulatory T-cell marker FOXP3 (r = 0.42) in the TCGA data.
FIG 5.
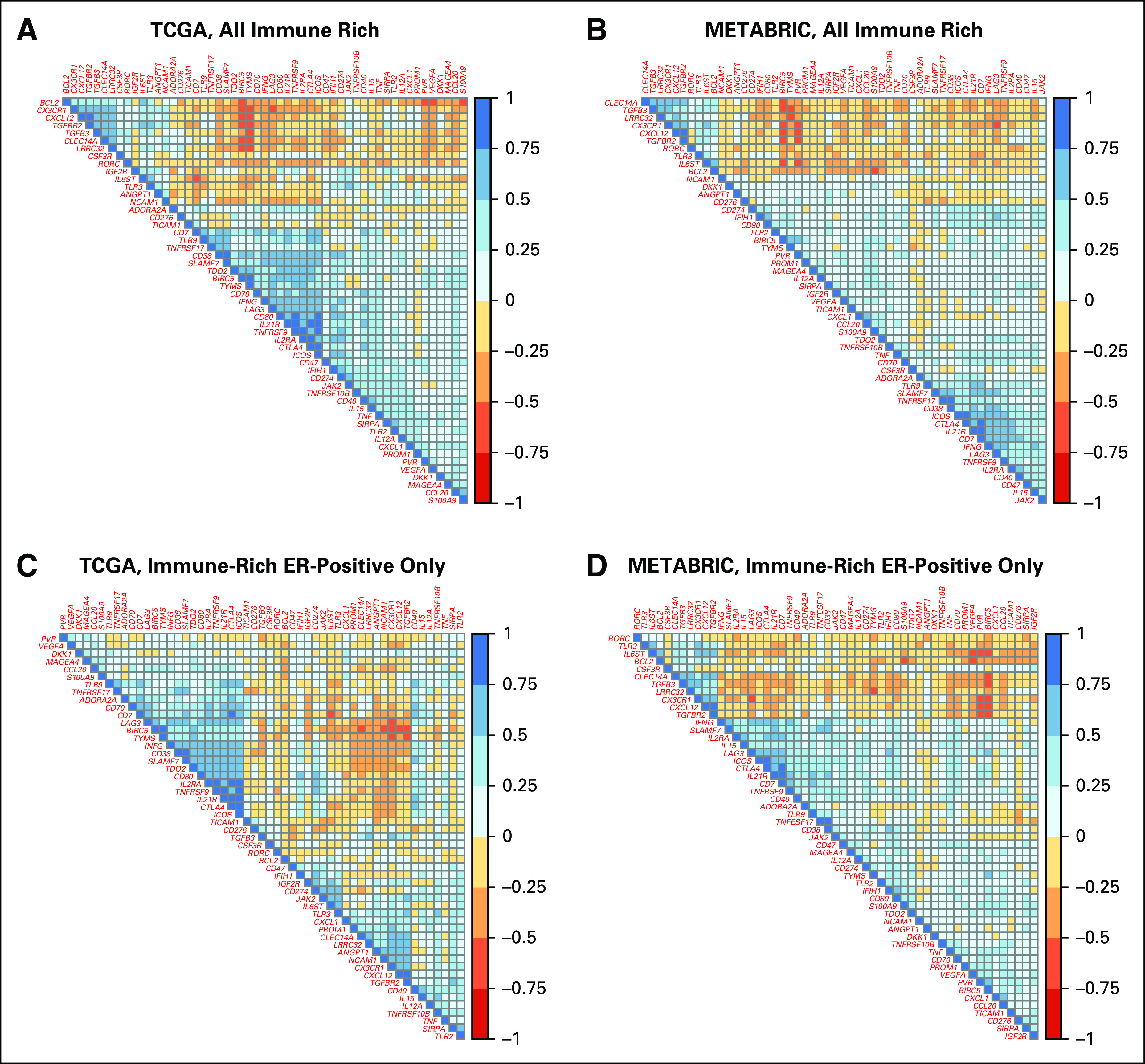
Correlation between expression levels of 53 differentially expressed immuno-oncology drug targets in immune-rich patients. For (A) The Cancer Genome Atlas (TCGA) and (B) Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) patients, correlation matrices display the Pearson correlation coefficients between expression levels of 53 immuno-oncology drug targets across patients with immune-rich estrogen receptor–positive breast cancers (ER-positive BCs) and triple-negative BCs (TNBCs). These 53 genes were selected for being significantly differentially expressed between patients with immune-rich ER-positive BCs and TNBCs in both TCGA and METABRIC databases. Blue boxes reflect positive correlations, and red boxes reflect negative correlations. For TCGA, the number of patients with immune-rich ER-positive BCs was 119, and for TNBCs, it was 86; for METABRIC, the number of patients with immune-rich ER-positive BCs was 225, and for TNBCs, it was 140. For (C) TCGA and (D) METABRIC patients, correlation matrices display the Pearson correlation coefficients between expression levels of 53 immuno-oncology drug targets in patients with immune-rich ER-positive BC alone. Blue boxes reflect positive correlations, and red boxes reflect negative correlations. The number of patients with immune-rich ER-positive BCs in TCGA was 119, and in METABRIC, it was 225.
DISCUSSION
In this report, we used a gene expression signature of total TILs to identify patients with immune-rich ER-positive BCs and TNBCs in 2 publicly available databases to compare the tumor immune microenvironment of these subtypes. The patients with immune-rich ER-positive BCs had significantly higher TMB than patients with non–immune-rich ER-positive BCs, but no difference in TMB was seen between immune-rich and non–immune-rich TNBCs. These results are consistent with previous reports.11,36 Approximately 70% of the immune-rich ER-positive BCs we identified were luminal A molecular subtype, and the PAM50 subtype distribution and Ki-67 expression were similar between immune-rich and non–immune-rich ER-positive BCs. An OS difference was detected between patients with non–immune-rich ER-positive BCs relative to non–immune-rich TNBCs, but no similar statistically significant difference was seen between patients with immune-rich ER-positive BCs and immune-rich TNBCs (both groups had excellent long-term survival). By using deconvolution methods, we observed differences in the relative fractions of immune cell subpopulations between immune-rich ER-positive BCs and immune-rich TNBCs. Immune-rich ER-positive BCs had higher relative fractions of M2-like macrophages, CD8+ T cells, and resting mast cell populations, whereas immune-rich TNBCs had overall higher macrophage content and higher M0-like and M1-like macrophage populations. These findings were largely consistent across 2 independent data sets. Higher overall CD68 protein expression in TNBCs was also confirmed by immunohistochemistry.
Interestingly, immune-rich ER-positive BCs had higher expression of a TGF-β signaling metagene and showed coordinated overexpression of TGF-β3, TGF-βR2, and LRRC32 compared with immune-rich TNBCs. The TGF-β pathway is activated by ligands TGF-β1, TGF-β2, and TGF-β3 that bind to membrane-bound serine/threonine protein receptor kinases composed of subunits TGF-βR1 and TGF-βR2. Although the TGF-β ligand isoforms are more than 75% similar in amino acid structure, TGF-βR2 has increased affinity for TGF-β1 and TGF-β3 relative to TGF-β2.37 LRRC32 encodes the GARP protein that tethers TGF-β ligands to the cell membrane to prime its activation.38 The simultaneous upregulation of the ligand, receptor, and signaling components suggests a functional role for the TGF-β pathway in immune-rich ER-positive BCs.
The immune-attenuating function of TGF-β signaling in the tumor microenvironment has been extensively studied in vitro and in vivo. In early in tumorigenesis, TGF-β acts as a tumor suppressor, inducing apoptosis in premalignant cells and inhibiting proliferation.39 After tumor formation, TGF-β promotes cancer progression through suppressing the host antitumor immune response.40 Among other roles, TGF-β inhibits CD8+ effector T-cell function, inhibits the Th1 helper T-cell phenotype, activates FOXP3-positive regulatory T cells, drives M2-like macrophage polarization, and excludes immune cells from the tumor compartment.41-45 The distinct roles of the different TGF-β isoforms in cancer remain undefined. Among the 3 isoforms, TGF-β3 is the most potent stimulator of neovascularization in models of wound healing and the most potent inhibitor of granulocyte-macrophage colony-stimulating factor–mediated hematopoiesis in the bone marrow. TGF-β1 and TGF-β3 are equally potent stimulators of extracellular matrix collagen deposition in models of pulmonary fibrosis.46 Overall, high TGF-β signaling is also associated with lesser response to immune checkpoint inhibitors.41,42 On an expression level, we found higher relative fractions of CD8+ T cells in immune-rich ER-positive BCs as well as higher TGF-β signaling; these CD8+ T cells in immune-rich ER-positive patients may therefore lack robust effector cytotoxic function or be excluded from the tumor compartment by augmented TGF-β signaling.
TGF-β can be released by cancer cells as well as by M2-like macrophages in a positive feedback loop to attenuate antitumor immunity.47 In several studies, the presence of M2-like macrophages in BC was associated with poor prognosis, and reprogramming of macrophages toward the M1-like phenotype has been shown to increase responsiveness to immune checkpoint therapy in experimental models.48,49 A recent study found that in a macrophage-dependent autochthonous mouse model of luminal B–type BC, inhibition of class IIa histone deacetylase (HDAC) by TMP195 induces the recruitment and differentiation of antitumor-type macrophages, reduces tumor burden and metastases, and enhances the durability of tumor reduction when combined with chemotherapy regimens or T-cell checkpoint blockade.50 This observation in an animal model of BC is consistent with our findings in human tissues. We found a high proportion of protumor M2-like macrophages in immune-rich ER-positive METABRIC and TCGA BC patients, so reprogramming macrophages toward the M1-like phenotype may be a viable therapeutic approach for immune-rich ER-positive tumors. Our analyses also showed that expression of TGF-β pathway members correlated negatively with the expression of many immune activation markers including interferon-γ (IFN-γ), granzymes, and perforin, but correlated positively with M2-like macrophage markers IL4, IL10, MMP9, and regulatory T-cell marker FOXP3. Although M2-like macrophages in the tumor microenvironment may be the source of TGF-β signaling, high levels of apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like–associated mutational signatures that have been previously identified in patients with immune-rich ER-positive BCs may also contribute to upregulation of the TGF-β pathway in tumor cells and downstream recruitment of M2-like macrophages.36
Deconvolution of the leukocyte compartment also revealed a higher relative fraction of mast cells in immune-rich ER-positive BCs; this was corroborated by overexpression of the mast cell markers CPA3 and MS4A2 in patients with immune-rich ER-positive BCs compared with those who have immune-rich TNBCs. How these cells influence antitumor immunity is unknown, but high mast cell infiltration in BC has been associated with ER expression and favorable prognosis in earlier studies.51,52
In summary, our analysis of gene expression data is consistent with a more attenuated immune microenvironment mediated by increased TGF-β signaling and M-2 macrophage presence in immune-rich ER-positive BCs. In this subset of ER-positive BCs, we observed high relative fractions of CD8+ T cells, mast cells, and M2-like macrophages with lower expression of granzyme-B, perforin, and IFN-γ. Future work is needed to explore the molecular mechanisms driving the distinct immune activation patterns in immune-rich ER-positive BCs and immune-rich TNBCs. In addition, practical assays that can distinguish patients with immune-rich and TGF-β-high from patients with TGF-β-low ER-positive BCs are needed before these results can be applied in the clinic. Nonetheless, the immunologic differences identified between immune-rich ER-positive BCs and TNBCs support the exploration of distinct immunotherapy strategies in these different molecular subtypes. Our results pose the hypothesis that therapeutic drugs targeting the TGF-β pathway (eg M7824, PF-06952229, NIS793, ABBV151) and M2-like macrophages (SNDX-6352) may be particularly effective in ER-positive BCs with high immune cell infiltration.53,54
Presented in part at the European Society for Medical Oncology Annual Congress, Barcelona, Spain, September 27-October 1, 2019.
SUPPORT
Supported by National Cancer Institute Grant No. R01CA219647 (L.P.), Susan Komen Foundation Leadership Award 190076 (L.P.), and Grant No. BCRF 19-133 from the Breast Cancer Research Foundation (L.P. and D.L.R.).
AUTHOR CONTRIBUTIONS
Conception and design: Tess O’Meara, Lajos Pusztai
Financial support: Lajos Pusztai
Provision of study materials or patients: David L. Rimm
Collection and assembly of data: Tess O’Meara, Vesal Yaghoobi, Kimberly Cole, Vasiliki Pelekanou, David L. Rimm, Lajos Pusztai
Data analysis and interpretation: Tess O’Meara, Michal Marczyk, Tao Qing, Kim Blenman, Vasiliki Pelekanou, Lajos Pusztai
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Vasiliki Pelekanou
Employment: Sanofi
David L. Rimm
Stock and Other Ownership Interests: Pixel Gear
Honoraria: Amgen, Bristol Myers Squibb, Ventana Medical Systems
Consulting or Advisory Role: Perkin Elmer, Bristol Myers Squibb, AstraZeneca, Agendia, Ultivue, Merck, Daiichi Sankyo, GlaxoSmithKline, Konica Minolta, NanoString Technologies, NextCure, Cell Signaling Technology, Roche, Paige, Cepheid, Sanofi
Research Funding: Cepheid (Inst), Perkin Elmer (Inst), AstraZeneca/MedImmune (Inst), NextCure (Inst), Eli Lilly (Inst), NanoString Technologies (Inst), Navigate Biopharma (Inst), Ultivue (Inst), Konica Minolta (Inst), Amgen (Inst)
Patents, Royalties, Other Intellectual Property: Rarecyte circulating tumor cells; Quantitative immunofluorescence (AQUA) (Inst)
Travel, Accommodations, Expenses: Ventana Medical Systems, NextCure, Bristol-Myers Squibb
Lajos Pusztai
Consulting or Advisory Role: H3 Biomedicine, Merck, Novartis, Seattle Genetics, Syndax Pharmaceuticals, Athenex, AstraZeneca, Genentech, Bristol Myers Squibb, Clovis Oncology, Immunomedics, Eisai, Almac Diagnostics
Research Funding: Merck (Inst), Genentech (Inst), Seattle Genetics (Inst), AstraZeneca (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Uncompensated Relationships: NanoString Technologies
Open Payments Link: https://openpaymentsdata.cms.gov/physician/110878/summary
No other potential conflicts of interest were reported.
REFERENCES
- 1.Bianchini G, Qi Y, Alvarez RH, et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol. 2010;28:4316–4323. doi: 10.1200/JCO.2009.27.2419. [DOI] [PubMed] [Google Scholar]
- 2.Karn T, Pusztai L, Holtrich U, et al. Homogeneous datasets of triple negative breast cancers enable the identification of novel prognostic and predictive signatures. PLoS One. 2011;6:e28403. doi: 10.1371/journal.pone.0028403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams S, Gray RJ, Demaria S, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32:2959–2966. doi: 10.1200/JCO.2013.55.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loi S, Sirtaine N, Piette F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31:860–867. doi: 10.1200/JCO.2011.41.0902. [DOI] [PubMed] [Google Scholar]
- 5.Iwamoto T, Bianchini G, Booser D, et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J Natl Cancer Inst. 2011;103:264–272. doi: 10.1093/jnci/djq524. [DOI] [PubMed] [Google Scholar]
- 6.Denkert C, von Minckwitz G, Brase JC, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33:983–991. doi: 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 7.Wimberly H, Brown JR, Schalper K, et al. PD-L1 expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res. 2015;3:326–332. doi: 10.1158/2326-6066.CIR-14-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loi S, Giobbe-Hurder A, Gombos A, et al. Phase Ib/II study evaluating safety and efficacy of pembrolizumab and trastuzumab in patients with trastuzumab-resistant HER2-positive metastatic breast cancer: Results from the PANACEA (IBCSG 45-13/BIG 4-13/KEYNOTE-014) study. Cancer Res. 2018;78 abstr GS2-06. [Google Scholar]
- 9.Buisseret L, Garaud S, de Wind A, et al. Tumor-infiltrating lymphocyte composition, organization and PD-1/ PD-L1 expression are linked in breast cancer. OncoImmunology. 2016;6:e1257452. doi: 10.1080/2162402X.2016.1257452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Narang P, Chen M, Sharma AA, et al. The neoepitope landscape of breast cancer: Implications for immunotherapy. BMC Cancer. 2019;19:200. doi: 10.1186/s12885-019-5402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safonov A, Jiang T, Bianchini G, et al. Immune gene expression is associated with genomic aberrations in breast cancer. Cancer Res. 2017;77:3317–3324. doi: 10.1158/0008-5472.CAN-16-3478. [DOI] [PubMed] [Google Scholar]
- 12.Adams S, Diamond JR, Hamilton E, et al. Atezolizumab plus nab-paclitaxel in the treatment of metastatic triple-negative breast cancer with 2-year survival follow-up: A phase 1b clinical trial. JAMA Oncol. 2019;5:334–342. doi: 10.1001/jamaoncol.2018.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 14.Tolaney SM, Barroso-Sousa R, Keenan T, et al. Randomized phase II study of eribulin mesylate (E) with or without pembrolizumab (P) for hormone receptor-positive (HR+) metastatic breast cancer (MBC) J Clin Oncol. 2019;37 suppl; abstr 1004. [Google Scholar]
- 15.Rugo HS, Delord JP, Im SA, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin Cancer Res. 2018;24:2804–2811. doi: 10.1158/1078-0432.CCR-17-3452. [DOI] [PubMed] [Google Scholar]
- 16.Esteva FJ, Hubbard-Lucey VM, Tang J, et al. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20:e175–e186. doi: 10.1016/S1470-2045(19)30026-9. [DOI] [PubMed] [Google Scholar]
- 17.American Cancer Society Breast Cancer Facts and Figures, 2017-2018. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2017-2018.pdf
- 18.van Maaren MC, de Munck L, Strobbe LJA, et al. Ten-year recurrence rates for breast cancer subtypes in the Netherlands: A large population-based study. Int J Cancer. 2019;144:263–272. doi: 10.1002/ijc.31914. [DOI] [PubMed] [Google Scholar]
- 19.Danaher P, Warren S, Dennis L, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer. 2017;5:18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499–1509. doi: 10.1158/1078-0432.CCR-15-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luen S, Virassamy B, Savas P, et al. The genomic landscape of breast cancer and its interaction with host immunity. Breast. 2016;29:241–250. doi: 10.1016/j.breast.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 22.Thomas A, Routh ED, Pullikuth A, et al. Tumor mutational burden is a determinant of immune-mediated survival in breast cancer. OncoImmunology. 2018;7:e1490854–e1490854. doi: 10.1080/2162402X.2018.1490854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schröder MS, Culhane AC, Quackenbush J, et al. survcomp: An R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. 2011;27:3206–3208. doi: 10.1093/bioinformatics/btr511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teschendorff AE, Gomez S, Arenas A, et al. Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules. BMC Cancer. 2010;10:604. doi: 10.1186/1471-2407-10-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrade JM, Estévez-Pérez MG. Statistical comparison of the slopes of two regression lines: A tutorial. Anal Chim Acta. 2014;838:1–12. doi: 10.1016/j.aca.2014.04.057. [DOI] [PubMed] [Google Scholar]
- 31.Pelekanou V, Villarroel-Espindola F, Schalper KA, et al. CD68, CD163, and matrix metalloproteinase 9 (MMP-9) co-localization in breast tumor microenvironment predicts survival differently in ER-positive and -negative cancers. Breast Cancer Res. 2018;20:154. doi: 10.1186/s13058-018-1076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang J, Pearce L, O’Donnell-Tormey J, et al. Trends in the global immuno-oncology landscape. Nat Rev Drug Discov. 2018;17:783–784. doi: 10.1038/nrd.2018.167. [DOI] [PubMed] [Google Scholar]
- 33.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei T, Simko V. R package “corrplot”: Visualization of a correlation matrix. 2017 https://github.com/taiyun/corrplot
- 36.Panda A, Betigeri A, Subramanian K, et al. Identifying a clinically applicable mutational burden threshold as a potential biomarker of response to immune checkpoint therapy in solid tumors. JCO Precis Oncol. doi: 10.1200/PO.17.00146. epub ahead of print on December 7, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massagué J. Receptors for the TGF-β family. Cell. 1992;69:1067–1070. doi: 10.1016/0092-8674(92)90627-o. [DOI] [PubMed] [Google Scholar]
- 38.Tran DQ, Andersson J, Wang R, et al. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guasch G, Schober M, Pasolli HA, et al. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313–327. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 41.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 42.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly A, Gunaltay S, McEntee CP, et al. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8-mediated TGFβ activation. J Exp Med. 2018;215:2725–2736. doi: 10.1084/jem.20171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sledzińska A, Hemmers S, Mair F, et al. TGF-β signalling is required for CD4+ T cell homeostasis but dispensable for regulatory T cell function. PLoS Biol. 2013;11:e1001674. doi: 10.1371/journal.pbio.1001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laverty HG, Wakefield LM, Occleston NL, et al. TGF-β3 and cancer: A review. Cytokine Growth Factor Rev. 2009;20:305–317. doi: 10.1016/j.cytogfr.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YS, Park JS, Kim JH, et al. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat Commun. 2011;2:460. doi: 10.1038/ncomms1469. [DOI] [PubMed] [Google Scholar]
- 48.Cassetta L, Kitamura T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front Cell Dev Biol. 2018;6:38. doi: 10.3389/fcell.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gan L, Qiu Z, Huang J, et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway. Int J Biol Sci. 2016;12:1533–1543. doi: 10.7150/ijbs.15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerriero JL, Sotayo A, Ponichtera HE, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajput AB, Turbin DA, Cheang MC, et al. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: A study of 4,444 cases. Breast Cancer Res Treat. 2008;107:249–257. doi: 10.1007/s10549-007-9546-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sang J, Yi D, Tang X, et al. The associations between mast cell infiltration, clinical features and molecular types of invasive breast cancer. Oncotarget. 2016;7:81661–81669. doi: 10.18632/oncotarget.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50:924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cannarile MA, Weisser M, Jacob W, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:53. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]