

Abstract
Purpose
To evaluate the feasibility of using circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) for the management of uveal melanoma (UM).
Patients and Methods
Low-coverage whole-genome sequencing was used to determine somatic chromosomal copy number alterations (SCNAs) in primary UM tumors, ctDNA, and whole-genome amplified CTCs. CTCs were immunocaptured using an antimelanoma-associated chondroitin sulfate antibody conjugated to magnetic beads and immunostained for melanoma antigen recognised by T cells 1 (MART1)/glycoprotein 100 (gp100)/S100 calcium-binding protein β (S100β). ctDNA was quantified using droplet digital polymerase chain reaction assay for mutations in the GNAQ, GNA11, PLCβ4, and CYSLTR2 genes.
Results
SCNA analysis of CTCs and ctDNA isolated from a patient with metastatic UM showed good concordance with the enucleated primary tumor. In a cohort of 30 patients with primary UM, CTCs were detected in 58% of patients (one to 37 CTCs per 8 mL of blood), whereas only 26% of patients had detectable ctDNA (1.6 to 29 copies/mL). The presence of CTCs or ctDNA was not associated with tumor size or other prognostic markers. However, the frequent detection of CTCs in patients with early-stage UM supports a model in which CTCs can be used to derive tumor-specific SCNA relevant for prognosis. Monitoring of ctDNA after treatment of the primary tumor allowed detection of metastatic disease earlier than 18F-labeled fluorodeoxyglucose positron emission tomography in two patients.
Conclusion
The presence of CTCs in localized UM can be used to ascertain prognostic SCNA, whereas ctDNA can be used to monitor patients for early signs of metastatic disease. This study paves the way for the analysis of CTCs and ctDNA as a liquid biopsy that will assist with treatment decisions in patients with UM.
INTRODUCTION
Uveal melanoma (UM) is the most common intraocular malignancy.1 Despite successful control of the primary tumor within the eye, metastatic disease ultimately develops in up to 50% of patients, predominantly in the liver. Currently, there are limited therapeutic options for metastatic UM, and as a result there is a high mortality rate.2 Extensive analysis of primary UMs has defined molecular features of the tumor cells that predict, with a high degree of accuracy, a patient’s risk for development of metastases. Biomarkers of poor prognosis include histopathological features of the tumor; somatic copy number alterations (SCNAs), such as loss of chromosome 3, 6q, and 8q3,4; BAP1 mutations5,6; and the differential expression of marker genes that include well-characterized cancer-associated factors.7,8 Other features, such as gain in 6p and mutations in EIF1AX and SF3B1, are associated with better prognosis.4,6
Given that metastasis in UM arises from hematogenous dissemination, investigation of circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) could provide a unique opportunity for genetic analysis of the patient’s tumor through a simple and safe blood test. Previous research has indicated that CTCs harbor genetic profiles representative of the primary tumor9 and can be used to detect tumor-specific mutations in other cancers.9-14 In UM, amplification of DNA from a single CTC has been reported, with comparative genomic hybridization array showing copy number abnormalities associated with poor prognosis.15 A recent study described the detection of loss in chromosome 3 in CTCs using a modified fluorescence in situ hybridization technique as a method to aid prognostication of UM patients likely to metastasize.16
An additional blood-based marker, commonly used to evaluate tumor burden and tumor-specific genetic features, is ctDNA.17,18 The high proportion of recurrent hot spot mutations in UM enables the opportune detection of ctDNA using droplet digital polymerase chain reaction (PCR). UMs carry mutually exclusive activating mutations in GNAQ, GNA11, PLCβ4, and CYSLTR2, encompassing more than 90% of UM patients.4,19-22 Bidard et al18 detected ctDNA in 84% of patients with UM with metastatic disease and found ctDNA levels to be an independent prognostic factor for both progression-free survival and overall survival.18 However, the presence and prognostic significance of ctDNA in patients with primary UM without detectable metastatic disease has yet to be evaluated.
Given that UM harbors distinct SCNAs that correlate with poor prognosis (L1p, L3, L6q, G8q) and good prognosis (G6p),4 their detection in CTCs may offer a minimally invasive method for prognostication. Furthermore, because ctDNA is highly correlated with tumor volume,18 it may offer a minimally invasive method for detection of metastatic disease.
Here, we evaluated the viability of CTC and ctDNA as suitable biomarkers to derive prognostic information in UM. Whole-genome SCNAs were derived from CTCs and ctDNA from a patient with metastatic UM and compared with those in the primary tumor. The blood of patients with primary UM was analyzed for both the number of CTCs immunomagnetically captured using melanoma-associated chondroitin sulfate proteoglycan (MCSP) and the level of plasma ctDNA. Finally, we showed that ctDNA monitoring allowed early detection of metastatic disease in two patients with UM.
PATIENTS AND METHODS
Patients and Sample Processing
Thirty patients with primary UM from the Lions Eye Institute and Royal Perth Hospital, Perth, Western Australia, were enrolled in the study between March 2014 and November 2016. UM was diagnosed by clinical and ultrasound examination performed by a specialist ophthalmologist to evaluate the size and location of the intraocular tumor, including the presence of ciliary body involvement. Peripheral blood samples were taken from 30 patients before radiation plaque insertion or enucleation. Eight patients with metastatic UM were recruited from oncology outpatient clinics at Sir Charles Gardner and Fiona Stanley Hospitals. Written informed consent was obtained from all patients under approved Human Research Ethics Committee protocols from Edith Cowan University (No. 11543) and Sir Charles Gardner Hospital (No. 2013-246), Western Australia. For CTC quantification, blood was collected in Vacutainer K2 EDTA tubes (BD Biosciences, Franklin Lakes, NJ), stored at 4°C, and processed within 24 hours. Plasma was isolated by double centrifugation at 1,600 g for 10 minutes and stored at −80°C.
CTC Capture, Quantification, and Low-Pass Whole-Genome Sequencing
CTC isolation was adapted from a previously described protocol,23 detailed in the Data Supplement. One hundred nanograms of whole-genome amplified (WGA) DNA (Data Supplement) was used to construct 200-bp sequencing libraries using an NEB Next Ultra Fragment Library Kit (New England Biosciences, Ipswich, MA) and barcoded using the Ion Xpress Barcode Adapters 1-96 Kit (Life Technologies, Carlsbad, CA). Libraries were sequenced for 520 flows on a P1 sequencing chip using an Ion Proton Sequencer (Life Technologies). Sequencing depths ranged from 0.26 to 0.59×. Somatic mutations and SCNAs were analyzed using Ion Reporter 4.6 (Life Technologies).
ctDNA Quantification
Cell-free DNA (cfDNA) was extracted from 1 to 5 mL of plasma using a QIAamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and stored at −80°C. ctDNA was quantified using droplet digital PCR and PrimePCR assays as described in the Data Supplement.
Statistical Analysis
The Spearman rank correlation coefficient was used to test the correlation between the level of ctDNA, number of CTCs, and tumor size. Tumor volume was calculated using a formula previously described.24 The numbers of captured CTCs in patients with and without monosomy of chromosome 3 were compared using a nonparametric Mann-Whitney U test. Statistical analyses were performed using Graphpad Prism version 5.0 (GraphPad Software, La Jolla, CA).
RESULTS
Analysis of Somatic Copy Number Alterations in CTCs From a Patient With UM With Metastatic Disease
CTCs were isolated from peripheral venous blood of a patient with metastatic UM (patient 640). The patient was originally diagnosed with UM in February 2015 and underwent plaque brachytherapy; however, response was suboptimal, and the eye was subsequently enucleated. The clinical history of this patient is detailed in the Data Supplement. In March 2017, the patient was diagnosed with metastatic UM, with bone marrow activity in the spine, pelvis, ribs, and both femora (Fig 1A). Analysis of a blood sample obtained in March 2017, before initiation of pembrolizumab therapy, showed the presence of 33 CTCs in 8 mL of blood. A total of three CTCs were separated and subjected to WGA (Fig 1B). A single peripheral blood mononuclear cell (PBMC) subjected to WGA was used as negative control. The accuracy and reliability of this WGA method for the analysis of single UM for assessment of SCNA were first demonstrated using individual cells from three UM cell lines (Data Supplement).
Fig 1.
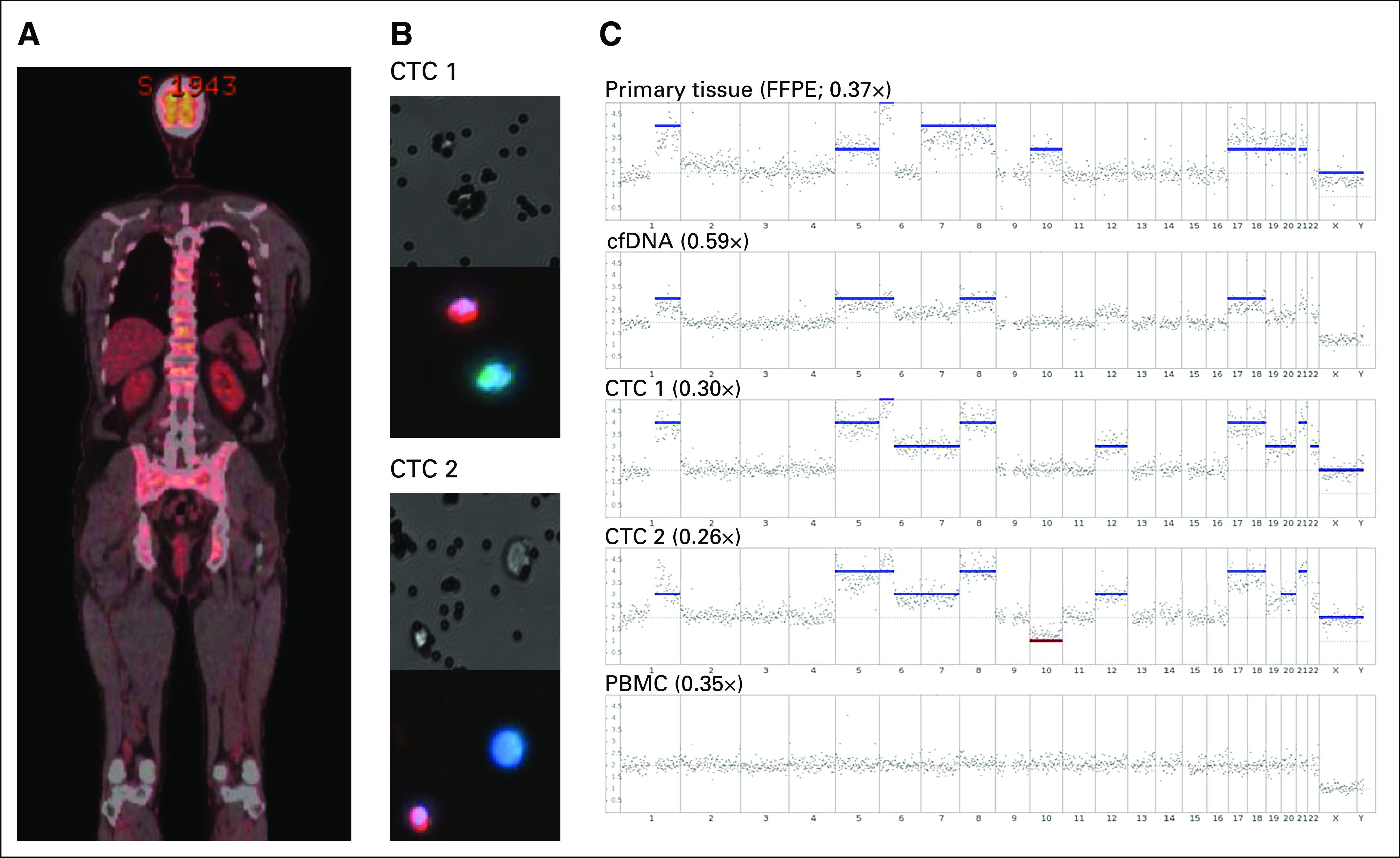
Comparison between the genetic profile of the primary tumor, cell-free DNA (cfDNA), two circulating tumor cells (CTCs), and a single peripheral blood mononuclear cell (PBMC) in a patient with metastatic uveal melanoma. (A) Whole-body fluorodeoxyglucose–positron emission tomography scan of a patient with uveal melanoma at the time of blood collection. (B) Brightfield and florescent images of the two CTCs used for somatic chromosomal copy number alteration analysis. Cells were stained with a combination of antibodies against the melanoma markers melanoma antigen recognised by T cells 1 (MART1)/glycoprotein 100(gp100)/S100 calcium-binding protein β (S100β; green), CD45 (red), and 4',6-diamidino-2-phenylindole (DAPI; blue), taken at ×200 magnification. (C) Whole-genome sequencing somatic chromosomal copy number alteration profile of primary formalin-fixed paraffin-embedded tumor, cfDNA, two CTCs, and a single PBMC. The obtained sequence depth is indicated for each plot. Red and blue bars represent chromosomal losses or gains, respectively.
Two CTCs and a PBMC provided suitable WGA-DNA material for whole-genome sequencing. In addition, we sequenced DNA extracted from the patient’s archived formalin-fixed paraffin-embedded primary tumor (with > 80% tumor cellularity) taken 2 years before isolation of CTCs. We also assessed cfDNA extracted from the same blood draw as that for CTC isolation. The primary tumor had large copy number gains in chromosome 1q, 5, 6p, 7, 8, 10, 17, 18, 19, 20, 21, X, and Y (Fig 1C). Multiplex ligand-dependent probe amplification (MLPA) analysis of the primary tumor confirmed the chromosomal gains of 6p and 8p/8q and the lack of evidence for SCNAs of 1p and 3p/3q (Data Supplement). The two isolated CTCs also showed overlapping chromosomal gains and losses in comparison with the primary tumor, despite the primary tumor being removed 2 years earlier (Fig 1C). DNA was unable to be recovered from the bone metastases because of acid decalcification of the specimen before embedding. Additional alterations found in CTCs comprised a gain in 6q in both cells, a gain of chromosome 22 in CTC1, and a loss of chromosome 10 in CTC2. The PBMC analyzed did not harbor any SCNAs (Fig 1C), same as multiple PBMCs used as negative controls in the validation experiments (Data Supplement). The GNA11 Q209L mutation was detectable in DNA derived from the primary tumor and in WGA CTCs.
Sequencing of cfDNA from the same blood sample from which CTCs were isolated also showed similar chromosome gains, with trends toward gains in some chromosomal alterations found in the primary tissue; however, these did not reach the threshold to be called a true gain (Fig 1C). cfDNA analysis could not detect all of the changes found in the primary tumor, most likely because of a high abundance of normal DNA. Droplet digital PCR targeting the GNA11 Q209L mutation in the cfDNA indicated the presence of 15,460 copies/mL of mutant DNA in plasma, with a 20.2% frequency abundance relative to normal DNA. Thus, despite the significant abundance of ctDNA, this compartment is not as sensitive as CTCs for the analysis of SCNAs.
These results illustrate that tumor-associated SCNA can be ascertained through genetic analysis of CTCs, providing important prognostic information. Because prognostication is critical for the clinical management of patients diagnosed with early-stage UM, it was important to determine whether CTCs and ctDNA were readily detectable in these patients.
CTCs in Blood of Patients With Primary UM
To determine the UM CTC detection rate in patients with localized disease, we analyzed the blood from 30 patients with primary UM, without the presence of clinically identifiable metastatic disease, obtained before radiation plaque insertion or enucleation (Table 1). CTCs were identified by positive staining for melanoma antigen recognised by T cells 1 (MART1)/glycoprotein 100(gp100)/S100 calcium-binding protein β (S100β) and negative staining for CD45 (Fig 2A). A total of 15 of 26 (58%) individuals with assessable results had at least one CTC in 8 mL of blood, with a range of one to 37 CTCs detected, and 14 (54%) patients had two or more detectable CTCs. Only single cells, rather than clusters, were detected in all patients. The presence or quantity of CTCs captured using MCSP did not correlate with the tumor basal and apical sizes or tumor volume (Figs 2B-2D). Nevertheless, among the 10 patients who underwent tumor biopsy, no significant difference was found between the number of CTCs and monosomy or loss of chromosome 3 in the tumor (Fig 2E; P = .062).
Table 1.
Clinical, Genotypic, and Histologic Characteristics of Patients Undergoing Circulating Tumor Cell Capture

Fig 2.
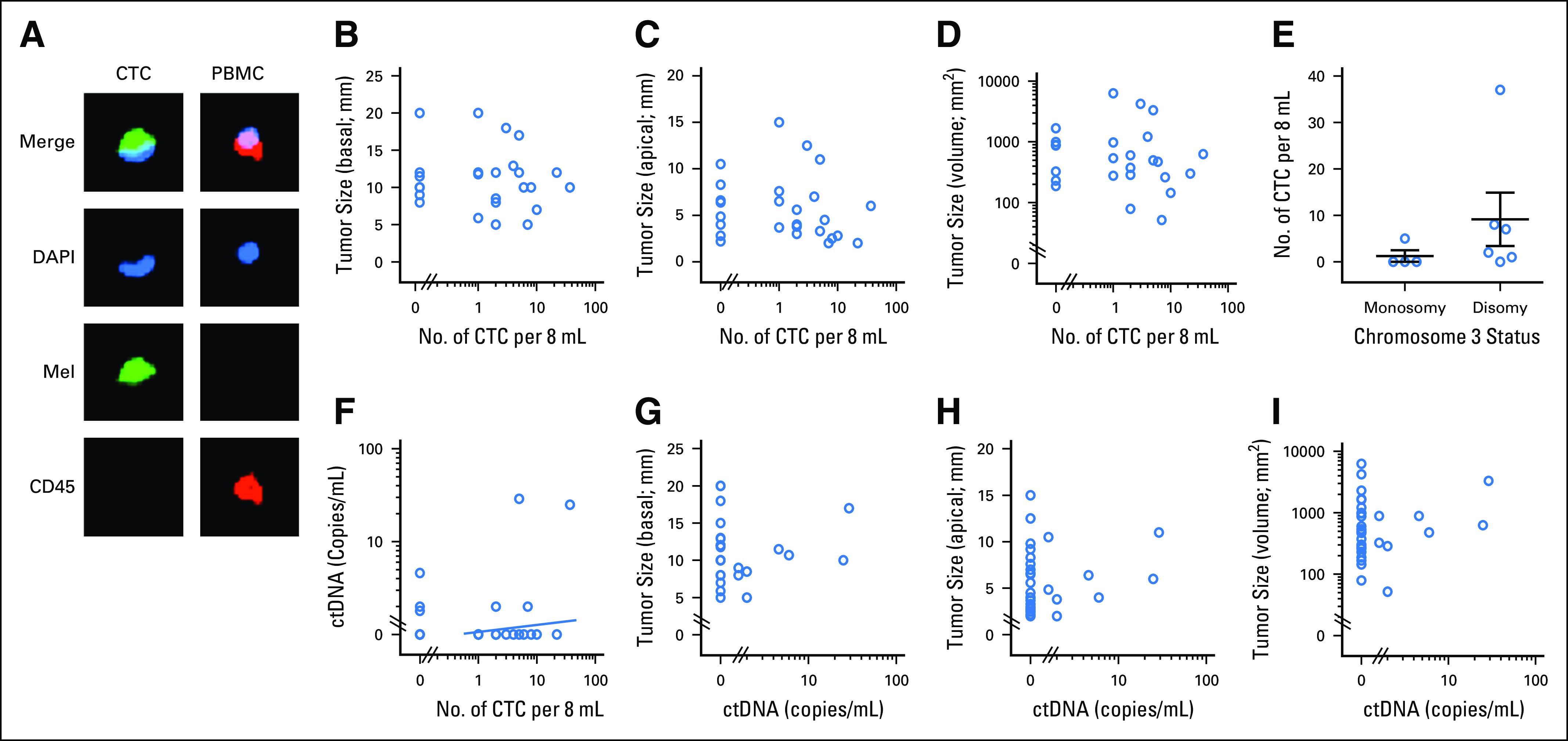
Circulating tumor cell (CTC) and circulating tumor DNA (ctDNA) quantification in a primary uveal melanoma (UM) cohort. (A) Example of immunocytochemical staining of a UM CTC and peripheral blood mononuclear cell (PBMC). Green fluorescence (AF488, Mel) indicates staining with a combination of antibodies against the melanoma markers MART1/gp100/S100β; red fluorescence (phycoerythrin [PE]) indicates CD45 positivity; and blue fluorescence (4',6-diamidino-2-phenylindole, DAPI) indicates the presence of a nucleus. CTCs were identified as Mel-positive and DAPI-positive and CD45-negative cells. (B-E) Graphs illustrate (B) CTC count versus basal median diameter (n = 26; P = .874; r = −0.034), (C) tumor size as apical height (n = 26; P = .237; r = −0.250), or (D) tumor volume (n = 26; P = .338; r = −0.244). Spearman rank r and P values are indicated. (E) Comparison of CTC counts in patients with UM with and without chromosome 3 monosomy (n = 10). Mann-Whitney U test P value is indicated. (F-I) Graphs illustrate (F) ctDNA copies/mL versus CTC count in 8 mL of blood (n = 26), (G) ctDNA copies/mL versus basal median diameter (n = 30; P = .787; r = 0.053), (H) tumor size as apical height (n = 30; P = .384; r = −0.170), or (I) tumor volume (n = 30; P = .982; r = −0.004). Spearman rank r and P values are indicated. No correlation was found between CTC, ctDNA, and tumor size.
ctDNA in the Blood of Patients With Primary UM
The plasma from all 30 patients with UM described above was also analyzed for the presence of common UM-associated mutations GNAQ/GNA11 Q209L/P, GNAQ/GNA11 R183Q/C, PLCβ4 D630Y, and CYSLTR2 L129Q (Table 1; Fig 2). Screening for all of these mutations was necessary in our study, because for most patients fine-needle aspirate biopsies were either not performed or provided limited amounts of DNA that was used for MLPA testing and, therefore, we were unable to determine the tumor’s driver mutations before testing for ctDNA. Instead, all patient blood samples were tested for these mutations, because they have been reported to occur in > 90% of UMs.19,20,22 We detected ctDNA in eight of the 30 patients tested (23%; range, 1.6 to 29 copies); two patients had a GNAQ Q209L mutation, two had a GNA11 Q209L mutation, one had a GNA11 Q209P mutation, one had a GNA11 R183C mutation, one had a PLCβ4 D630Y mutation, and one had a CYSLTR2 L129Q mutation. Only four of 30 patients had simultaneous detection of ctDNA and CTCs (Fig 2F). In those with detectable mutations, ctDNA levels were correlated with tumor size (largest basal/apical diameter/volume; Figs 2G-2I).
ctDNA for Detection of Metastatic UM
We also analyzed ctDNA in eight patients with metastatic UM. In contrast to those with localized disease, all patients with metastatic UM had detectable ctDNA (Table 2; Fig 3A; P < .001). Retrospective analysis of longitudinal samples collected from two patients indicated that detection of ctDNA preceded radiologic recognition of liver metastases.
Table 2.
Characteristics of Patients with Confirmed Metastatic Disease

Fig 3.

Analysis of circulating tumor DNA (ctDNA) in patients with liver metastasis. (A) Comparison of ctDNA levels in patients with primary (n = 27) and metastatic (n = 8) uveal melanoma (UM) showed statistically significant differences (Student t test, P < .001). (B) Plasma ctDNA levels in longitudinally collected samples from patient 656 with UM. (C) Plasma ctDNA levels in longitudinally collected samples from patient 433 with UM, before and after the development of overt metastatic disease as shown by fluorodeoxyglucose–positron emission tomography imaging of the liver. NAD, no active disease; PD, progressive disease. (*) Resection of a solitary liver metastasis.
Patient 656 had an enucleation 3 months before enrollment and a history of low-grade lymphoproliferative disorder and pulmonary embolism. Pathology of the enucleated tumor confirmed a choroidal melanoma, with Callender classification mixed and no angiolymphatic invasion. MLPA classified this patient as high risk (loss of chromosome 3p/q plus gain of 8q). A positron emission tomography (PET) scan 4 weeks before enrollment showed mildly fluorodeoxyglucose (FDG)-avid bilateral pelvic and inguinofemoral lymphadenopathy consistent with her history of a low-grade lymphoproliferative disorder. Surveillance computed tomography scans performed 6 months later did not show any evidence of metastatic melanoma. However, the baseline liquid biopsy indicated the presence of mutant GNA11 R183C ctDNA at 13 copies/mL (Fig 3B). A second blood test at week 13 showed a similar low level of ctDNA (20 copies/mL). A concurrent PET scan showed small-volume FDG-avid foci evident near the right hilum and at the right lower lung. An early follow-up computed tomography scan at week 24 confirmed multiple small moderate FDG-avid liver and lung metastases. The closest blood analysis, at week 26, before initiation of pembrolizumab therapy, still indicated low ctDNA concentrations (10.8 copies/mL). A final blood collection at week 33 indicated the presence of 17.2 copies of ctDNA, with a recent scan showing no improvement on disease burden.
Similarly, in another patient (patient 433), 1.8 copies/mL of ctDNA encoding a GNA11 Q209L mutation was detected 55 weeks after brachytherapy (Fig 3C). A concurrent PET scan did not indicate the presence of metastatic growth. By week 75, a PET scan and MRI scan detected an isolated liver metastasis. Plasma analysis indicated an increase in ctDNA to 9 copies/mL. The metastatic lesion was surgically removed, and a slight decrease in ctDNA was observed. However, ctDNA levels significantly increased to 1,380 copies/mL by week 96, with the concurrent imaging indicating new extensive metastases to the left lobe of the liver and new bone metastases. These results illustrate that for UM, ctDNA can be used to track disease burden and has the potential to provide a complementary measure in addition to imaging.
DISCUSSION
Our results provide the proof of concept for using blood-based biomarkers for prognostication and routine monitoring of patients with UM. First, we showed that SCNA significant for UM prognostication can be detected in CTCs. Second, we found that CTCs can be detected, albeit at a low quantity, in most patients with UM with primary localized disease. Finally, although ctDNA was commonly undetectable in localized UM, monitoring of ctDNA allowed for early detection of metastatic disease. These preliminary findings warrant additional clinical studies to validate the use of these two biomarkers for the management of UM.
Previously, SCNA profiles of the primary tumor have been shown to accurately identify patients with UM at risk for developing metastatic disease.4,25 Although fine-needle aspirate biopsies are performed worldwide, genetic testing of UM primary tumors can be hampered by patients declining biopsy because of the perceived invasiveness of the procedure and the preferred use of sight-conserving therapies. Nevertheless, up to 50% of patients with UM will develop metastatic tumors after either short or long latency periods.26 Thus, development of pre-emptive adjuvant therapies may be an important strategy for improving survival. In this context, the provision of a blood test for the identification of prognostic SCNA from CTCs would allow identification of patients at risk, aiding triaging of patients for clinical trials and more frequent systemic surveillance for metastatic disease.
Most CTC studies in UM have been limited to CTC quantification. However, they have failed to find significant association between the levels of CTCs and disease prognosis.27,28 Similarly, we found that most patients have detectable CTCs irrespective of the predicted propensity of their tumor to metastasize, indicating that the presence of these CTCs may not be associated with metastatic disease risk. However, our CTC detection was restricted to the detection of MCSP-expressing cells, and thus we cannot exclude that CTCs expressing other cell surface markers may also provide prognostic information, as shown in some studies.28,29 Nevertheless, the opportunity to examine the genomic features in CTCs may offer a more accurate indication of patient metastatic risk, in comparison with simple quantification of CTCs. On the basis of the results presented here, methodologies to enhance CTC capture16,28 are needed for the optimal implementation of CTCs as a viable alternate source of tumor genetic material from which prognosis can be derived for most patients.
Sequencing of the matching cfDNA revealed several large chromosomal gains, but because of the high abundance of normal cfDNA, we could only detect trends toward chromosomal amplifications found in the CTCs and primary tumor. Patient 640 (Fig 1) had a high disease volume, with 15,460 copies/mL of GNA11 Q209L ctDNA with a fractional abundance of 20%, whereas by contrast the highest ctDNA level in our primary UM study was found to be 29 copies/mL, with a fractional abundance of < 1%; therefore, sequencing of patients with localized disease for SCNA profiles may prove ineffective. Previous studies investigating SCNA in cfDNA similarly required the presence of a large fraction of ctDNA present to obtain results similar to the tissue of origin.30 Thus, although ctDNA is much easier to isolate, analysis of SCNA in CTCs should prove to be a more effective means of analyzing prognostic SCNA.
We also showed that ctDNA is not commonly detectable in blood of patients with localized UM. In contrast, most patients with metastatic UM we have tested have detectable ctDNA, consistent with a previous report.18 Nonetheless, longitudinal monitoring of two patients with primary UM who later developed metastatic disease showed undetectable levels of ctDNA at baseline, but ctDNA was detected around the time of clinical confirmation of disease progression by PET scan. Our findings in this study indicate that the low levels of detectable ctDNA in patients with primary disease are not suitable for screening of patients at a high risk of developing metastasis. However, given the high proportion of hotspot mutations in UM,4 ctDNA analysis may be a feasible minimally invasive method of monitoring metastatic disease burden and disease progression, as we have exemplified here.
In conclusion, our study underscores the potential clinical use of liquid biopsy for UM. Possible clinical applications involve the use of CTCs to derive SCNA for stratification of UMs into low- or high-risk categories and ctDNA to monitor disease progression, pending clinical validation of our findings through future prospective studies.
ACKNOWLEDGMENT
We thank all the participants, patients, and healthy volunteers for their assistance with the study. We also thank Sergio Roman-Roman, MD, from the Institut Curie, France, for kindly supplying the uveal melanoma cell lines. We also thank Pauline Zaenker, Ashleigh McEvoy, and Danielle Bartlett for their phlebotomy services, William Cunningham, MD, and Rian Vijoen, MD, for assisting with patient recruitment at Royal Perth Hospital, and Karen Shakespeare for gathering the clinical data.
Footnotes
Supported by an Edith Cowan University Postgraduate Scholarship and a Cancer Council Western Australia Honours Scholarship (A.B.); a fellowship from the Cancer Research Trust (E.S.G.); Raine Medical Research Foundation Priming grant, Ophthalmic Research Institute of Australia grant, and Western Australia Cancer Council Grant No. 1100249 (E.S.G.); and National Health and Medical Research Grant No. 1046711 (M.R.Z.).
AUTHOR CONTRIBUTIONS
Conception and design: Aaron Beasley, Muhammad A. Khattak, Fred K. Chen, Michael Millward, Melanie R. Ziman, Elin S. Gray
Financial support: Melanie R. Ziman, Elin S. Gray
Administrative support: Fred K. Chen, Melanie R. Ziman
Provision of study material or patients: Muhammad A. Khattak, Fred K. Chen, Jaqueline Bentel, Tersia Vermulen, Michael Millward, Elin S. Gray
Collection and assembly of data: Aaron Beasley, Timothy Isaacs, Muhammad A. Khattak, James B. Freeman, Richard Allcock, Fred K. Chen, Michelle R. Pereira, Kyle Yau, Tersia Vermulen, Leslie Calapre, Michael Millward, Elin S. Gray
Data analysis and interpretation: Aaron Beasley, Muhammad A. Khattak, Richard Allcock, Fred K. Chen, Jaqueline Bentel, Tersia Vermulen, Leslie Calapre, Michael Millward, Elin S. Gray
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Aaron Beasley
No relationship to disclose
Timothy Isaacs
Travel, Accommodations, Expenses: Pfizer
Muhammad A. Khattak
Honoraria: MSD Oncology, Novartis, Merck Serono
Consulting or Advisory Role: Bristol-Myers Squibb, Merck Serono
Speakers' Bureau: Merck Serono, MSD Oncology, Novartis
Research Funding: MSD Oncology
Travel, Accommodations, Expenses: MSD Oncology, Amgen, Merck Serono
James B. Freeman
No relationship to disclose
Richard Allcock
No relationship to disclose
Fred K. Chen
Speakers' Bureau: Bayer HealthCare Pharmaceuticals
Research Funding: Novartis (Inst)
Travel, Accommodations, Expenses: Bayer HealthCare Pharmaceuticals, Allergan
Michelle R. Pereira
No relationship to disclose
Kyle Yau
No relationship to disclose
Jacqueline Bentel
No relationship to disclose
Tersia Vermeulen
No relationship to disclose
Leslie Calapre
No relationship to disclose
Michael Millward
Consulting or Advisory Role: Roche, Bristol-Myers Squibb, AstraZeneca, Merck Sharp & Dohme, Novartis, Boehringer Ingelheim
Travel, Accommodations, Expenses: Roche, Merck Sharp & Dohme, Bristol-Myers Squibb, AstraZeneca
Melanie R. Ziman
Research Funding: Merck Sharp & Dohme
Elin S. Gray
Research Funding: Merck Sharp & Dohme
Patents, Royalties, Other Intellectual Property: Provisional patent on a blood test to detect melanoma based on auto-antibody detection
Travel, Accommodations, Expenses: Bio-Rad Laboratories
REFERENCES
- 1.McLaughlin CC, Wu XC, Jemal A, et al. : Incidence of noncutaneous melanomas in the US. Cancer 103:1000-1007, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Blum ES, Yang J, Komatsubara KM, et al. : Clinical management of uveal and conjunctival melanoma. Oncology (Williston Park) 30:29-32, 34-43, 48, 2016 [PubMed] [Google Scholar]
- 3.Damato B, Dopierala JA, Coupland SE: Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res 16:6083-6092, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Robertson AG, Shih J, Yau C, et al. : Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 32:204-220.e15, 2017 [Erratum: Cancer Cell 33:151, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Decatur CL, Ong E, Garg N, et al. : Driver mutations in uveal melanoma: Associations with gene expression profile and patient outcomes. JAMA Ophthalmol 134:728-733, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Field MG, Durante MA, Anbunathan H, et al. : Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat Commun 9:116, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harbour JW, Roberson ED, Anbunathan H, et al. : Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45:133-135, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Onken MD, Worley LA, Ehlers JP, et al. : Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res 64:7205-7209, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heitzer E, Auer M, Gasch C, et al. : Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 73:2965-2975, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Gasch C, Bauernhofer T, Pichler M, et al. : Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem 59:252-260, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Maheswaran S, Sequist LV, Nagrath S, et al. : Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 359:366-377, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter L, Rothwell DG, Mesquita B, et al. : Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med 23:114-119, 2017 [DOI] [PubMed] [Google Scholar]
- 13.Dago AE, Stepansky A, Carlsson A, et al. : Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS One 9:e101777, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidary M, Auer M, Ulz P, et al. : The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res 16:421, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulmer A, Schmidt-Kittler O, Fischer J, et al. : Immunomagnetic enrichment, genomic characterization, and prognostic impact of circulating melanoma cells. Clin Cancer Res 10:531-537, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Tura A, Merz H, Reinsberg M, et al. : Analysis of monosomy-3 in immunomagnetically isolated circulating melanoma cells in uveal melanoma patients. Pigment Cell Melanoma Res 29:583-589, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Bettegowda C, Sausen M, Leary RJ, et al. : Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6:224ra24, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bidard FC, Madic J, Mariani P, et al. : Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int J Cancer 134:1207-1213, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Johansson P, Aoude LG, Wadt K, et al. : Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 7:4624-4631, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore AR, Ceraudo E, Sher JJ, et al. : Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet 48:675-680, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Raamsdonk CD, Bezrookove V, Green G, et al. : Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457:599-602, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Raamsdonk CD, Griewank KG, Crosby MB, et al. : Mutations in GNA11 in uveal melanoma. N Engl J Med 363:2191-2199, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman JB, Gray ES, Millward M, et al. : Evaluation of a multi-marker immunomagnetic enrichment assay for the quantification of circulating melanoma cells. J Transl Med 10:192, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richtig E, Langmann G, Müllner K, et al. : Calculated tumour volume as a prognostic parameter for survival in choroidal melanomas. Eye (Lond) 18:619-623, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Damato B, Dopierala J, Klaasen A, et al. : Multiplex ligation-dependent probe amplification of uveal melanoma: Correlation with metastatic death. Invest Ophthalmol Vis Sci 50:3048-3055, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Kujala E, Mäkitie T, Kivelä T: Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci 44:4651-4659, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Suesskind D, Ulmer A, Schiebel U, et al. : Circulating melanoma cells in peripheral blood of patients with uveal melanoma before and after different therapies and association with prognostic parameters: A pilot study. Acta Ophthalmol 89:17-24, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Tura A, Lüke J, Merz H, et al. : Identification of circulating melanoma cells in uveal melanoma patients by dual-marker immunoenrichment. Invest Ophthalmol Vis Sci 55:4395-4404, 2014 [DOI] [PubMed] [Google Scholar]
- 29.Mazzini C, Pinzani P, Salvianti F, et al. : Circulating tumor cells detection and counting in uveal melanomas by a filtration-based method. Cancers (Basel) 6:323-332, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heitzer E, Auer M, Hoffmann EM, et al. : Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int J Cancer 133:346-356, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]