

INTRODUCTION
Pancreatic cancer is the seventh leading cause of cancer death among men and women worldwide, with > 430,000 deaths in 2018.1 Despite advances in surgical techniques, radiation, and systemic treatment in the past few decades, overall survival for pancreatic cancer is extremely poor, with a 5-year survival rate of 9%.2 Current systemic treatment regimens for metastatic pancreatic cancer include FOLFIRINOX (fluorouracil, folic acid, oxaliplatin, and irinotecan), gemcitabine plus nab-paclitaxel, and liposomal irinotecan with fluorouracil.3-6
Approximately 88%-95% of pancreatic adenocarcinomas harbor KRAS driver mutations. However, there have been no US Food and Drug Administration (FDA)–approved targeted therapies for KRAS.7-9 In KRAS wild-type tumors, alternate oncogenic drivers have been identified, including BRAF, ROS, NRG1, GNAS, CTNNB1, and ALK gene fusions.10,11 ALK gene fusions were first described in pancreatic adenocarcinoma in 2017.12,13 Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor family, and ALK fusion genes have been characterized in multiple solid tumors, including thyroid, breast, and colorectal cancers and non–small-cell lung cancer (NSCLC).14,15 ALK translocations are seen in 3%-7% of NSCLC, causing constitutive activation of ALK and mediating oncogenesis through various signal transduction pathways, including the MAPK pathway.16 ALK inhibitors include crizotinib, the first-in-class FDA-approved targeted therapy with a greater response rate (65% v 20%) and longer progression-free survival (PFS; 7.7 months v 3 months) than standard chemotherapy in a phase III trial of previously treated ALK-positive NSCLC.17,18 In a phase III trial in treatment-naïve patients with NSCLC, crizotinib-associated PFS was notably greater than PFS with chemotherapy (10.9 months v 7.0 months).19 In the phase III ALEX trial, second-generation the ALK inhibitor alectinib greatly increased PFS compared with crizotinib in treatment-naïve patients with NSCLC—with a response rate of 82.9%—and generally is used as first-line therapy in ALK-positive NSCLC.20 However, patients can acquire resistance to ALK inhibitors through development of ALK resistance mutations.21 ALK G1202R, V1180L, and I1171 T/N/S are known alectinib-resistant mutations that were seen in 53% of patients who acquired resistance to alectinib.22,23 In a phase II trial, lorlatinib, a third-generation ALK inhibitor, was active in both treatment-naïve and previously ALK inhibitor–treated patients with ALK-positive NSCLC (objective responses of 90% and 47%, respectively).24 Additionally, lorlatinib can overcome ALK G1202R and V1180L resistance mutations that are seen in acquired resistance to alectinib.25,26
To date, only 6 occurrences of ALK-positive pancreatic cancer have been identified. In a study of > 3,100 patients with pancreatic cancer, only 5 patients had ALK translocation, none of whom had KRAS mutations.12 We present a case of a young woman with a novel ALK fusion partner, PPFIBP1-ALK, in metastatic pancreatic ductal adenocarcinoma who initially experienced a response to alectinib, then experienced progression with acquired alectinib-resistant mutations, which were stabilized with lorlatinib.
CASE REPORT
A 41-year-old woman with no past medical history presented with abdominal pain and bloating for 3 weeks. She had no family history of cancer and denied smoking or alcohol use. AST, ALT, and alkaline phosphatase levels were elevated at 139, 88, 257 u/L, respectively. γ-glutamyl transferase and lactate dehydrogenase levels were elevated at 317 and 1,886 u/L, respectively; however, the CA 19-9 measure was normal at 3.2 u/L. The initial ultrasound showed multiple liver lesions, and follow-up positron emission tomography/computed tomography (CT) was remarkable for a 2.1 × 2.7 cm lesion in the uncinate process of the pancreas and several lesions in the liver, the largest lesion being 7.9 cm.
Biopsies of the pancreas and liver lesions were consistent with pancreatic ductal adenocarcinoma. Molecular profiling of the tumor was remarkable for PPFIBP1-ALK translocation, confirmed by immunohistochemistry (D5F3 companion diagnostic assay; Ventana, Roche Diagnostics, Indianapolis, IN). Fluorescence in situ hybridization (FISH) was negative. It has been shown that FISH may have different sensitivities of detecting ALK compared with next-generation sequencing or immunohistochemistry.27 Of note, molecular profiling was negative for KRAS, microsatellite instability, NF1 and p53 inactivating mutations, and CDKN2A/B loss. Profiling was positive for BRCA2 c.8007A>G (silent) and CDKN1B 407A>G, both of unknown clinical significance. Patient was started on FOLFIRINOX with radiographic response. However, because of the adverse effects of chemotherapy, including nausea and neuropathy, the patient was switched to alectinib 600 mg twice daily, given her ALK fusion status. The 2-month follow-up CT imaging showed continuing response (Fig 1). However, she experienced progression on alectinib after 5 months and was placed back on FOLFIRINOX. Subsequent cell-free plasma (Guardant360; Guardant Health, Redwood City, CA) showed newly acquired ALK mutations G1202R and V1180L in addition to the PPFIBP1-ALK translocation. The patient experienced progression quickly on FOLFIRINOX and was transitioned to lorlatinib. The patient’s disease has been stable on lorlatinib on 2-month follow-up imaging, and she continues to be treated with lorlatinib (Figs 2 and 3).
FIG 1.

Computed tomography imaging of hepatic metastasis before and after alectinib. In row 1, the circled hepatic lesion in the right lobe of the liver at baseline (A) before alectinib decreased in size (B) 2 months after alectinib. Similarly, row 2 shows a different metastatic hepatic lesion (C) before alectinib with continued response seen (D) after 2 months of alectinib. The patient experienced progression during alectinib treatment after 5 months and experienced progression during fluorouracil, folic acid, oxaliplatin, and irinotecan treatment shortly thereafter.
FIG 2.
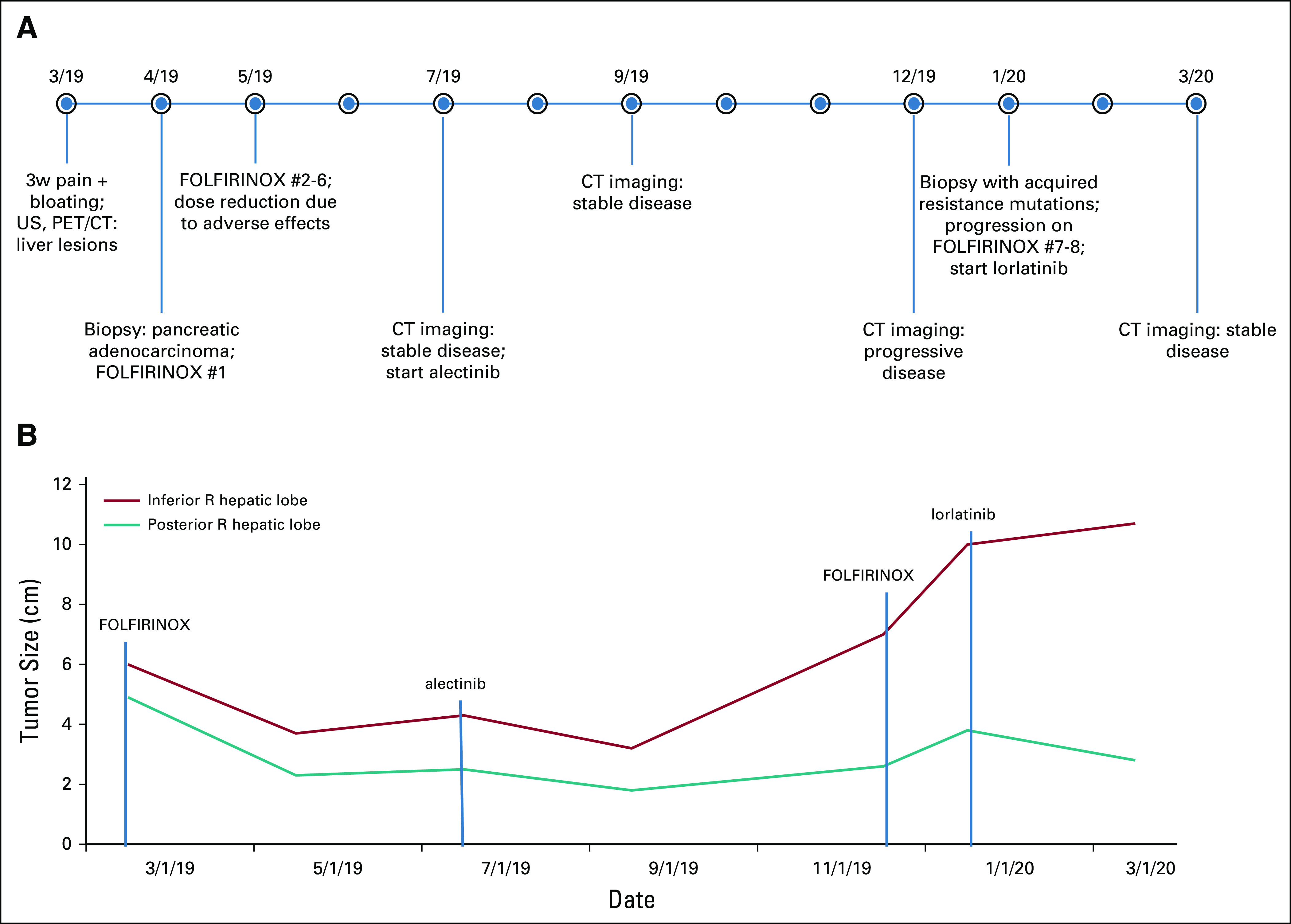
(A) Chronologic timeline of events including dates of initiation of anaplastic lymphoma kinase (ALK) inhibitor and radiographic imaging. (B) Graphic description of change in tumor size over time with respect to type of therapy. #, dose number; 3w, 3-week; CT, computed tomography; FOLFIRINOX, fluorouracil, folic acid, oxaliplatin, and irinotecan; PET, positron emission tomography; R, right; US, ultrasound.
FIG 3.

Computed tomography imaging of hepatic metastasis before and after lorlatinib. In row 1, the circled hepatic lesion after disease progression on fluorouracil, folic acid, oxaliplatin, and irinotecan (A) before lorlatinib was stable in size (B) 2 months after lorlatinib treatment. Similarly, row 2 shows a different metastatic hepatic lesion (C) before lorlatinib with stable disease (D) after 2 months of lorlatinib. The patient currently continues to be treated with lorlatinib.
DISCUSSION
Currently, there are no FDA-approved targeted therapies for pancreatic cancer, and standard chemotherapy (FOLFIRINOX) for metastatic pancreatic cancer is quite toxic, with a median overall survival of approximately 11 months.3 To our knowledge, we present the seventh case of ALK-positive metastatic pancreatic cancer with a novel PPFIBP1-ALK fusion gene and the first patient to be treated with alectinib as first-line targeted therapy. The patient ultimately acquired alectinib-resistant ALK mutations G1202R and V1180L; however, the disease has been stable on the third-generation ALK inhibitor lorlatinib.
ALK translocation in pancreatic cancer was first described in 2017, and there have been only 6 documented cases of ALK-positive pancreatic adenocarcinoma through literature review (Table 1). Although the prevalence of the ALK fusion gene is rare, at 0.16%, the prevalence increases to 1.3%.among patients < 50 years old12 The majority of ALK fusion partners seen in NSCLC includes EML4, but other partners include STRN, KCNQ, KLC1, KIF5B, PPM1B, and TGF genes.28 PPFIBP1-ALK gene fusion was first described in 2011 in a patient with pulmonary inflammatory myofibroblastic tumor29 and subsequently was described in epithelioid fibrous histiocytoma,30 but it has never been described in NSCLC or pancreatic cancer. Of the 6 patients with ALK-positive pancreatic cancer, 4 patients had EML4-ALK translocation, 1 patient had STRN-ALK, and 1 patient had a DCTN1-ALK translocation (Table 1). This patient demonstrates a novel PPFIBP1-ALK fusion gene, of which the tumorigenicity of PPFIBP1-ALK was elucidated in pulmonary inflammatory myofibroblastic tumor.29 PPFIBP1 is involved in cell adhesion and migration, and the different ALK fusion partners may be responsible for various invasive and proliferative capabilities, ultimately leading to activation of different signaling pathways.28
TABLE 1.
Clinical Characteristics of Patients With ALK-Positive Pancreatic Cancer

In NSCLC, ALK fusion is an oncogenic driver that is generally mutually exclusive of KRAS mutation16; however, there are reports of concomitant KRAS and ALK fusion double alteration that may confer worse prognosis.31 All 7 patients with ALK-positive pancreatic cancer were KRAS wild type, suggesting, albeit in a small sample size, that ALK translocation drives oncogenesis and that these 2 oncogenic drivers are mutually exclusive. Overall survival ranged from 5-52 months with crizotinib treatment in ALK-positive pancreatic cancer (Table 1). This case demonstrates the first time, to our knowledge, that alectinib has been used as the first targeted therapy, and the development of known alectinib-resistant ALK mutations suggests that ALK-positive pancreatic cancer acquires resistance in a fashion similar to that of NSCLC.
There is a clear unmet medical need for novel therapeutic approaches in pancreatic cancer. A recent study in metastatic pancreatic cancer with germ-line BRCA mutations (7% prevalence) regardless of KRAS mutation status showed increased PFS with the poly ADP ribose polymerase inhibitor olaparib after first-line platinum-based chemotherapy.4 In a recent phase II basket study, 2 of 9 patients who had pancreatic cancer with HER2 amplification/overexpression experienced responses to HER2-targeted therapies trastuzumab plus pertuzumab. Additionally, a patient who had pancreatic cancer and BRAF gene fusion (CUX1-BRAF) had a partial response to the BRAF-targeted therapy vemurafenib.32 Last, 3 patients who had pancreatic cancer with NTRK1 and ROS1 gene fusions experienced responses to entrectinib, a targeted inhibitor of these genes.33 These studies are additional proof of concept that targeted therapies can be used successfully when paired with the right genomic alteration and that ALK inhibitors may be of substantial clinical benefit in the right patient. In patients with resected pancreatic cancer, KRAS mutation is associated with worse overall survival than KRAS wild-type tumors.34 Additionally, prevalence of KRAS mutation is notably less (80% v 89%) in patients < 50 years old, and 38% of KRAS wild-type tumors have genomic alterations that could activate the MAPK signaling cascade.9 Identification of molecular alterations in patients with KRAS wild-type status that may drive oncogenesis will help guide therapeutic intervention in a small but clinically relevant population.
ALK translocations are rare in pancreatic cancer. This is, to our knowledge, the first report of PPFIBP1-ALK rearrangement in pancreatic or NSCLC, the first reported patient with pancreatic cancer to be treated with alectinib as the first targeted therapy, and the first patient to be treated with lorlatinib after acquired resistance to alectinib with stabilization of disease. The molecular drivers of this tumor behave similarly to ALK-positive NSCLC. All 7 occurrences of ALK-positive pancreatic cancer have been KRAS wild type; given that 6 of the 7 patients were < age 50 years, there may be clinical benefit to screen young patients with pancreatic cancer who are KRAS negative for the ALK gene fusion, as they may benefit from ALK inhibitors, which may lead to improved overall survival. Last, noninvasive sampling of cell-free DNA can be used for monitoring resistance to targeted therapies in ALK-positive pancreatic cancer.
SUPPORT
Supported by the Samuel Oschin Cancer Center at Cedars Sinai Medical Center.
AUTHOR CONTRIBUTIONS
Conception and design: Arjan Gower, Andrew Eugene Hendifar
Provision of study material or patients: Aatur D. Singhi, Andrew Eugene Hendifar
Collection and assembly of data: Arjan Gower, Barry Golestany, Andrew Eugene Hendifar
Data analysis and interpretation: Arjan Gower, Jun Gong, Aatur D. Singhi, Andrew Eugene Hendifar
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Jun Gong
Honoraria: Amgen, Astellas Pharma, Clinical Congress Consultants, QED Therapeutics, Exelixis, Elsevier
Consulting or Advisory Role: Amgen, Astellas Pharma, Clinical Congress Consultants, QED Therapeutics, Exelixis, Elsevier
Aatur D. Singhi
Honoraria: Foundation Medicine
Andrew Eugene Hendifar
Consulting or Advisory Role: Novartis, Ipsen, Perthera, Celgene, AbbVie
Research Funding: Ipsen
Travel, Accommodations, Expenses: Halozyme
No other potential conflicts of interest were reported.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang-Gillam A, Li CP, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet. 2016;387:545–557. doi: 10.1016/S0140-6736(15)00986-1. [DOI] [PubMed] [Google Scholar]
- 7.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singhi AD, George B, Greenbowe JR, et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology. 2019;156:2242–2253.e4. doi: 10.1053/j.gastro.2019.02.037. [DOI] [PubMed] [Google Scholar]
- 10.Heining C, Horak P, Uhrig S, et al. NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov. 2018;8:1087–1095. doi: 10.1158/2159-8290.CD-18-0036. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singhi AD, Ali SM, Lacy J, et al. Identification of targetable ALK rearrangements in pancreatic ductal adenocarcinoma. J Natl Compr Canc Netw. 2017;15:555–562. doi: 10.6004/jnccn.2017.0058. [DOI] [PubMed] [Google Scholar]
- 13.Shimada Y, Kohno T, Ueno H, et al. An oncogenic ALK fusion and an RRAS mutation in KRAS mutation-negative pancreatic ductal adenocarcinoma. Oncologist. 2017;22:158–164. doi: 10.1634/theoncologist.2016-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin E, Li L, Guan Y, et al. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non–small-cell lung cancers. Mol Cancer Res. 2009;7:1466–1476. doi: 10.1158/1541-7786.MCR-08-0522. [DOI] [PubMed] [Google Scholar]
- 15.Kelly LM, Barila G, Liu P, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci USA. 2014;111:4233–4238. doi: 10.1073/pnas.1321937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gower A, Wang Y, Giaccone G. Oncogenic drivers, targeted therapies, and acquired resistance in non–small-cell lung cancer. J Mol Med (Berl) 2014;92:697–707. doi: 10.1007/s00109-014-1165-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 19.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 20.Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N Engl J Med. 2017;377:829–838. doi: 10.1056/NEJMoa1704795. [DOI] [PubMed] [Google Scholar]
- 21.Camidge DR, Doebele RC. Treating ALK-positive lung cancer: Early successes and future challenges. Nat Rev Clin Oncol. 2012;9:268–277. doi: 10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noé J, Lovejoy A, Ignatius Ou S-H, et al. ALK mutation status before and after alectinib treatment in locally advanced or metastatic ALK-positive NSCLC: Pooled analysis of two prospective trials. J Thorac Oncol. 2019;15:601–608. doi: 10.1016/j.jtho.2019.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20:5686–5696. doi: 10.1158/1078-0432.CCR-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solomon BJ, Besse B, Bauer TM, et al. Lorlatinib in patients with ALK-positive non–small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018;19:1654–1667. doi: 10.1016/S1470-2045(18)30649-1. [DOI] [PubMed] [Google Scholar]
- 25.Dagogo-Jack I, et al. Tracking the evolution of resistance to ALK tyrosine kinase inhibitors through longitudinal analysis of circulating tumor DNA. JCO Precis Oncol. doi: 10.1200/PO.17.00160 [epub on January 23, 2018] [DOI] [PMC free article] [PubMed]
- 26.Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28:70–81. doi: 10.1016/j.ccell.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin C, Shi X, Yang S, et al. Comparison of ALK detection by FISH, IHC, and NGS to predict benefit from crizotinib in advanced non–small-cell lung cancer. Lung Cancer. 2019;131:62–68. doi: 10.1016/j.lungcan.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Della Corte CM, Viscardi G, Di Liello R, et al. Role and targeting of anaplastic lymphoma kinase in cancer. Mol Cancer. 2018;17:30. doi: 10.1186/s12943-018-0776-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeuchi K, Soda M, Togashi Y, et al. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: Reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res. 2011;17:3341–3348. doi: 10.1158/1078-0432.CCR-11-0063. [DOI] [PubMed] [Google Scholar]
- 30.Dickson BC, Swanson D, Charames GS, et al. Epithelioid fibrous histiocytoma: Molecular characterization of ALK fusion partners in 23 cases. Mod Pathol. 2018;31:753–762. doi: 10.1038/modpathol.2017.191. [DOI] [PubMed] [Google Scholar]
- 31.Ulivi P, Chiadini E, Dazzi C, et al. Nonsquamous, non–small-cell lung cancer patients who carry a double mutation of EGFR, EML4-ALK or KRAS: Frequency, clinical-pathological characteristics, and response to therapy. Clin Lung Cancer. 2016;17:384–390. doi: 10.1016/j.cllc.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: Results from MyPathway, an open-label, Phase IIa multiple basket study. J Clin Oncol. 2018;36:536–542. doi: 10.1200/JCO.2017.75.3780. [DOI] [PubMed] [Google Scholar]
- 33.Pishvaian MJ, Garrido-Laguna I, Liu SV, et al. Entrectinib in TRK and ROS1 fusion-positive metastatic pancreatic cancer. JCO Precis Oncol. doi: [epub on July 25, 2018]. [DOI] [PubMed]
- 34.Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4:e173420. doi: 10.1001/jamaoncol.2017.3420. [DOI] [PMC free article] [PubMed] [Google Scholar]