

Abstract
PURPOSE
Atypical, non-V600 BRAF (aBRAF) mutations represent a rare molecular subtype of metastatic colorectal cancer (mCRC). Preclinical data are used to categorize aBRAF mutations into class II (intermediate to high levels of kinase activity, RAS independent) and III (low kinase activity level, RAS dependent). The clinical impact of these mutations on anti-EGFR treatment efficacy is unknown.
PATIENTS AND METHODS
Data from 2,084 patients with mCRC at a single institution and from an external cohort of 5,257 circulating tumor DNA (ctDNA) samples were retrospectively analyzed. Overall survival (OS) was calculated using Kaplan-Meier and log-rank tests. Statistical tests were two-sided.
RESULTS
BRAF mutations were harbored by 257 patients, including 36 with aBRAF mutations: 22 class III, 10 class II, four unclassified. For patients with aBRAF mCRC, median OS was 36.1 months, without a difference between classes, and median OS was 21.0 months for patients with BRAFV600E mCRC. In contrast to right-sided predominance of tumors with BRAFV600E mutation, 53% of patients with aBRAF mCRC had left-sided primary tumors. Concurrent RAS mutations were noted in 33% of patients with aBRAF mCRC, and 67% of patients had microsatellite stable disease. Among patients with aBRAF RAS wild-type mCRC who received anti-EGFR antibodies (monotherapy, n = 1; combination therapy, n = 10), no responses to anti-EGFR therapy were reported, and six patients (four with class III aBRAF mutations, one with class II, and one unclassified) achieved stable disease as best response. Median time receiving therapy was 4 months (range, 1 to 16). In the ctDNA cohort, there was an increased prevalence of aBRAF mutations and subclonal aBRAF mutations (P < .001 for both) among predicted anti-EGFR exposed compared with nonexposed patients.
CONCLUSION
Efficacy of anti-EGFR therapy is limited in class II and III aBRAF mCRC. Detection of aBRAF mutations in ctDNA after EGFR inhibition may represent a novel mechanism of resistance.
INTRODUCTION
BRAFV600E missense mutations are present in 6% to 10% of patients with metastatic colorectal cancer (mCRC).1,2 Within the BRAF kinase domain, substitution of a valine to glutamic acid at position 600 manifests as constitutive activation and oncogenic signaling along the mitogen-activated protein kinase (MAPK) pathway.1 BRAFV600E mCRC represents an aggressive molecular subtype of colorectal cancer inherently refractory to standard chemotherapy; thus, tremendous research focus has been directed toward novel therapeutic development.2 Because of increased use of next-generation sequencing (NGS) in the management of mCRC, various mutational hotspots of clinical significance have emerged within genes of interest, such as expanded RAS testing. However, with such broad testing including circulating tumor DNA (ctDNA), alterations of evolving significance without clear predictive, prognostic, or therapeutic implications have also been identified. Atypical, non-V600 BRAF (aBRAF) mutations that occur outside of codon 600 are one example of an emerging molecular subtype in colorectal cancer.
aBRAF mutations were retrospectively studied at two large centers comprehensively describing the clinical, pathologic, and survival implications of these mutations in patients with mCRC.3 A total of 9,643 patients with mCRC underwent NGS testing and 208 patients with aBRAF mutations were identified, representing 2.2% of all patients tested. Interestingly, these patients had distinct clinical features from those with traditional BRAFV600E mCRC, with median overall survival (mOS) significantly longer (60.7 months) than that of patients with BRAFV600E (11.4 months) mCRC or wild-type BRAF mCRC (43.0 months).3 Although this represents an excellent prognosis comparatively, these patients still ultimately succumb to the disease. In addition, the chronicity of their disease suggests a need for improved, novel, targeted therapeutics that can be used sequentially throughout the course of the disease.
Context
Key Objective
To determine if atypical, non-V600 BRAF (aBRAF) mutations, stratified by class, affect outcomes with anti-EGFR therapy and, secondarily, if acquisition of aBRAF mutations imparts resistance to EGFR inhibition.
Knowledge Generated
Clinical outcomes on the basis of functional class and their effect on anti-EGFR efficacy for aBRAF metastatic colorectal cancer (mCRC) have not been defined. In patients with aBRAF wild-type RAS mCRC who received anti-EGFR antibodies, there were no responses among class II or III aBRAF mCRC, with stable disease as best response in our internal cohort. Evaluation of a large external cohort of patients with circulating tumor DNA interrogated by an anti-EGFR exposure score revealed increased prevalence of aBRAF and subclonal aBRAF mutations among predicted anti-EGFR exposed compared with nonexposed patients, suggesting an acquired mechanism of resistance to EGFR inhibition.
Relevance
Anti-EGFR therapy is limited in aBRAF mCRC, with class II aBRAF mutations emerging as a negative predictive biomarker. Detection of aBRAF mutations in ctDNA may represent a novel mechanism of resistance warranting additional investigation.
Although BRAFV600E mCRC is known to be predictive of poor response to anti-EGFR therapy, the clinical utility of EGFR inhibition in aBRAF mCRC remains unclear.4,5 Of note, previous retrospective work investigating a cohort of 150 patients with refractory mCRC identified seven patients with aBRAF mutations and reported poor progression-free survival when they were exposed to anti-EGFR therapy in comparison with a cohort with RAS/RAF wild-type (RAS/RAFWT) mCRC.6 Preclinical work has highlighted unique biology among traditional BRAFV600E mutations (class I) and aBRAF mutations (classes II and III).7,8 BRAFV600E mutations, designated as class I, result in feedback inhibition of GTP-bound RAS, are RAS independent, and signal as active monomers. In contrast, class II aBRAF mutations have intermediate to high levels of kinase activity, are RAS independent, signal as constitutive dimers, and are resistant to vemurafenib, whereas class III aBRAF mutations have low or absent kinase activity and are primarily RAS dependent and sensitive to ERK-dependent feedback of RAS.7,8 These tumors bind more tightly to RAS-GTP than do wild-type BRAF tumors, and binding to wild-type CRAF is enhanced, resulting in increased ERK signaling.8 These stark differences in underlying tumor biology may explain the varying response to therapy, specifically the potential limitations to anti-EGFR inhibition in aBRAF mCRC. To date, clinical outcomes on the basis of functional class designation and the potential effect on anti-EGFR efficacy for aBRAF mCRC have not been well defined.
In this study, we aimed to describe the prevalence of aBRAF mutations in a large data set of patients who underwent NGS and disease treatment at a single institution; highlight the landscape of aBRAF by reviewing the clinical, pathologic, and molecular characteristics of tumors with these mutations; and analyze the survival outcomes of patients with aBRAF mCRC exposed to anti-EGFR therapy to define the efficacy of EGFR inhibition in this rare subset and define the frequency of aBRAF mutations in patients who underwent ctDNA testing to elucidate the potential role of the mutations in EGFR inhibition resistance.
PATIENTS AND METHODS
This study used an internal cohort created retrospectively with institutional review board approval at the University of Texas MD Anderson Cancer Center (MDACC), and a waiver of written consent was obtained. All databases used were deidentified, and the study adhered to MDACC institutional review board guidelines for deidentified databases. Specific clinico-pathologic, mutational, molecular, and outcomes data for patients with aBRAF mCRC were reviewed and collected from this cohort. Cohort 1 included patients with stage IV colorectal cancer who were seen at MDACC between March 1, 2012, and June 27, 2017, and underwent targeted exome sequencing. Biopsy samples for sequencing were obtained from primary tumor (n = 18 patients) and metastatic sites (n = 18 patients). In regard to timing of the samples, 26 patients underwent molecular profiling at initial stage IV diagnosis (before therapy) and 10 patients were profiled subsequently. All clinical and outcomes data were derived from this cohort.
Cohort 2 included patients with mCRC who underwent a targeted NGS ctDNA assay (Guardant360; Guardant Health, Redwood City, CA) as part of their care between June 1, 2014, and December 26, 2017, at multiple institutions. Details of the assay, including detection limit of sensitivity and coverage (covers all exons of BRAF), have been published.9,10 Treatment histories were not known for this cohort; therefore, we used a previously validated and highly specific score to define EGFR-exposed patients.11 We calculated the prevalence and relative mutational allele frequencies for both cohorts of aBRAF mutations: predicted anti-EGFR exposed and nonexposed.
Statistical Analysis
Clinicopathologic features were compared by χ2 test or Fisher’s exact test for categorical variables and assessment of odds ratios for associations as appropriate, as well as by t test to compare continuous variables. Overall survival (OS) was calculated using Kaplan-Meier method and log-rank tests. OS was defined as the time from diagnosis with stage IV disease until death. Matched analysis adjusted by age and sidedness was undertaken to compare OS among all molecular classes. χ2 or Fisher’s exact tests, as well as t test, were used to evaluate the adequacy of the matching. Statistical analyses were performed using GraphPad Prism, version 6.07 (GraphPad Software, La Jolla, CA), and SPSS, version 23.0 (IBM, Armonk, NY). All statistical tests were two sided, and P < .05 was considered statistically significant.
RESULTS
In cohort 1 (n = 2,084), aBRAF mutations occurred in 36 patients with mCRC (prevalence, 1.7%; 95% CI, 1.2% to 2.4%) compared with 221 patients with V600E mutations (prevalence 10.6%; 95% CI, 9.3% to 12.0%; Fig 1; Table 1). In terms of functional class designation, there were 22 patients with class III aBRAF mCRC, 10 with class II, and four unclassified patients. The most common class II and class III mutated BRAF codons were 469 (60%) and 594 (59%), respectively (Appendix Fig A1). mOS for aBRAF mCRC was 36.1 months (95% CI, 19.9 to 52.3), without difference between class III and II BRAF mCRC (Fig 2). Patients with BRAFV600E mCRC had an mOS of 21.0 months (95% CI, 18.6 to 22.9) and patients with RAS/RAFWT mCRC (n = 438) had a mOS of 42.3 months (95% CI, 36.9 to 47.8; Fig 2). Statistical significance was noted when aBRAF mCRC was compared with BRAFV600E mCRC (P < .001) and when RAS/RAFWT mCRC was compared with BRAFV600E mCRC (P < .001). Among 36 patients with aBRAF mCRC, 19 (53%) had left-sided primary tumors and 24 (67%) had microsatellite stable (MSS) disease and were a median age of 56 years (Table 1). Of note, in contrast to BRAFV600E mCRC, which is mutually exclusive with RAS mutations, 12 patients with aBRAF mCRC had RAS mutations (class III, n = 7 of 21 [33%]; class II, n = 5 of 10 [50%]). Among these, one patient with class II aBRAF mCRC also had microsatellite instability–high (MSI-H) disease.
FIG 1.

Study CONSORT diagram. aBRAF, atypical, non-V600 BRAF; ctDNA, circulating tumor DNA; mCRC, metastatic colorectal cancer; MDACC, MD Anderson Cancer Center; MT, mutant type.
TABLE 1.
Baseline Characteristics of Cohort 1

FIG 2.

Overall survival according to BRAF mutation status. aBRAF, atypical, non-V600 BRAF.
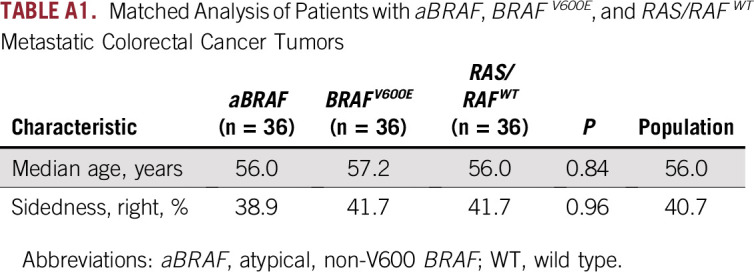
For a more accurate comparison of survival outcomes among respective molecular classes, we performed a matched analysis by age and sidedness among the 36 patients with aBRAF mCRC compared with 36 patients with BRAFV600E mCRC and 36 with RAS/RAFWT disease (Fig 3; Appendix Table A1). OS was 36.1 months for patients with aBRAF mCRC (95% CI, 19.9 to 52.3 months), 18.9 months for those with BRAFV600E mCRC (95% CI, 15.4 to 22.5 months), and 48.0 months for those with RAS/RAFWT mCRC (95% CI, 15.4 to 80.6 months). Statistical significance was noted when aBRAF mCRC was compared with BRAFV600E mCRC (P = .003) and when RAS/RAFWT mCRC was compared with BRAFV600E mCRC (P < .001).
FIG 3.

Overall survival with matched analysis. aBRAF, atypical, non-V600 BRAF.
Clinical Efficacy of Anti-EGFR Monoclonal Antibodies in Patients With aBRAF mCRC
To describe the clinical efficacy of EGFR inhibition in aBRAF mCRC, we assessed survival outcomes for patients exposed to anti-EGFR therapy in their treatment course. Among patients with aBRAF RASWT mCRC, 11 (50%) received anti-EGFR monoclonal antibodies (mAbs; monotherapy, n = 1, combination therapy, n = 10; class III, n = 7 of 14 [50%], class II, n = 3 of 5 [60%], unclassified, n = 1). There were no responses reported, and six patients (class III, n = 4; class II, n = 1; unclassified, n = 1) achieved stable disease as best response. Median time receiving anti-EGFR therapy was 4 months (range, 1 to 16). Patients with class II RASWT mCRC treated with anti-EGFR mAbs had an mOS of 31.7 months versus 46.8 months for those not exposed (hazard ratio, 2.0; 95% CI, 0.3 to 15.9). Median time receiving therapy for patients with class II aBRAF mCRC was 3 months. Patients with class III RASWT mCRC treated with anti-EGFR mAbs had an mOS of 44.2 months versus 45.7 months for those not treated (hazard ratio, 0.80; 95% CI, 0.2 to 2.6). Median time receiving therapy for patients with class III aBRAF mCRC was 5 months. In line with previous reports, patients with class I or BRAFV600E mCRC had worse survival outcomes when exposed to EGFR inhibition (Fig 4).
FIG 4.

Overall survival among BRAF mCRC classes according to anti-EGFR exposure.
aBRAF Mutations as a Potential Mechanism of Resistance to Anti-EGFR Therapy
To understand our institutional findings of limited efficacy of anti-EGFR therapy in aBRAF mCRC, we analyzed an external ctDNA cohort of 5,257 samples (cohort 2) from 4,465 patients with mCRC. Of 277 aBRAF mutations identified, 24 were class II, 56 were class III, and 197 unclassified. Of note, 62% of aBRAF mutations were subclonal, compared with only 18% of the BRAFV600E mutations. Considering the lack of treatment history in this cohort, after excluding RAS and BRAFV600E mutations, we applied a previously validated anti-EGFR exposure score to divide patients into predicted prior exposure to anti-EGFR (n = 644) versus predicted nonexposed (n = 2,880).10 This revealed that the prevalence of aBRAF mutations increased from 3.99% among patients predicted to have no prior anti-EGFR exposure to 8.38% among those with predicted anti-EGFR exposure (P < .001; Fig 5A).
FIG 5.

Circulating tumor DNA (ctDNA) analysis of atypical, non-V600 BRAF (aBRAF) in cohort 2. (A) ctDNA prevalence of aBRAF (3.99%) among non-exposed patients (n = 2,880) v ctDNA prevalence of aBRAF (8.38%) among predicted anti-EGFR exposed (n = 644). (B) Relative mutational allele frequency (rMAF) of aBRAF among predicted anti-EGFR exposed v nonexposed patients, P < .001. (C) Frequency and location of mutations in the BRAF protein in mCRC patients predicted to be exposed to anti-EGFR antibodies. Schematic figure of the conserved region-3 (CR3) of the BRAF protein, also known as the BRAF kinase domain (amino acids 457 to 717), and the location of the class II and III hotspot mutations. The kinase domain contains the P-loop (amino acids 464-471), the αC helix (amino acids 492-504), the dimerization interface (DIF, amino acids 504-511), the catalytic loop (CL, amino acids 574-581), the DFG motif (amino acids 594-596), and the activation segment (AS, amino acids 594-623). class II, activating G469A mutations (P < .001) and class III, D594N (P < .001) were the most common, respectively, among predicted anti-EGFR exposed patients. Figure adapted.12 rMAF, relative mutational allele frequency.
As a sensitivity analysis, we evaluated the prevalence of aBRAF mutations among patients with EGFR ectodomain mutations, compared with those without, because EGFR ectodomain mutations are unequivocally present only after exposure to EGFR inhibition. We found that the prevalence of aBRAF mutations increased from 6.6% among patients without EGFR ectodomain mutations to 12.8% among those with EGFR ectodomain mutations (P = .039).
We then calculated the relative mutational allele frequencies for both cohorts of aBRAF mutations (anti-EGFR exposed and nonexposed) and found an increase in subclonal aBRAF mutations among patients with predicted anti-EGFR exposure (P < .001; Fig 5B). When stratified by class designation, both class II and class III aBRAF mutations were found to increase in prevalence among patients with predicted anti-EGFR exposure compared with patients with predicted nonexposure. Patients with class II aBRAF mCRC had a prevalence of 0.34% among patients predicted to not have anti-EGFR therapy exposure versus 1.24% among those with predicted anti-EGFR exposure (P = .009). The prevalence of patients with class III aBRAF mCRC increased from 0.94% among nonexposed patients to 2.48% among those predicted to have anti-EGFR exposure (P = .004). Class II activating G469A mutations (P < .001) and class III D594N (P < .001) were the most common alterations among patients with predicted anti-EGFR exposure (Fig 5C). Furthermore, we observed multiple aBRAF mutations present in ctDNA samples of individual patients, with the prevalence of multiple aBRAF mutations increasing from 0.5% among nonexposed patients to 3.5% among those with predicted anti-EGFR exposure (P < .001).
DISCUSSION
Patients with aBRAF mCRC have class-specific biochemical signaling mechanisms with a unique clinical phenotype.3,7,8 In fact, unlike BRAFV600E mCRC, patients with aBRAF mutations tend to be younger, male, and more often have left-sided colon tumors without MSI-H.3,12-15 These distinct differences highlight the importance of understanding tumor biology in aBRAF mCRC to provide a more personalized therapeutic approach moving forward.
Although activation of the MAPK pathway is critical for both BRAFV600E and aBRAF mCRC, preclinical work suggests these mutations differ in their signaling mechanisms, so much so that aBRAF tumors can be categorized into respective classes.8 For instance, aBRAF tumors can have impaired kinase activity but still activate the signaling pathway through dimerization, whereas BRAFV600E tumors activate as monomers. As a result, inhibiting BRAF is not as effective for aBRAF tumors. Agents such as vemurafenib are not active, because the drug is designed to target the shape of the V600E-mutated protein and is more than 10-fold less active against nonmutated forms. This fundamental difference highlights why traditional BRAF monomer inhibitors are ineffective in the management of aBRAF mCRC. Dimerization sets aBRAF classes II and III apart from traditional BRAFV600E (class I) mutations and is a critical distinction that must be appreciated to best target these alterations in the future.
Aligning with earlier reports, we found that aBRAF mCRC had an underwhelming response to anti-EGFR therapy.6,16-18 Specifically, class II RASWT aBRAF mCRC seems to have a BRAFV600E-like phenotype in its response to anti-EGFR exposure, which may be explained by the similarity of RAS-independent MAPK signaling between these subtypes. Our cohort highlights that those patients with class II aBRAF mCRC exposed to anti-EGFR therapy may have inferior outcomes than those who are nonexposed. This is suggestive of class II aBRAF mutations reflecting a potential negative predictive biomarker for EGFR inhibition. Although previous reports have suggested class III aBRAF mCRC may achieve responses with anti-EGFR therapy owing to RAS dependency, our cohort, albeit small, only revealed stable disease as best response.6,8,12-15,19 These results support the findings of the BREAC study, which revealed no responses in six patients with aBRAF mCRC (n = 2 each with class II, class III, and unclassified).6
Of note, the finding of concomitant RAS mutations with aBRAF mCRC in tissue, confirmed in our external ctDNA cohort, clearly precludes the use of EGFR inhibition for all patients, highlighting the need for novel combination strategies that consider this distinct signaling biology.20,21 In our internal and external cohorts, we did not find a difference between patients with class II and class III aBRAF mCRC in regard to the presence of concurrent RAS mutations. Therefore, continued investigation is warranted to understand the physiologic role of concomitant RAS mutations in patients with aBRAF mutations. Also, whether the actionability of aBRAF mutations in patients with MSI-H disease would be less than in those with MSS disease warrants additional investigation; conclusions could not be made due to the sample size of our internal cohort.
In our MDACC internal cohort, four patients had unclassified disease (Y656D, A415V, K752R, F109I) and had poor survival outcomes. These mutations have not yet been functionally annotated; therefore, understanding their role in tumorigenesis and survival outcomes is critical. Furthermore, co-mutation status in unclassified patients should be investigated because it is possible these may represent codriver events. In the external ctDNA cohort, we found numerous unclassified aBRAF mutations that are yet to be functionally described; most were commonly co-mutated with RAS, PIK3CA, and EGFR.
Development of acquired RAS and subclonal EGFR ectodomain mutations are known mechanisms of resistance to anti-EGFR therapy in RASWT mCRC.22-26 Considering the limitation of EGFR inhibition noted in our internal cohort, whether the emergence of aBRAF potentially reflects a novel mechanism of acquired resistance to EGFR inhibition was investigated. Analysis of our ctDNA cohort showed statistically significant increases in the prevalence of aBRAF mutations, subclonal aBRAF mutations, and multiple aBRAF mutations in individual ctDNA samples among patients predicted to be exposed to EGFR inhibition versus those predicted to be nonexposed. It is important to note that the rate of aBRAF mutation in our ctDNA cohort was higher than in our internal cohort, as well as higher than what has been published in tissue-based testing.3 These findings highlight the potential of aBRAF mutations as a novel acquired resistance mechanism to EGFR inhibition and warrant additional prospective investigation. Of note, in patients with EGFR-mutant non–small-cell lung cancer, aBRAF mutations have been reported as a mechanism of acquired resistance to EGFR tyrosine kinase inhibition.27
Our results underscore the need for innovative combinatorial targeted approaches for aBRAF mutations exploiting class biology. Data suggest that downstream signaling with novel inhibitors of MEK or ERK may be useful, because the pathway is still active.20 Furthermore, because both class II and class III mutations signal through BRAF and/or CRAF dimers, next-generation RAF inhibitors that target dimerization is of interest in combination with these agents as a rationale next step for therapeutic intervention.21 Of note, safety lead-in data for a novel triplet regimen using encorafenib, binimetinib, and cetuximab for refractory BRAFV600E mCRC have been published showing impressive response rates and significant improvement in survival outcomes.28 Subsequent studies will need to investigate whether this novel triplet combination strategy can also enhance optimal MAPK inhibition and provide durable benefit in patients with aBRAF mCRC.
There are limitations to this study. First, this is a nonrandomized, retrospective cohort analysis at a single institution. Second, considering the rarity of aBRAF mCRC, we were only able to identify 36 patients in our internal cohort, of whom 30% received EGFR inhibition therapy. Third, detailed clinical annotation is not available for the external ctDNA cohort, and we were unable to confirm that these patients had received prior anti-EGFR therapy. Therefore, we applied a previously validated and published anti-EGFR exposure score to identify patients predicted to have anti-EGFR exposure versus patients predicted to be nonexposed to conduct our ctDNA analysis.
Our results suggest selection criteria for the use of anti-EGFR therapy in aBRAF-mutated mCRC should be based not only on RASWT status but also the functional class of the atypical variant. In our cohort, although limited by sample size, findings continue to generate more hypotheses in that class II aBRAF mutations may represent a negative predictive biomarker of EGFR inhibition, and exposure to anti-EGFR mAbs may adversely affect survival outcomes, whereas efficacy of EGFR inhibition was limited in patients with class III aBRAF mCRC. Increased subclonal aBRAF mutations and prevalence of class II and class III variants in ctDNA among patients with predicted anti-EGFR exposure suggest a novel mechanism of resistance warranting additional prospective investigation. Also, our data support the clinical utility of comprehensive genomic profiling (both tissue and ctDNA) in the management of advanced mCRC, because standard hotspot polymerase chain reaction testing would not capture emerging clinically significant alterations. Of note, many aBRAF alterations remain unclassified to date, highlighting the need for continued functional annotation. Efforts are needed to establish the predictive impact of functional classes on anti-EGFR efficacy and to design novel therapeutic strategies for aBRAF mCRC.
Appendix
FIG A1.
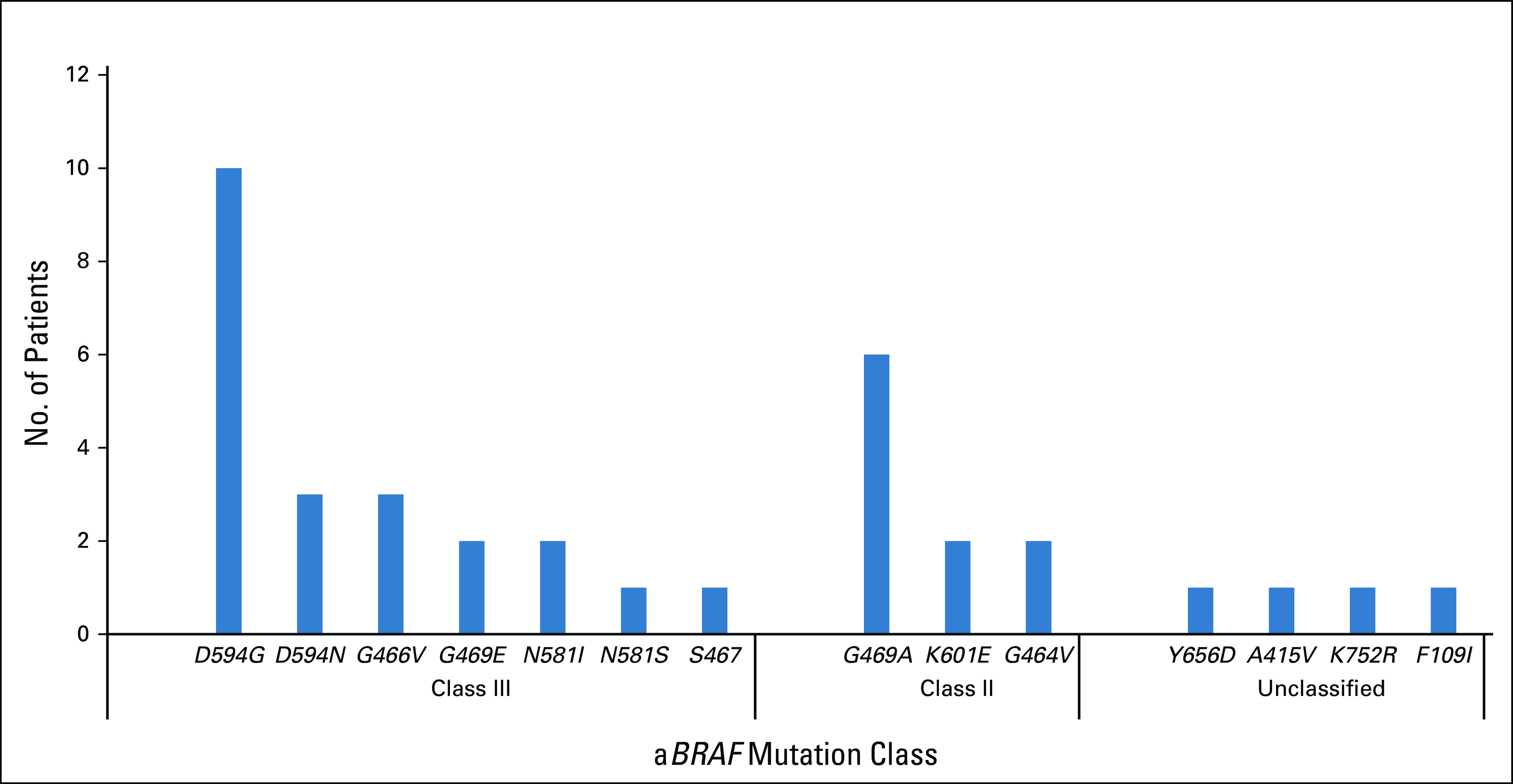
Distribution of aBRAF (atypical, non-V600 BRAF) mutations in cohort 1 (n = 36).
TABLE A1.
Matched Analysis of Patients with aBRAF, BRAFV600E, and RAS/RAFWT Metastatic Colorectal Cancer Tumors
Footnotes
Supported by National Institutes of Health Cancer Center Gore Grant No. NCI P30 CA016672.
AUTHOR CONTRIBUTIONS
Conception and design: Benny Johnson, Jonathan M. Loree, Kanwal Raghav, Scott Kopetz
Provision of study material or patients: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: Benny Johnson, Alexandre A. Jacome, Kanwal Raghav, Scott Kopetz
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Jonathan M. Loree
Consulting or Advisory Role: Taiho Pharma Canada, Ipsen, Novartis, Bayer, Amgen
Shehara Mendis
Travel, Accommodations, Expenses: Merck
Van K. Morris II
Consulting or Advisory Role: Clinical Genomics
Travel, Accommodations, Expenses: Array BioPharma
Arvind Dasari
Consulting or Advisory Role: Ipsen, AbbVie/Stemcentrx, Novartis, Voluntis, Lexicon
Research Funding: Novartis, eFFECTOR Therapeutics, Eisai
Shubham Pant
Honoraria: 4D Pharma
Consulting or Advisory Role: TYME
Research Funding: Mirati Therapeutics (Inst), Eli Lilly (Inst), RedHill Biopharma (Inst), Xencor (Inst), Five Prime Therapeutics (Inst), Novartis (Inst), Rgenix (Inst), Sanofi/Aventis (Inst), ArQule (Inst), Bristol-Myers Squibb (Inst), Onco Response (Inst), GlaxoSmithKline (Inst)
Victoria M. Raymond
Employment: Trovagene, Guardant Health
Stock and Other Ownership Interests: Trovagene, Guardant Health
Eduardo Vilar
Consulting or Advisory Role: Janssen Research & Development
Michael Overman
Consulting or Advisory Role: Bristol-Myers Squibb, Roche, Gritstone Oncology, MedImmune, Novartis, Promega
Research Funding: Bristol-Myers Squibb, Merck, Roche, MedImmune
Bryan Kee
Stock and Other Ownership Interests: Medtronic
Cathy Eng
Honoraria: Roche, Bayer
Consulting or Advisory Role: Roche, Bayer Schering Pharma, Taiho
Travel, Accommodations, Expenses: Roche, Bayer, Sirtex Medical
Kanwal Raghav
Honoraria: Bayer, Eisai
Consulting or Advisory Role: Bayer (Inst)
Travel, Accommodations, Expenses: TRACON Pharma
Scott Kopetz
Stock and Other Ownership Interests: MolecularMatch, Navire
Consulting or Advisory Role: Roche, Genentech, EMD Serono, Merck, Karyopharm Therapeutics, Amal Therapeutics, Navire Pharma, Symphogen, Holy Stone, Biocartis, Amgen, Novartis, Eli Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health
Research Funding: Amgen (Inst), Sanofi (Inst), Biocartis (Inst), Guardant Health (Inst), Array BioPharma (Inst), Roche (Inst), EMD Serono (Inst), MedImmune (Inst), Novartis (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Tie J, Gibbs P, Lipton L, et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer. 2011;128:2075–2084. doi: 10.1002/ijc.25555. [DOI] [PubMed] [Google Scholar]
- 2.Morris V, Overman MJ, Jiang ZQ, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13:164–171. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones JC, Renfro LA, Al-Shamsi HO, et al. Non-V600 BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol. 2017;35:2624–2630. doi: 10.1200/JCO.2016.71.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 5.Pietrantonio F, Petrelli F, Coinu A, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur J Cancer. 2015;51:587–594. doi: 10.1016/j.ejca.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 6.Shinozaki E, Yoshino T, Yamazaki K, et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: The Biomarker Research for Anti-EGFR Monoclonal Antibodies by Comprehensive Cancer Genomics (BREAC) study. Br J Cancer. 2017;117:1450–1458. doi: 10.1038/bjc.2017.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–383. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234–238. doi: 10.1038/nature23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strickler JH, Loree JM, Ahronian LG, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018;8:164–173. doi: 10.1158/2159-8290.CD-17-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10:e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parseghian CM, Loree JM, Morris VK, et al. Anti-EGFR resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann Oncol. 2019;30:243–249. doi: 10.1093/annonc/mdy509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dankner M, Rose AAN, Rajkumar S, et al. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene. 2018;37:3183–3199. doi: 10.1038/s41388-018-0171-x. [DOI] [PubMed] [Google Scholar]
- 13.De Roock W, De Vriendt V, Normanno N, et al. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Jones JC, Kipp BR, et al. Activity of EGFR antibody in non-V600 BRAF mutant metastatic colorectal cancer. Ann Oncol. 2019;30:147–149. doi: 10.1093/annonc/mdy477. [DOI] [PubMed] [Google Scholar]
- 15.Cremolini C, Di Bartolomeo M, Amatu A, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015;26:2092–2097. doi: 10.1093/annonc/mdv290. [DOI] [PubMed] [Google Scholar]
- 16.Sullivan RJ, Infante JR, Janku F, et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018;8:184–195. doi: 10.1158/2159-8290.CD-17-1119. [DOI] [PubMed] [Google Scholar]
- 17.Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33:125–136.e3. doi: 10.1016/j.ccell.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rankin A, Klempner SJ, Erlich R, et al. Broad detection of alterations predicted to confer lack of benefit from EGFR antibodies or sensitivity to targeted therapy in advanced colorectal cancer. Oncologist. 2016;21:1306–1314. doi: 10.1634/theoncologist.2016-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schirripa M, Biason P, Cortiula F, et al. Clinico-pathological and molecular characterization of BRAF mutant metastatic colorectal cancer (mCRC): Are all mutations created equal? J Clin Oncol. 2018;36(suppl; 3590) [Google Scholar]
- 20.Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 21.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17:676–691. doi: 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thierry AR, Pastor B, Jiang ZQ, et al. Circulating DNA demonstrates convergent evolution and common resistance mechanisms during treatment of colorectal cancer. Clin Cancer Res. 2017;23:4578–4591. doi: 10.1158/1078-0432.CCR-17-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pietrantonio F, Vernieri C, Siravegna G, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23:2414–2422. doi: 10.1158/1078-0432.CCR-16-1863. [DOI] [PubMed] [Google Scholar]
- 24.Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21:2157–2166. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 25. doi: 10.1038/nm.3870. , 2015] [DOI] [Google Scholar]
- 26.Van Emburgh BO, Arena S, Siravegna G, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun. 2016;7:13665. doi: 10.1038/ncomms13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu HA, Suzawa K, Jordan E, et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res. 2018;24:3108–3118. doi: 10.1158/1078-0432.CCR-17-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Cutsem E, Huijberts S, Grothey A, et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAF V600E-mutant metastatic colorectal cancer: Safety lead-in results from the phase III BEACON Colorectal Cancer Study. J Clin Oncol. 2019;37:1460–1469. doi: 10.1200/JCO.18.02459. [DOI] [PMC free article] [PubMed] [Google Scholar]