

Abstract
PURPOSE
Conversion of tumor subtype frequently occurs in the course of metastatic breast cancer but is a poorly understood phenomenon. This study aims to compare molecular subtypes with subsequent lung or pleural metastasis.
PATIENTS AND METHODS
In a cohort of 57 patients with breast cancer and lung or pleural metastasis (BCLPM), we investigated paired primary and metastatic tissues for differential gene expression of 269 breast cancer genes. The PAM50 classifier was applied to identify intrinsic subtypes, and differential gene expression and cluster analysis were used to further characterize subtypes and tumors with subtype conversion.
RESULTS
In primary breast cancer, the most frequent molecular subtype was luminal A (lumA; 49.1%); it was luminal B (lumB) in BCLPM (38.6%). Subtype conversion occurred predominantly in lumA breast cancers compared with other molecular subtypes (57.1% v 27.6%). In lumA cancers, 62 genes were identified with differential expression in metastatic versus primary disease, compared with only 10 differentially expressed genes in lumB, human epidermal growth factor receptor 2 (HER2)–enriched, and basal subtypes combined. Gene expression changes in lumA cancers affected not only the repression of the estrogen receptor pathway and cell cycle–related genes but also the WNT pathway, proteinases (MME, MMP11), and motility-associated cytoskeletal proteins (CK5, CK14, CK17). Subtype-switched lumA cancers were further characterized by cell proliferation and cell cycle checkpoint gene upregulation and dysregulation of the p53 pathway. This involved 83 notable gene expression changes.
CONCLUSION
Our results indicate that gene expression changes and subsequent subtype conversion occur on a large scale in metastatic luminal A–type breast cancer compared with other molecular subtypes. This underlines the significance of molecular changes in metastatic disease, especially in tumors of initially low aggressive potential.
INTRODUCTION
Visceral metastases often occur as late events in the course of metastatic breast cancer and are generally followed by a rapidly fatal outcome. Despite recent advances in our understanding of molecular events that occur during disease progression,1,2 details of the tumor biology of visceral metastases in comparison with primary breast cancer (PBC), and their relationship to tumor subtype, have not been fully elucidated. Molecular evolution is believed to be linked to the heterogeneity and molecular plasticity of primary breast cancer, which is reflected in gene expression.3
CONTEXT
Key Objective
Tumor metastasis involves a series of molecular changes that may result in a remarkably different tumor phenotype. We examined changes in PAM50 gene signatures in a matched-pair series of 57 breast cancer primaries and corresponding lung or pleural metastases to determine the nature and extent of tumor subtype conversion.
Knowledge Generated
Subtype conversion to PAM50 luminal B subtype was observed in the majority of luminal A–type breast cancers. This involved notable alterations in tumor proliferation and estrogen receptor signaling and was paralleled by an almost doubling of the calculated risk of recurrence score. Only few cases of primary luminal B, human epidermal growth factor receptor 2–enriched, and basal-like subtypes converted to another PAM50 category in metastasis.
Relevance
PAM50-based subtype classification of breast cancer lung and pleural metastases may be different than that seen in the primary tumor, especially in primary luminal A cancers. This change in PAM50 classification between primary and metastatic disease may affect response to systemic therapy and must be investigated.
Lungs (23%) and pleura (12%) are the most common sites of visceral metastasis in breast cancer, followed by liver (10%) and brain (2%).4 In a large autopsy series, the incidence of lung metastases from breast primaries was as high as 71%.5 As a first site of tumor progression during the first 5 years of follow-up, the lungs rank third at 20%, after bone (38%) and liver (23%).6 When interpreting these figures, it must be noted that lung metastases are referred to as either including or excluding pleural metastasis.
The site of metastasis in breast cancer is not random, but there is clinical and pathologic evidence of distinct patterns of disease relapse.7 Brain metastasis is often associated with the hormone receptor–negative/human epidermal growth factor receptor 2–positive (HER2-positive) phenotype, and liver metastasis also was more frequently observed in the HER2-positive subtypes compared with HER2-negative subtypes.8 In lung metastasis, there is no clear preference of a particular tumor subtype, but the luminal A subtype had a lower rate of lung relapse compared with the other three subtypes by tissue microarray analysis,7 and basal subtypes in particular were more frequent than expected.9
In this study, we included patients with invasive breast cancer of all molecular subtypes and metachronous breast cancer with lung or pleural metastasis (BCLPM) to provide insights into the up- and downregulation of genes during the course of the disease. Therefore, we retrospectively examined gene expression profiles in a well-characterized, prospectively assembled group of patients with breast cancer and metachronous lung or pleural metastasis (Data Supplement 2, Fig S1). Data were interpreted to provide information on which genes or gene groups undergo notable changes during the course of BCLPM regarding up- or downregulation.
PATIENTS AND METHODS
Study Population
Patient samples were recruited from a series of female patients with metastatic PBC and biopsy-confirmed BCLPM in 2003 to 2014. Patients had received both primary sur-gery and metastasis biopsies at the University Hospital, Heidelberg. Tumor histology of both sites and clinical records were reviewed (Data Supplement 2, Table S1), and pertinent data were updated retrospectively using current tumor classification criteria.10,11 No follow-up after metastasis was available.
Formalin-fixed and paraffin-embedded (FFPE) tissue sam-ples were provided by the Tissue Bank at the National Center for Tumor Diseases (Heidelberg, Germany) in accordance with the regulations of the Tissue Bank and approval of the ethics committee of the medical faculty of the University of Heidelberg (approval No. S-716/2018). The final cohort of this study included 57 paired samples of PBC and BCLPM and was selected from 81 patients with BCLPM and PBC. Exclusion criteria included no availability or insufficiency of tumor tissue from PBC or BCLPM tumor tissue (n = 11), primary lung cancer after review of patient records (n = 2), and low RNA content or not meeting quality control criteria of RNA data (n = 11).
Gene Expression Analysis
For the selection of tumor tissue for RNA extraction, FFPE tissue blocks from the PBC and metastatic lesions were selected after reviewing all original tissue slides and were recut for hematoxylin & eosin sections, to be used for reference and to determine tumor cell content (tumor surface area). RNA was extracted using 5-10 unstained FFPE slides for each tumor. Microdissection was performed in most cases to avoid normal breast tissue contamination. A minimum of approximately 50 ng of total RNA was used. Hybridization time per cartridge was 16 hours before measurement.
RNA expression analysis was performed by measuring a custom panel of 269 breast cancer–related genes and 11 housekeeping genes (Data Supplement 2, Table S2), using the nCounter platform (Nanostring Technologies, Seattle CA) according to the manufacturer's instructions. Two cases were excluded, because quality control criteria of RNA measurements were not met. The gene panel includes 25 published gene signatures with prognostic or predictive properties in luminal-type breast cancer with the aim to determine their role in metastatic disease (Data Supplement 2, Table S3). The selection of gene signatures covered by this list of genes includes nine proliferation-related signatures, four estrogen receptor–related signatures, one immune-related signature, and 11 signatures related to cancer pathways. In reporting this study, we have adhered to the recommendations for reporting tumor marker studies (REMARK guidelines).12
Statistics
For molecular subtyping, the PAM50 subtype clustering model was fitted and risk of recurrence scores (ROR-S) were calculated.13 Differential expression analysis was carried out using paired moderated t statistic in limma,14,15 and nominal P values were corrected for multiple comparisons using Benjamini and Hochberg’s method.16 All genes with an adjusted false discovery rate of P < .05 and a fold change of < 0.66 or > 1.5 were considered differentially expressed. For gene function, DAVID Bioinformatics Resources were used (version 6.8),17,18 and functional annotation analysis in the Biological Process category and KEGG pathway enrichment were computed. For two-dimensional visualization of the data, the UMAP method of multidimensional scaling was used.19 Time to metastasis was calculated using the Kaplan-Meier method. All statistical calculations, except gene function analysis, were done using R, version 3.6.1.20 Additional information is available in Data Supplement 1.
RESULTS
Patient Characteristics
A total of 57 patients with BCLPM were included in this study. The metastatic biopsy site was mostly pleura (n = 48; 84.2%) and less often intrapulmonary (n = 9; 15.8%). Three lung lesions presented as solitary metastasis, and 54 cases presented as multiple metastatic lesions. Kaplan-Meier analysis revealed significant differences for time intervals to BCLPM depending on the subtype of PBC (Data Supplement 2, Fig S2). The median interval between breast cancer diagnosis and BCLPM was 62.9 months.
PAM50 Subtypes of PBC and BCLPM
The most frequent subtypes were luminal A for PBC (49.1%) and luminal B for BCLPM (38.6%; Fig 1A). Two tumors with normal-like subtypes were identified in PBC, and none were identified in BCLPM. Tumor content in these samples was > 50%; therefore, these samples were regarded as representative, and neither case was excluded. A low-risk class was assigned to these cases, in concordance with the literature.21 Luminal B, HER2-enriched, and basal-like subtypes were regarded as high-risk subtypes.13 When comparing molecular subtypes in PBC with BCLPM, discordant PAM50 (subtype switching) was observed in 24 cases (42.1%). PAM50 molecular subtype conversion occurred in the majority of luminal A–type PBC (n = 16; 57.1%), evolving mostly into luminal B–type or HER2-type metastasis (Table 1; Fig 1B). In the other subtypes, a subtype switch occurred in only four of 14 luminal B–type tumors. Also, one HER2 enriched–type tumor recurred as luminal B metastasis, and one basal-like PBC recurred as a HER2-enriched subtype. Overall, subtype conversion resulted in increased numbers of high-risk subtypes in BCLPM versus PBC (73.7% v 47.4%; P = .007; Data Supplement 2, Table S4).
FIG 1.

(A) Frequencies of PAM50 subtypes in primary breast cancer and subsequent lung metastasis. For luminal A (LumA) breast cancers, the change in PAM50 subtype is significant (P = .02, Fisher’s exact test). (B) Alluvial plot showing molecular subtype conversion and number of cases with and without conversion (without normal-like [Normal] subtype). The majority of cases with subtype switching affected the LumA category. LumB, luminal B; HER2, human epidermal growth factor receptor 2 (HER2)–enriched; Basal, basal-like; n.s., not significant.
TABLE 1.
Clinical and Molecular Characteristics of Luminal-Type Breast Cancers, Comparing LumA Subtypes With and Without Subtype Conversion to LumB-Type Tumors

PAM50 subtypes were clearly separated in UMAP mul-tidimensional scaling on the basis of all 269 genes, and subtype mapping revealed differences in gene expression of PAM50 clusters with regard to primary or metastatic site. Gene expression clustering of BCLPM was clearly different from PBC in the low-risk category but not with cancers of high-risk subtypes (Figs 2A-2B). This indicates a similar tumor biology of BCLPM and PBC for tumors of high-risk subtypes but a different biology in the low-risk (luminal A) category. In particular, this method revealed that two distinct clusters of luminal A–type tumors were associated with either PBC or metastatic breast cancer. Similarly, heatmap clustering confirmed separate luminal A clusters for primary tumors and metastases (Fig 3).
FIG 2.

Multidimensional scaling (UMAP) of gene expression, showing (A) gene expression in primary versus metastatic cancers, and (B) PAM50 subtypes of the primary and metastatic tumors. For luminal A (LumA) subtype, two subtype clusters can be distinguished, (bottom) one for primary breast cancers, and (top) the smaller one representing lung metastases. LumB, luminal B; HER2, Human epidermal growth factor rector 2–enriched; Basal, basal-like; Normal, normal-like subtype.
FIG 3.
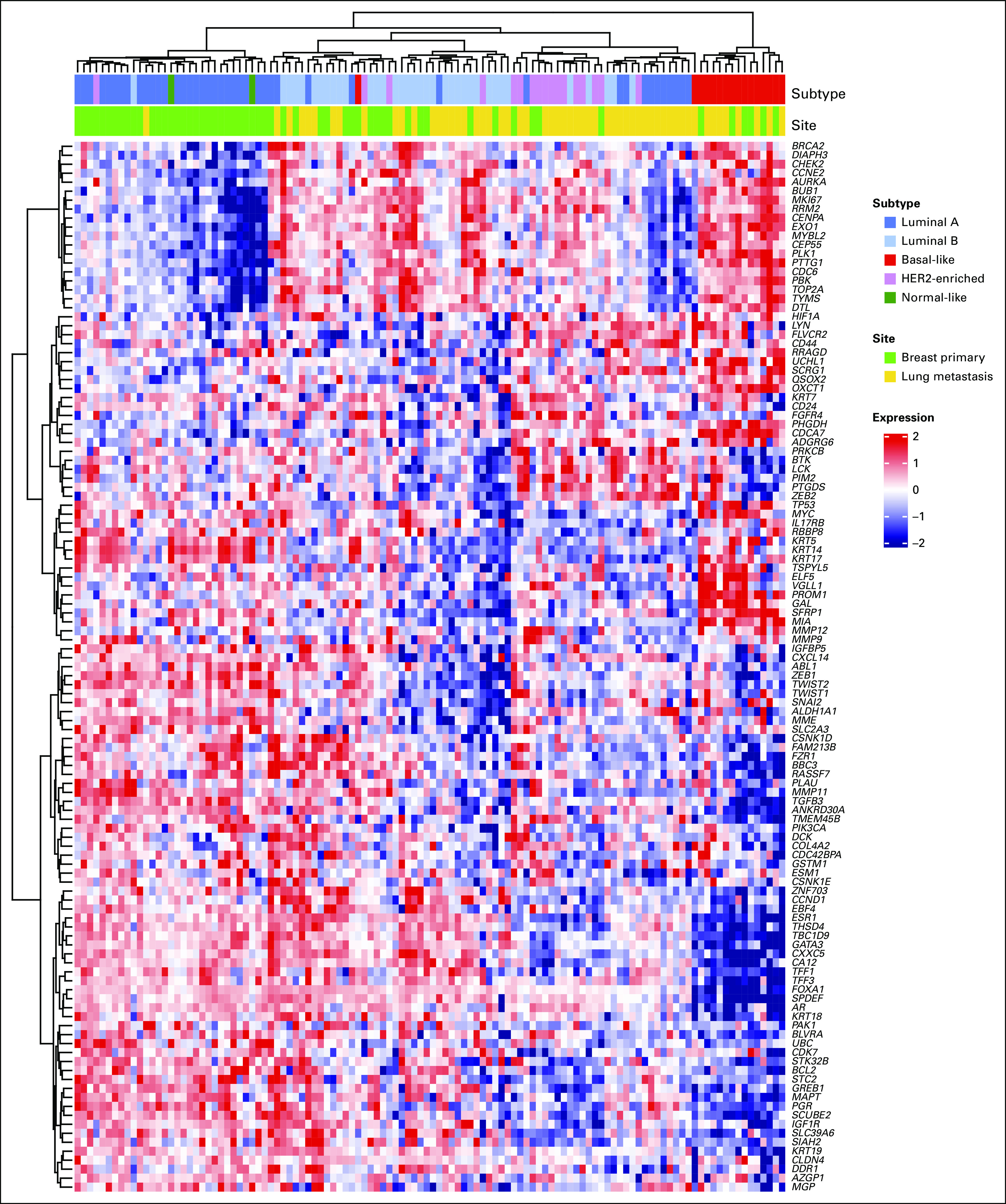
Unsupervised clustering heatmap of significantly differentially expressed genes in primary breast cancer and lung metastasis (P < .05, any fold change). Tumors are discriminated according to their molecular subtype, with separation of metastatic luminal A tumors from primary luminal A cancers (shown as blue bars across top). HER2, human epidermal growth factor receptor 2.
Differential Gene Expression Analysis
To characterize which genes are involved in the process of BCLPM, differential gene expression analysis was performed. For this purpose, a logistic model was fitted using a design matrix for paired samples comparisons between BCLPM and PBC. Across all subtypes, 41 genes were downregulated in BCLPM and 5 genes were upregulated (Data Supplement 2, Fig S3 and Table S5). Six genes were downregulated with a fold change of < 0.25. These included 3 cytoskeletal proteins (KRT14, KRT17, KRT5), 2 matrix metalloproteinases (MMP11, MME), and ANKRD30A. The latter is a breast differentiation gene that is frequently expressed in the breast and in receptor-positive breast cancer.22 Other molecular changes in metastases were linked to proliferation (CDC6, CCNB1, MKI67, TOP2A, AURKB, PTTG1), cell cycle checkpoints (BRCA2, BUB1, BUB1B, CHEK1), and epithelial-mesenchymal transition (EMT) genes. Only one gene, SCRG1, was upregulated in BCLPM with a fold change of > 2, which is a marker of mesenchymal stem cells,23 pointing toward EMT in BCLPM. Among the genes most relevant for therapy in breast cancer (ESR1, PGR, HER2, MKI67), the expressions of ESR1 and HER2 were unaffected in BCLPM, but PGR was significantly downregulated (P = .0015, Wilcoxon rank-sum test), and MKI67 was not significantly increased (Data Supplement 2, Fig S5).
Subtype-Specific Changes
The PAM50 subtypes of PBC showed marked variation with regard to the extent of differential gene expression of this gene panel in BCLPM. The greatest number of differentially expressed genes was seen in luminal A–type breast cancer, with 47 and 11 down- and upregulated genes, respectively, compared with fewer expression changes in metastatic high-risk tumors (luminal B, HER2-enriched, basal-like), with six and four down- and upregulated genes, respectively (Figs 4A-4B). Only 1 gene was downregulated in BCLPM that was HER2 enriched, and no genes reached significance after adjustment for multiple testing in the basal-like subtype, which in part may be attributed to the comparably small sample sizes of 6 and 7, respectively (Data Supplement 2, Table S6). Of the genes repressed in luminal A metastases, several are associated with EMT. Four of these (TWIST2, TWIST1, ZEB1, TGFB3) are key regulators of EMT. In addition, STC2 promotes EMT via AKT and ERK, and PROM1 facilitates EMT.
FIG 4.
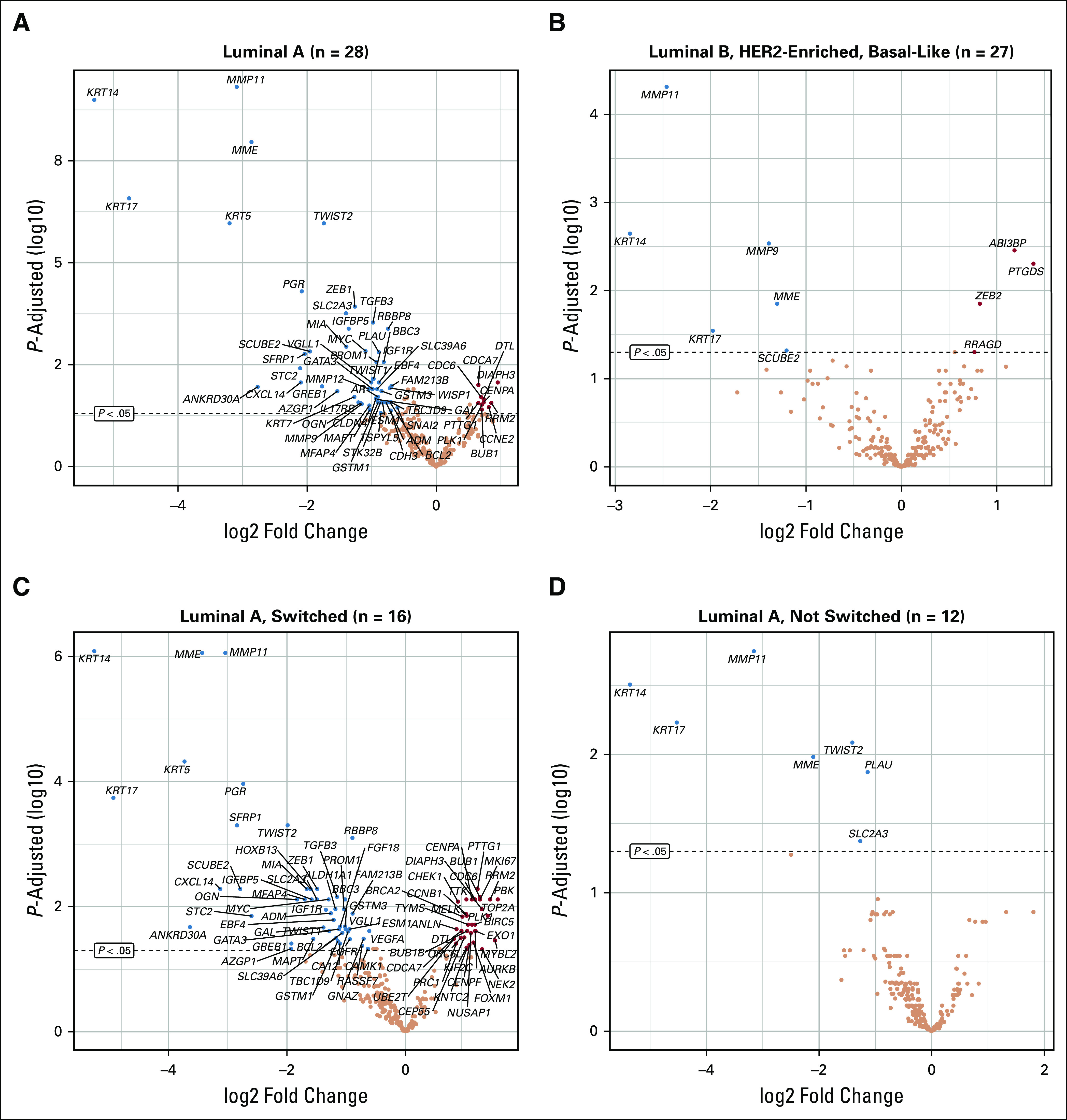
Differential gene expression for breast cancer lung metastasis versus primary breast cancer. Genes with significant down- and upregulation are indicated in blue and red, respectively (adjusted P < .05). Different patterns of gene expression changes are observed, with (A) significantly more changes in the luminal A subtype compared with (B) high-risk tumors (lumB, Human epidermal growth factor receptor 2 (HER2)–enriched, Basal-like). Within the luminal A subtype, gene expression changes were mostly confined to (C) tumors with subtype switch in lung metastasis compared with (D) nonswitched metastases.
Subtype Switching in Luminal A–Type Breast Cancers
Luminal A–type breast cancer demonstrated a unique behavior with regard to the frequency of subtype switching (Fig 1B), the phenotype in breast compared with BCLPM (Figs 2A-2B), and the magnitude and type of gene expression changes in lung metastasis (Table 1). Thirty-four and 49 genes were significantly up- and downregulated, respectively, in BCLPM from luminal A cancers with subtype switch (adjusted P < .05) compared with only 7 genes downregulated and none upregulated in the non–subtype switch group (Figs 4C-4D). Interestingly, the time to progression to metastatic events was similar for tumors undergoing subtype switch or not (median, 118.4 v 104.9 months), suggesting that the events leading to higher-grade metastasis in luminal A cancers are late events in the metastatic pro-cess. In luminal A cancers, subtype switch affected cell cycle and cell proliferation genes (mitotic cell cycle checkpoint, sister chromatid cohesion, mitotic nuclear division, cell division) and the p53-signaling as well as progesterone-signaling pathways (Data Supplement 2, Figs 4A and 4B).
Clustering of Gene Expression in Luminal A–Type Cancers
To better characterize the phenomenon of subtype conversion in luminal A cancers, we set up a logistic model for the assessment of differential gene expression with and without subtype conversion. In the group of 28 luminal A–subtype tumors, a subtype conversion was more likely to occur when genes involved in certain growth factors, nuclear proteins, and signaling molecules were upregulated (GNAZ, HOXB13, VEGFA) and some genes involved in immune response and inflammation (FOXP3, CD8A, CD68, CSF1R) were downregulated (Fig 5A). In the metastatic setting, a much larger number of genes were clustered according to subtype switching of luminal A–subtype cancers. This included 10 genes of various functions with downregulation in metastases and 52 genes, also of diverse functions, that were upregulated after subtype switch (Fig 5B). This upregulation of a substantial number of genes suggests a common mechanism of gene regulation that is involved in subtype switching.
FIG 5.
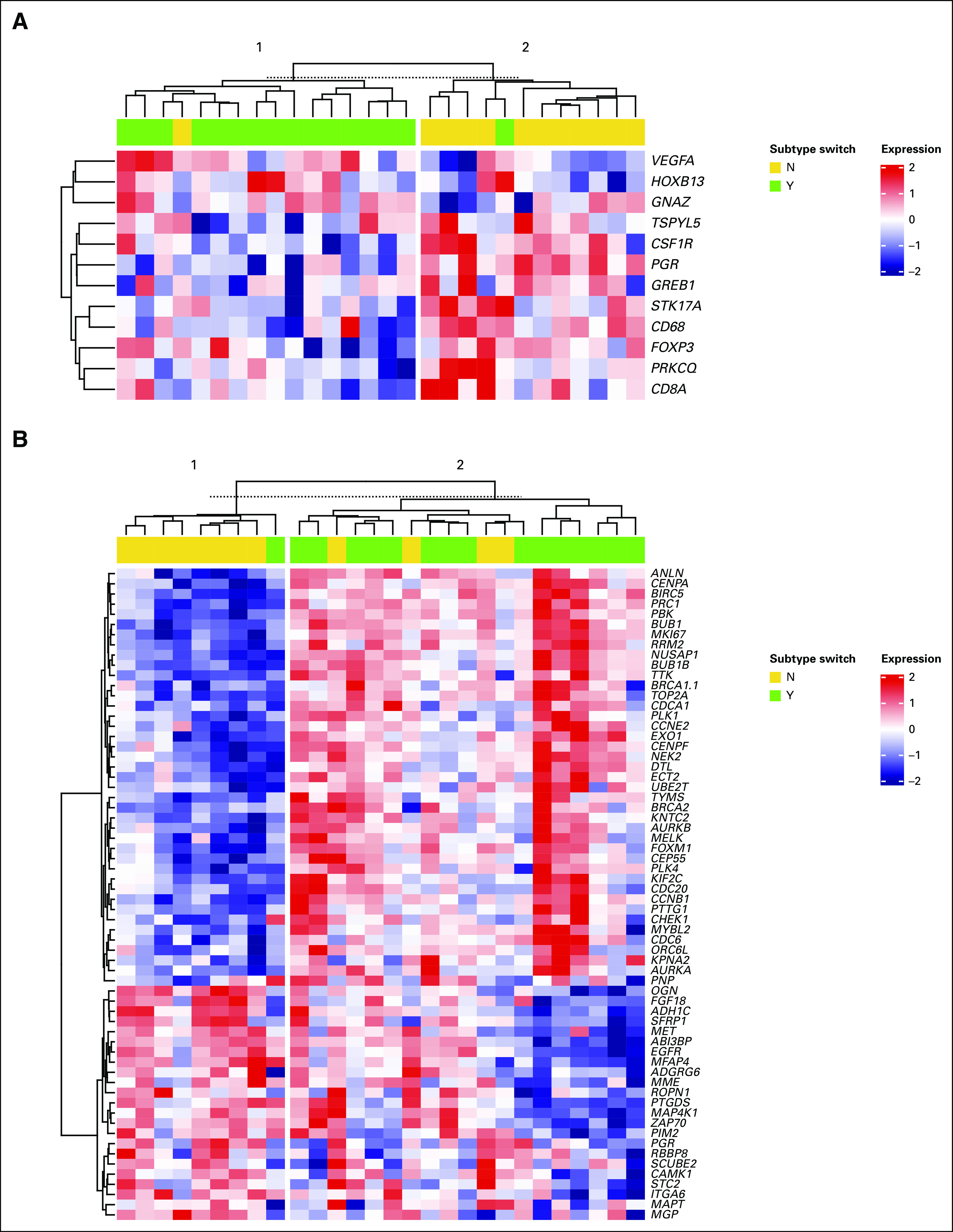
Heatmaps of luminal A–type breast cancer genes (P < .05, any fold change) in breast cancer lung metastasis. Gene expression changes related to subtype switch are evident in both (A) primary breast cancers and (B) lung metastasis, each associated with alterations in various pathways, including proliferation-, estrogen receptor pathway–, and inflammation-associated genes.
Comparison of Gene Signatures in Luminal A– and B–Type Cancers
For additional insight into the classes of genes involved in subtype switching, a couple of well-characterized gene signatures for estrogen receptor signaling, tumor proliferation, and hallmarks of cancer were analyzed in BCLPM (Data Supplement 2, Figs S6a and S6B). Interestingly, and despite the different genes involved in these signatures, all 4 tumor proliferation signatures tested showed similar patterns of gene expression in metastases derived from luminal A–type tumors with and without subtype switch. In particular, all proliferation signatures were significantly upregulated in subtype-switched tumors, with similar levels compared with luminal B cancers. Conversely, estrogen receptor–related signatures were downregulated in subtype-switched metastases of former luminal A–type breast cancers. However, no such changes in genes expression were seen in several gene signatures that are attributed to hallmarks of cancer.
Data Availability
The datasets generated and analyzed during this study are available in the GEO repository, GSE145752.
DISCUSSION
Metastatic breast cancer often belongs to a prognostically more aggressive category than the primary tumor.24 The molecular changes that are acquired by the tumor cell during disease progression have been summarized as molecular evolution25 and with respect to changes in intrinsic tumor subtype as subtype conversion or subtype switch.26 Our results indicate that evolution within cancer cell populations during metastasis leads to more transcriptomic and phenotypic changes than might be expected from the acquisition of genetic events. The magnitude of these changes and their relationships to molecular subtypes have not been studied systematically in visceral metastasis yet, but overall approximately 40% of organ metastases were shown to represent a different PAM50 subtype.27
Subtype conversion only partly reflects the changes occurring in tumor evolution, and our data indicate that, even without change of the PAM50 subtype, the molecular phenotype may still be different in the metastasis, especially in low-grade tumors. In multidimensional scaling, tumors of luminal A subtype in BCLPM form a different cluster than luminal A subtype in the primary (Fig 2), and, in the differential expression heatmap, they cluster with more aggressive subtypes (Fig 3). Along that line, relevant gene expression changes in brain metastases were found without change of PAM50 subtype.28 When PAM50 subtype conversion occurs, it can be regarded as an indicator of genetic remodeling in metastasis and as a risk factor of additional disease progression. This is also evident by the change in the risk of recurrence score, which almost doubled in luminal A–type cancers. Of note, although estrogen receptor and HER2 statuses as predictive clinical markers remained unchanged in this group, the proliferation rate greatly increased, and breast hormone receptor signaling declined. As such, although therapeutically relevant markers were unaltered upon subtype switch, remarkable changes occurred that are generally associated with a worsened malignant behavior and prognostic impact.
The gene expression changes that were evident across all subtypes included evidence for increased mobility and invasive behavior of the metastatic tumor cell through downregulation of cytokeratins (KRT5/14)29; tumor activation through downregulation of MMP1130,31; and deactivation of estrogen receptor–dependent pathways, as indicated by the downregulation of the progesterone receptor and the ankyrin repeat-domain gene (ANKRD30A). Conversely, genes involved in EMT and growth signaling were upregulated in all molecular subtypes (Data Supplement 2, Fig S3). As far as datasets are comparable, most changes found in BCLPM have been reported to occur also in other metastatic sites,27,32 with the exception of HER2, which was demonstrated to be upregulated in the brain28; however, it was not in our dataset.
Luminal A cancers undergoing a subtype switch were characterized by a much higher variability in gene expression in BCLPM compared with high-risk carcinomas. This variability affected various molecular pathways. From gene expression data alone, it appears not possible to name a common denominator for the changes in metastasis of luminal A cancers that affected several dozens of genes. When trying to dissect the alterations that occur in subtype conversion, we found that primary tumors with low expression of inflammation-related genes but increased expression of growth signaling–associated genes were more likely to undergo subtype conversion. In the metastatic tumor cell, the subtype switch was characterized by quite extensive gene expression changes of various molecular pathways, similar to the finding of important gene expression changes in > 100 genes in brain metastasis.29 No hypothesis about the root cause of changes for subtype conversion can be put forward, but the manifold changes of expression status in BCLPM from luminal A cancers do indicate a major reprogramming of the tumor cell.
Limitations of this study include the use of a curated gene list. As such, in contrast to genome-wide analyses, significant alterations in other, unrepresented pathways cannot be excluded in this study. Another limitation is that the selection of cases that underwent diagnostic biopsy was driven by clinical needs and may not be representative for metastatic breast cancer in general. Moreover, the molecular changes occurring in BCLPM were studied only on the RNA expression level, and no genomic assays were performed. Last but not least, although the discrimination of patients with breast cancer into specific cancer subtypes is a widely applied clinical concept with profound prognostic implications, luminal A and B cancers may much more represent a continuum rather than true distinct subtypes from a biologic point of view.
In conclusion, we have shown that luminal A–type breast cancer is most affected by subtype conversion and differential gene expression in matched metastases to lung or pleura involving but not limited to estrogen receptor signaling (downregulation) and tumor proliferation (upregulation). This supports additional molecular subtyping in luminal A–type breast cancer to better understand metastatic biology and clinical implications.
SUPPORT
Supported by Research Grant No. 23011195 from the Dietmar Hopp Stiftung for Gene Expression Analysis in Breast Cancer.
AUTHOR CONTRIBUTIONS
Conception and design: Max Klebe, Carlo Fremd, Ralph Wirtz, Andreas Schneeweiss, Peter Sinn
Administrative support: Martina Kirchner
Provision of study material or patients: Nadine Volk, Andreas Schneeweiss, Hauke Winter, Peter Sinn
Collection and assembly of data: Max Klebe, Carlo Fremd, Verena Thewes, Martina Kirchner, Nadine Volk, Johannes Haag, Alexandra Schulz, Jörg Heil, Andreas Schneeweiss, Hauke Winter, Peter Sinn
Data analysis and interpretation: Max Klebe, Carlo Fremd, Mark Kriegsmann, Katharina Kriegsmann, Thomas Albrecht, Verena Thewes, Pornpimol Charoentong, Ralph Wirtz, Thordur Oskarsson, Alexandra Schulz, Andreas Schneeweiss, Hauke Winter, Peter Sinn
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Carlo Fremd
Honoraria: Roche, Pfizer, Celgene, AstraZeneca, Amgen
Consulting or Advisory Role: Roche, Pfizer
Travel, Accommodations, Expenses: Celgene, Roche
Ralph Wirtz
Employment: STRATIFYER Molecular Pathology GmbH
Stock and Other Ownership Interests: STRATIFYER Molecular Pathology GmbH
Consulting or Advisory Role: Janssen Oncology (Inst), BioNTech AG (Inst)
Research Funding: Janssen Research & Development (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from BioNTech (Inst); royalties from Qiagen (Inst)
Thordur Oskarsson
Stock and Other Ownership Interests: Neovasc, Cyclacel Pharmaceuticals, Windtree Therapeutics, Enzon Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Migrastatins and uses thereof. Inventors: Samuel J. Danishefsky, Joan Massague, Manuel Valiente Cortes, Thordur Oskarsson, Malcolm Moore, Nicolas Lecomte, Ouathek Ouerfelli, Guangli Yang. Publication date: January 1, 2017. Patent office: US Patent number: 9546146. Application number: 15065090. Description: The present invention provides compounds, pharmaceutically acceptable compositions thereof, and methods of using the same. Migrastatins and uses thereof. Inventors: Samuel J. Danishefsky, Joan Massague, Manuel Valiente Cortes, Thordur Oskarsson, Malcom Moore, Nicolas Lecomte, Ouathek Ouerfelli, Guangli Yang. Publication date: April 5, 2016. Patent Office: US Patent number: 9303009. Application number: 14110115. Description: The present invention provides compounds, pharmaceutically acceptable compositions thereof, and methods of using the same.
Other Relationship: Genentech
Jörg Heil
Honoraria: Roche, Siemens Healthcare Diagnostics, BARD
Consulting or Advisory Role: Roche, Siemens Healthcare Diagnostics
Research Funding: BARD
Travel, Accommodations, Expenses: Celgene
Andreas Schneeweiss
Honoraria: Roche Pharma AG, Celgene, AstraZeneca, Pfizer, Novartis, MSD Oncology, Lilly, Tesaro
Research Funding: Roche Pharma AG (Inst), Celgene (Inst), Abbvie, Molecular Partners
Expert Testimony: Roche, AstraZeneca
Travel, Accommodations, Expenses: Roche, Celgene
Hauke Winter
Expert Testimony: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca
Peter Sinn
Honoraria: NanoString Technologies, Roche Pharma AG
Research Funding: Roche Pharma AG
Travel, Accommodations, Expenses: NanoString Technologies
No other potential conflicts of interest were reported.
REFERENCES
- 1.Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell. 2013;24:410–421. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yates LR, Knappskog S, Wedge D, et al. : Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 32:169-184 e7, 2017
- 3.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamby C. The pattern of metastases in human breast cancer: Methodological aspects and influence of prognostic factors. Cancer Treat Rev. 1990;17:37–61. doi: 10.1016/0305-7372(90)90075-q. [DOI] [PubMed] [Google Scholar]
- 5.Lee YT. Breast carcinoma: Pattern of metastasis at autopsy. J Surg Oncol. 1983;23:175–180. doi: 10.1002/jso.2930230311. [DOI] [PubMed] [Google Scholar]
- 6.van den Hurk CJ, Eckel R, van de Poll-Franse LV, et al. Unfavourable pattern of metastases in M0 breast cancer patients during 1978-2008: A population-based analysis of the Munich Cancer Registry. Breast Cancer Res Treat. 2011;128:795–805. doi: 10.1007/s10549-011-1372-y. [DOI] [PubMed] [Google Scholar]
- 7.Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28:3271–3277. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 8.Wu Q, Li J, Zhu S, et al. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget. 2017;8:27990–27996. doi: 10.18632/oncotarget.15856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smid M, Wang Y, Zhang Y, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68:3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 10.2016. Brierley JD, Gospodarowicz MK, Wittekind C (eds): TNM Classification of Malignant Tumours, 8th Edition. Weinheim, Germany, Wiley-VCH, [Google Scholar]
- 11.: WHO Classification of Tumours of the Breast. Lyon, France: International Agency for Research on Cancer; 2012. Lakhani SR, Ellis IO, Schnitt SJ, et al. [Google Scholar]
- 12.McShane LM, Hayes DF. Publication of tumor marker research results: The necessity for complete and transparent reporting. J Clin Oncol. 2012;30:4223–4232. doi: 10.1200/JCO.2012.42.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smyth GK: Limma: Linear models for microarray data, in Gentleman R, Carey V, Dudoit S, et al (eds): Bioinformatics and Computational Biology Solutions using R and Bioconductor. Statistics for Biology and Health. Berlin, Heidelberg, New York, Springer, 2005,p 397-420. [Google Scholar]
- 15.Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 17.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McInnes L, Healy J, Melville J. 2018 UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv:1802.03426,
- 20.R Core Team http://www.R-project.org/ R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- 21.Kensler KH, Sankar VN, Wang J, et al. PAM50 molecular intrinsic subtypes in the Nurses’ Health Study cohorts. Cancer Epidemiol Biomarkers Prev. 2019;28:798–806. doi: 10.1158/1055-9965.EPI-18-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kosaloglu Z, Bitzer J, Halama N, et al. In silico SNP analysis of the breast cancer antigen NY-BR-1. BMC Cancer. 2016;16:901. doi: 10.1186/s12885-016-2924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aomatsu E, Takahashi N, Sawada S, et al. Novel SCRG1/BST1 axis regulates self-renewal, migration, and osteogenic differentiation potential in mesenchymal stem cells. Sci Rep. 2014;4:3652. doi: 10.1038/srep03652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson AM, Jordan LB, Quinlan P, et al. Prospective comparison of switches in biomarker status between primary and recurrent breast cancer: The Breast Recurrence In Tissues Study (BRITS) Breast Cancer Res. 2010;12:R92. doi: 10.1186/bcr2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yates LR, Gerstung M, Knappskog S, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stefanovic S, Wirtz R, Deutsch TM, et al. Tumor biomarker conversion between primary and metastatic breast cancer: mRNA assessment and its concordance with immunohistochemistry. Oncotarget. 2017;8:51416–51428. doi: 10.18632/oncotarget.18006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cejalvo JM, Martínez de Dueñas E, Galván P, et al. Intrinsic subtypes and gene expression profiles in primary and metastatic breast cancer. Cancer Res. 2017;77:2213–2221. doi: 10.1158/0008-5472.CAN-16-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Priedigkeit N, Hartmaier RJ, Chen Y, et al. Intrinsic subtype switching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA Oncol. 2017;3:666–671. doi: 10.1001/jamaoncol.2016.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seltmann K, Fritsch AW, Käs JA, et al. Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc Natl Acad Sci USA. 2013;110:18507–18512. doi: 10.1073/pnas.1310493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Huang S, Guo J, et al. Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics. Int J Oncol. 2016;48:1783–1793. doi: 10.3892/ijo.2016.3400. [DOI] [PubMed] [Google Scholar]
- 31.Noël A, Gutiérrez-Fernández A, Sounni NE, et al. New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Front Pharmacol. 2012;3:140. doi: 10.3389/fphar.2012.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JY, Park K, Lee E, et al. Gene expression profiling of breast cancer brain metastasis. Sci Rep. 2016;6:28623. doi: 10.1038/srep28623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analyzed during this study are available in the GEO repository, GSE145752.