

INTRODUCTION
Cowden syndrome is an autosomal dominant genetic disease with an estimated incidence of one in 200,000. Affected individuals develop multiple systemic hamartomas and have a cumulative lifetime risk of breast cancer of 85%.1 Approximately 80% of patients with Cowden syndrome have a germline inactivating mutation in PTEN (10q23.3).2 PTEN acts as a tumor suppressor gene via numerous mechanisms,3 one of which is antagonizing the PI3K/AKT/mTOR signaling pathway by dephosphorylating phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 functions as a secondary messenger in the PI3K pathway that binds and activates proteins that have a pleckstrin homology domain, such as AKT1, and triggers their activation and localization to the plasma membrane, promoting cellular proliferation and survival.4
Germline PTEN loss-of-function mutations may result in dominant AKT activation as a driving oncogenic event in Cowden-related breast tumors.5 Preclinical evidence suggests that cancers with AKT activation have increased sensitivity to AKT inhibition.6 Preliminary clinical evidence is derived from phase I and II trials in patients with breast cancers bearing somatic mutations in the PI3K/AKT/mTOR pathway.7-11
Capivasertib (AZD5363, AstraZeneca) is a potent and selective oral inhibitor of all three isoforms of the serine/threonine kinase AKT (ie, AKT1, 2, 3) and has preclinical evidence of efficacy as monotherapy or in combination with cytotoxic and targeted therapies.12-15 Despite the encouraging progression-free survival observed with capivasertib monotherapy in heavily pretreated patients with AKT1 E17K–mutant breast and gynecologic cancers,9 RECIST response rates in phase I studies only reached 22% (Table 1).8,9,16 Because PTEN loss activates AKT1, we hypothesized that tumors from patients with Cowden syndrome could be sensitive to this drug family.
TABLE 1.
Summary of Main Phase I Clinical Trials With Capivasertib Monotherapy

Case 1: SAFIR02 Trial
A 50-year-old woman with a family history of Cowden syndrome was diagnosed with a T3N3 breast cancer, which was estrogen (ER) and progesterone receptor negative, human epidermal growth factor receptor 2 (HER2) negative, and was designated grade III invasive carcinoma of no special type (NST). The patient received six cycles of neoadjuvant cyclophosphamide 600 mg/m2, epirubicin 75 mg/m2, and docetaxel 100 mg/m2 before a mastectomy with left axillary lymph node dissection (revealing residual disease in 10 of 18 lymph nodes) and adjuvant radiotherapy.
Eight months later, the patient experienced relapse with cutaneous disease and thoracic nodal involvement. After enrolling in SAFIR02 (ClinicalTrials.gov identifier: NCT02299999), targeted panel sequencing (Ion Torrent PGM; Thermo Fisher Scientific, Villebon, France) of a fresh tumor biopsy sample revealed the presence of a heterozygous germline PTEN mutation (c.389G>A, p.R130Q, SNP rs121909229) alongside other variants (Fig 1A; Data Supplement). Immunohistochemistry revealed lack of PTEN expression in the tumor (Fig 1B). The patient received six cycles of paclitaxel (90 mg/m2 on days 1, 8, and 15 of a 28-day cycle) with bevacizumab (10 mg/m2 on days 1 and 15) and carboplatin (AUC2, days 1, 8, and 15 of a 28-day cycle, ceased after 6 weeks).
FIG 1.
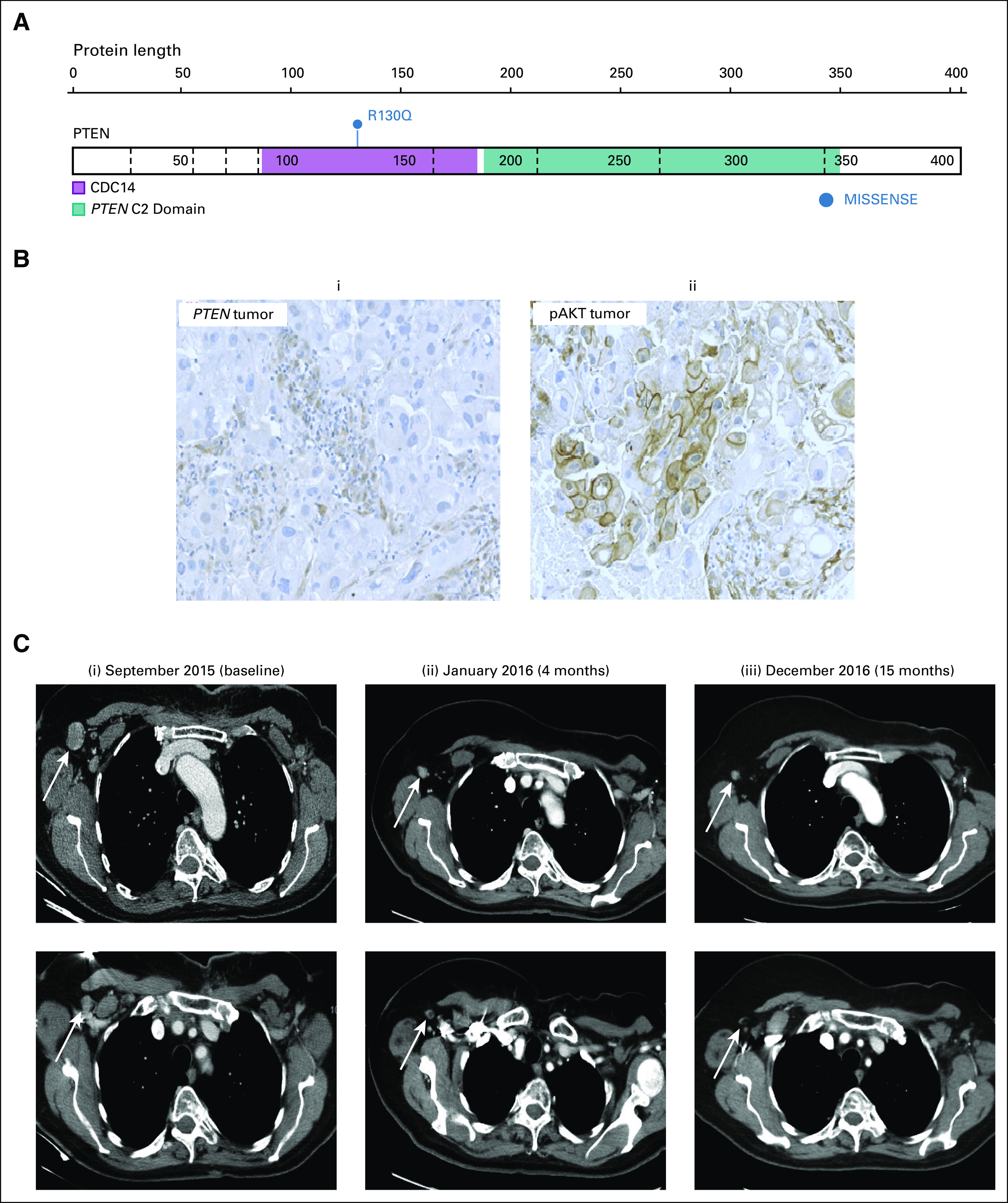
Exceptional response in patient with germline PTEN R130Q mutation. (A) Germline PTEN mutation c.389G>A, pR130Q. CDC14, phosphatase domain. Illustration from https://proteinpaint.stjude.org. (B) Immunohistochemistry demonstrating (i) absent PTEN staining in the tumor and (ii) cytoplasmic and membranous expression of pAKT in the 40% of tumor cells. (C) Computed tomography (CT) scans during the patient’s time on capivasertib, with white arrows indicating axillary disease: (i), September 2015, baseline CT showing two areas of axillary lymphadenopathy; (ii) January 2016, CT scans demonstrating partial response following 4 months of carboplatin-paclitaxel-bevacizumab chemotherapy; and (iii) December 2016, CT scans following 11 months of capivasertib monotherapy demonstrating persistent complete response before progression in February 2017.
Upon completion of chemotherapy, tumor evaluation demonstrated a partial response (by RECIST, version 1.1) with 60% reduction of target lesions (Fig 1C). In the context of SAFIR02, the patient was randomly assigned to maintenance targeted therapy and received capivasertib 480 mg twice per day, 4 days on and 3 days off. This treatment was well tolerated, with no grade 2 or greater toxicities. After 3 months of capivasertib monotherapy, a complete response was observed, which was maintained for 12 months before the patient experienced progression while on capivasertib.
Case 2: BEECH Trial
In March 2010, a 37-year-old woman with known Cowden syndrome and a history of a neck arteriovenous malformation, multinodular goiter, and rectal hamartomatous polyps was diagnosed with bilateral breast cancer. On the right side, she presented with a T3, ER-positive and progesterone receptor–positive, HER2-negative, grade II invasive carcinoma NST with 20 of 23 involved lymph nodes. On the left, she presented with a 4-mm, grade II invasive carcinoma NST, strongly ER positive and HER2 negative. After a bilateral mastectomy, exome sequencing (Hiseq2500, Illumina, Cambridge, UK) of the right breast cancer and germline DNA revealed the germline PTEN mutation (c.T68G:p.L23X), and a second-hit somatic stop-gain PTEN mutation (exon 1, c.T264A:p.Y88X) with an allele frequency (AF) of 25.5%, alongside other variants (Fig 2A; Data Supplement). PTEN immunohistochemistry revealed reduced PTEN staining in the tumor (Fig 2B). Postoperative staging revealed metastatic disease with mediastinal lymph node and lung metastases. In May 2010, the patient received six cycles of chemotherapy every 3 weeks (fluorouracil 600 mg/m2, epirubicin 75 mg/m2, and cyclophosphamide 600 mg/m2) before starting maintenance tamoxifen. In October 2011, she underwent a bilateral salpingo-oophorectomy.
FIG 2.

Exceptional response in patient with germline PTEN L23X mutation. (A) Germline PTEN mutation c.T68G:p.L23X. CDC14, phosphatase domain. Illustration from https://proteinpaint.stjude.org. (B) PTEN immunohistochemistry from (i) control tissue and (ii) noncancerous and (iii and iv) tumor-containing lymph node. The control tissue shows mainly cytoplasmic expression of PTEN. The noncancerous lymph node and the residual lymphatic tissue in the lymph node metastasis also show a PTEN expression comparable with the control. The tumor cells in the lymph node metastasis display weaker PTEN staining, which is mainly nucleolar. (C) Computed tomography (CT) scans during the patient’s time on capivasertib, with white arrows indicating disease in the liver: (i) October 2012, baseline CT demonstrating a liver deposit; (ii) January 2013, CT following two cycles of paclitaxel and capivasertib; (iii) CT demonstrating continued complete response on maintenance capivasertib 7 months after cessation of paclitaxel; and (iv) May 2014, final CT before progression June 2014.
After a 28-month progression-free period, the patient had new liver metastases. In November 2012, she enrolled in the phase I/II BEECH study (ClinicalTrials.gov identifier: NCT01625286) and was assigned to receive paclitaxel plus capivasertib (part A, schedule 2). She received eight cycles of paclitaxel (90 mg/m2 on days 1, 8, and 15 of a 28-day cycle) combined with capivasertib (360 mg twice per day on days 2-5, 9-12, and 16-18 of each 28-day cycle). In June 2013, a computed tomography scan showed a complete response of the liver metastases. The patient continued maintenance capivasertib alone with no grade 2 or greater toxicities. She had a confirmed maintained response in May 2014 until progression occurred in June 2014—a progression-free survival of 19 months—and a maintained complete response for 12 months on capivasertib alone (Fig 2C). Plasma circulating tumor DNA analysis from baseline and progression time points during the BEECH study showed no major changes (Data Supplement).
DISCUSSION
The AKT inhibitor capivasertib has been examined in early-phase trials (Table 1), and no complete responses have been noted, yet both patients with Cowden syndrome presented here had durable complete responses to capivasertib. Such outlier sensitivity likely reflects the germline, and therefore fundamentally clonal, nature of PTEN alteration. In vivo mice models with PTEN homozygous deletion have shown dramatic regression of the Cowden phenotype features of trichilemmomas on treatment with the mTOR (downstream from AKT) inhibitor rapamycin.17 In humans, Hyman et al9 demonstrated that tumor response to targeted treatment with capivasertib was proportional to AKT1 mutation clonality. Furthermore, three case reports in pediatric patients and a pilot study (n = 18) in adults with PTEN aberrations have shown regression of phenotypic changes associated with PTEN loss after treatment with mTOR inhibitor sirolimus,18-21 demonstrating sensitivity of even nonmalignant cells carrying a germline PTEN mutation to PI3K-pathway inhibition. Similar to the cases presented here, no severe toxicity was noted in the pilot study,21 with just one patient experiencing a grade 3 adverse event (hypophosphatemia/hypercholesterolemia).
In recent years, drugs targeting the PI3K/AKT/mTOR pathway have been developed. Trials have attempted to identify molecular predictors of response by identifying alterations in the PI3K/AKT/mTOR pathway (Data Supplement). Tumor PIK3CA mutations are not clear predictors of response to mTOR inhibition. In contrast, AKT inhibitors in combination with paclitaxel have been shown to be more active in patients with triple-negative breast cancers harboring a PIK3CA/PTEN/AKT1 pathway alteration.7,10 Similarly, PI3K inhibitors have demonstrated activity in patients with PIK3CA mutations.22,23
Of note in case 2 is the presence of the second-hit PTEN stop-gain mutation Y88X, present with an AF of 25.5%. However, the presence of a TP53 mutation at an AF of 51.9% indicates that this second-hit mutation is subclonal rather than a truncal driver mutation. Conversely, the patient in case 1 does not have a PTEN second-hit mutation or loss of heterozygosity (LOH) yet demonstrates lack of PTEN expression. Low tumor purity can make LOH difficult to detect, and, although the estimated purity was 40%, the true purity may have been lower. Explanations for the PTEN phenotype in case 1 include undetected LOH, PTEN promotor hypermethylation,24,25 complex PTEN genomic rearrangements,26 and post-translational modification.27
Contrary to the two-hit model by Knudson et al28 of tumorigenesis in tumor suppressor genes, PTEN aberrations appear to be protumorigenic in the absence of a second hit. In 2010, Alimonti et al29 demonstrated that PTEN hypermorphic mice (with 80% of the normal PTEN protein level) had a greater propensity to tumorigenesis than mice with two functional alleles but were less tumorigenic than PTEN heterozygous mice, supporting a haploinsufficiency model of tumorigenesis in PTEN aberrations.
A later in vivo study of PTEN knock-in mouse models suggested that the conformation of PTEN underlies the dominant-negative behavior of PTEN heterozygous mutants. Papa et al30 demonstrated that PTEN is catalytically active in PIP3 dephosphorylation and subsequent downstream PI3K/AKT/mTOR pathway regulation, after dimerization. Significantly, mutant PTEN protein was able to dimerize with wild-type PTEN, but the resultant hetero-dimers were less able to hydrolyze PIP3. Moreover, mutant PTEN outcompeted and displaced wild-type PTEN protein in dimerization and membrane localization. This supports the rationale that PTEN heterozygous mutants act in a dominant-negative manner to promote tumorigenesis.
The patients in the cases presented here were treated with combination chemotherapy and capivasertib followed by capivasertib monotherapy. The patient in case 1 had previously demonstrated tumor resistance to taxane therapy, with residual disease after neoadjuvant chemotherapy and a short progression-free survival after treatment of primary breast cancer. The second patient achieved a complete response with the combination of paclitaxel and capivasertib and maintained this complete response for a period of 12 months on capivasertib alone, suggesting that capivasertib was highly active in this patient.
In summary, these two patients with breast cancer and different germline PTEN mutations both showed a dramatic response to capivasertib superior to that seen in early trials of the drug. The excellent response in these two patients, despite their differing histology, is a promising indication that targeted AKT therapy is an effective approach in patients with germline PTEN mutations.
Footnotes
Supported by Foundation ARC (https://www.fondation-arc.org/the-fondation-arc) and Breast Cancer Now (https://breastcancernow.org/).
AUTHOR CONTRIBUTIONS
Conception and design: Belinda Kingston, Caroline Bailleux, Suzette Delaloge, Nicholas Turner, Fabrice André
Collection and assembly of data: Belinda Kingston, Caroline Bailleux, Suzette Delaloge, Gaia Schiavon, Veronique Scott, Magali Lacroix-Triki, Iwanka Kozarewa, Heidrun Gevensleben, Alex Pearson, Fabrice André
Data analysis and interpretation: Belinda Kingston, Caroline Bailleux, Suzette Delaloge, Magali Lacroix-Triki, T. Hedley Carr, Zoe Kemp, Nicholas Turner, Fabrice André
Provision of study material or patients: Suzette Delaloge, Veronique Scott, Nicholas Turner
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Suzette Delaloge
Consulting or Advisory Role: AstraZeneca
Research Funding: AstraZeneca (Inst), Pfizer (Inst), Roche (Inst), Genentech (Inst), Puma (Inst), Lilly (Inst), Novartis (Inst), Sanofi (Inst)
Travel, Accommodations, Expenses: Pfizer, AstraZeneca, Roche
Gaia Schiavon
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Magali Lacroix-Triki
Leadership: MyPL
Stock and Other Ownership Interests: MyPL
Honoraria: Myriad Genetics, Genomic Health
Consulting or Advisory Role: Roche
Travel, Accommodations, Expenses: Agendia
T. Hedley Carr
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Iwanka Kozarewa
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Zoe Kemp
Honoraria: Lilly
Honoraria: AstraZeneca
Nicholas Turner
Consulting or Advisory Role: Roche, Novartis, AstraZeneca, Pfizer, Bicycle Therapeutics, Bristol-Myers Squibb, Merck Sharp & Dohme, Tesaro, Lilly
Research Funding: Pfizer (Inst), Roche (Inst), AstraZeneca (Inst), Clovis Oncology (Inst), Bio-Rad (Inst), Guardant Health (Inst)
Fabrice André
Research Funding: AstraZeneca (Inst), Novartis (Inst), Pfizer (Inst), Lilly (Inst), Roche (Inst), Daiichi (Inst)
Travel, Accommodations, Expenses: Novartis, Roche, GlaxoSmithKline, AstraZeneca
No other potential conflicts of interest were reported.
REFERENCES
- 1.Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eng C. Will the real Cowden syndrome please stand up: Revised diagnostic criteria. J Med Genet. 2000;37:828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat Rev Mol Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 4.Miller TW, Pérez-Torres M, Narasanna A, et al. Loss of phosphatase and tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69:4192–4201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 6.Davies BR, Guan N, Logie A, et al. Tumors with AKT1E17K mutations are rational targets for single agent or combination therapy with AKT inhibitors. Mol Cancer Ther. 2015;14:2441–2451. doi: 10.1158/1535-7163.MCT-15-0230. [DOI] [PubMed] [Google Scholar]
- 7.Kim SB, Dent R, Im SA, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18:1360–1372. doi: 10.1016/S1470-2045(17)30450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerji U, Dean EJ, Pérez-Fidalgo JA, et al. A phase I open-label study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin Cancer Res. 2018;24:2050–2059. doi: 10.1158/1078-0432.CCR-17-2260. [DOI] [PubMed] [Google Scholar]
- 9.Hyman DM, Smyth LM, Donoghue MTA, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35:2251–2259. doi: 10.1200/JCO.2017.73.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmid P, Abraham J, Chan S, et al. AZD5363 plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (PAKT): A randomised, double-blind, placebo-controlled, phase II trial. J Clin Oncol. 2018;36 doi: 10.1200/JCO.2018.36.15_suppl.1007. (suppl; abstr 1007) [DOI] [PubMed] [Google Scholar]
- 11.Smyth L, Oliveira M, Ciruelos E, et al. AZD5363 in combination with fulvestrant in AKT1-mutant ER-positive metastatic breast cancer. Cancer Res 78, 2018 (suppl; abstr P5-21-32)
- 12.Patnaik A, Appleman LJ, Tolcher AW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016;27:1928–1940. doi: 10.1093/annonc/mdw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Xu B, Liu P. Addition of the p110α inhibitor BYL719 overcomes targeted therapy resistance in cells from Her2-positive-PTEN-loss breast cancer. Tumour Biol. 2016;37:14831–14839. doi: 10.1007/s13277-016-5381-7. [DOI] [PubMed] [Google Scholar]
- 14.Yu Y, Savage RE, Eathiraj S, et al. Targeting AKT1-E17K and the PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS One. 2015;10:e0140479. doi: 10.1371/journal.pone.0140479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal R, Liebe S, Turski ML, et al. Targeted therapy for genetic cancer syndromes: Von Hippel-Lindau disease, Cowden syndrome, and Proteus syndrome. Discov Med. 2015;19:109–116. [PubMed] [Google Scholar]
- 16.Tamura K, Hashimoto J, Tanabe Y, et al. Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77:787–795. doi: 10.1007/s00280-016-2987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden’s disease by mTOR inhibition with rapamycin. Cancer Res. 2008;68:7066–7072. doi: 10.1158/0008-5472.CAN-08-0922. [DOI] [PubMed] [Google Scholar]
- 18.Marsh DJ, Trahair TN, Martin JL, et al. Rapamycin treatment for a child with germline PTEN mutation. Nat Clin Pract Oncol. 2008;5:357–361. doi: 10.1038/ncponc1112. [DOI] [PubMed] [Google Scholar]
- 19.Schmid GL, Kässner F, Uhlig HH, et al. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: Case report and in vitro studies. Pediatr Res. 2014;75:527–534. doi: 10.1038/pr.2013.246. [DOI] [PubMed] [Google Scholar]
- 20.Iacobas I, Burrows PE, Adams DM, et al. Oral rapamycin in the treatment of patients with hamartoma syndromes and PTEN mutation. Pediatr Blood Cancer. 2011;57:321–323. doi: 10.1002/pbc.23098. [DOI] [PubMed] [Google Scholar]
- 21.Komiya T, Blumenthal GM, Ballas MS, et al. A pilot study of sirolimus (S) in subjects with Cowden syndrome (CS) with germ-line mutations in PTEN. J Clin Oncol. 2013;31:2532-2532. doi: 10.1634/theoncologist.2019-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juric D, Ciruelos E, Rubovszky G, et al. Alpelisib + fulvestrant for advanced breast cancer: Subgroup analyses from the phase III SOLAR-1 trial. Cancer Res 79, 2019 (suppl; abstr GS3-08)
- 23.Baselga J, Dent SF, Cortés J, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. J Clin Oncol. 2018;36 doi: 10.1200/JCO.2018.36.18_suppl.LBA1006. (suppl; abstr LBA1006) [DOI] [Google Scholar]
- 24.Luo S, Chen J, Mo X. The association of PTEN hypermethylation and breast cancer: A meta-analysis. OncoTargets Ther. 2016;9:5643–5650. doi: 10.2147/OTT.S111684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H-Y, Liang F, Jia Z-L, et al. PTEN mutation, methylation and expression in breast cancer patients. Oncol Lett. 2013;6:161–168. doi: 10.3892/ol.2013.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones N, Bonnet F, Sfar S, et al. Comprehensive analysis of PTEN status in breast carcinomas. Int J Cancer. 2013;133:323–334. doi: 10.1002/ijc.28021. [DOI] [PubMed] [Google Scholar]
- 27.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 28.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 29.Alimonti A, Carracedo A, Clohessy JG, et al. Subtle variations in PTEN dose determine cancer susceptibility. Nat Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papa A, Wan L, Bonora M, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157:595–610. doi: 10.1016/j.cell.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]