

Abstract
PURPOSE
Precision oncology connects highly complex diagnostic procedures with patient histories to identify individualized treatment options in interdisciplinary molecular tumor boards (MTBs). Detailed data on MTB-guided treatments and outcome with a focus on advanced GI cancers have not been reported yet.
PATIENTS AND METHODS
Next-generation sequencing of tumor and normal tissue pairs was performed between April 2016 and February 2018. After identification of relevant molecular alterations, available clinical studies or in-label, off-label, or matched experimental treatment options were recommended. Follow-up data and a response assessment that was based on radiologic imaging were recorded.
RESULTS
Ninety-six patients were presented to the MTB of Tuebingen University Hospital. Sixteen (17%) showed “pathogenic” or “likely pathogenic” germline variants. Recommendations on the basis of molecular alterations or tumor mutational burden were given for 41 patients (43%). Twenty-five received the suggested drug, and 20 were evaluable for best response assessment. Three patients (15%) reached a partial response (PR), and 6 (30%), stable disease (SD), whereas 11 (55%) had tumor progression (progressive disease). Median progression-free survival (PFS) for all treated and evaluable patients was 2.8 months (range, 1.0-9.0 months), and median overall survival (OS) of all treated patients was 5.2 months (range, 0.1 months to not reached). Patients with SD for ≥ 3 months or PR compared with progressive disease showed both a statistically significant longer median PFS (7.8 months [95% CI, 4.2 to 11.4 months] v 2.2 months [95% CI, 1.5 to 2.8 months], P < .0001) and median OS (18.0 months [95% CI, 10.4 to 25.6 months] v 3.8 months [95% CI, 2.3 to 5.4 months], P < .0001).
CONCLUSION
Next-generation sequencing diagnostics of advanced GI cancers identified a substantial number of pathogenic or likely pathogenic germline variants and unique individual treatment options. Patients with PR or SD in the course of MTB-recommended treatments seemed to benefit with respect to PFS and OS.
INTRODUCTION
The challenge of precision oncology is to connect highly complex diagnostic procedures with individual patient histories to identify optimal treatments. The complexity of this approach resulted in the implementation of interdisciplinary molecular tumor boards (MTBs) at academic centers. Detailed data on MTB-guided treatment suggestions, and specifically the outcomes of patients, have only been reported for nonselected patient groups, including for a variety of different tumors.1-4 However, each tumor entity, including GI cancers, harbors unique features that will have to be considered to identify the patients who will benefit the most from such an approach.4
Recent reports highlight the experiences of MTBs in everyday practice. A series from the MD Anderson Cancer Center that examined hot-spot regions in 11 to 50 genes for 2,000 cancer tissues found actionable mutations for 39% of patients.2 Only 11% of these selected patients could be enrolled in genotype-matched clinical trials.2 A report from the Mayo Clinic identified actionable mutations in 65% of 141 patients, but only 29 (21%) were subsequently treated in clinical trials or with targeted Food and Drug Administration (FDA)–approved drugs, with an objective response or stable disease (SD) for ≥ 4 months in 10 patients.3 Freiburg University reported 104 patients with recommendations that were based on diagnostic tests that ranged from an 8-gene panel to whole-exome sequencing (WES).1 Thirty-three patients received a recommended treatment; 11 had an objective response, and 8 additional patients reached SD at ≥ 10 weeks. Eight of 9 patients in that cohort with solid tumors and partial response (PR) received checkpoint inhibitiors.1
CONTEXT
Key Objectives
Academic molecular tumor boards (MTBs) bridge the gap between a growing complexity in diagnostic procedures and clinicians at the bedside. This retrospective study investigated the course of patients with advanced gastrointestinal (GI) cancers that have been considered for personalized treatment options, including the outcome receiving MTB-guided treatment.
Knowledge Generated
Next-generation sequencing (NGS) revealed a relevant number of germline variants, suggesting genetic counseling. More than one quarter of patients who did not have further established therapeutic options could be treated according to MTB recommendation. Responding patients with prolonged disease stabilization seemed to derive a meaningful survival benefit from cancer genome sequencing and matched treatments.
Relevance
Patients with GI cancers can benefit from currently available NGS sequencing procedures. In perspective, a continued improvement of MTB recommendations and the inclusion of additional complex diagnostic procedures might substantively enhance treatment opportunities in everyday clinical practice for these patients in the near future.
The sequencing of matched tumor and normal tissue is an important procedure for the precise identification of potentially actionable genes.5,6 This inevitably leads to the detection of germline alterations. A recent investigation in 10,389 patients across 33 cancer types detected pathogenic or likely pathogenic germline variants in 8% of all patients with cancer and in 8.8% of patients with GI tumors.7 The frequency of germline mutations varied greatly across cancer types in general but also across different GI cancers, ranging from 2.2% for cholangiocarcinoma to 14.1% for pancreatic cancer (PC).7
To our knowledge, our experience with GI tumors in an academic MTB is to date the largest reported series of this group with detailed information of comprehensive sequencing data, subsequent board recommendations, and documented outcome. MTB recommendations included approved drugs for the disease, clinical studies, approved drugs against the driver mutations in different indications (off-label use), and matched experimental treatments.
PATIENTS AND METHODS
Patients and MTB Organization
All 96 patients with GI tumors at the University Hospital Tuebingen referred to the MTB between April 2016 and February 2018 were included in this retrospective, open-label, histology-agnostic analysis, which was reviewed and approved by the local ethics committee (511/2018BO). Before patients consented to next-generation sequencing (NGS) of their tumor tissue, they were informed by a specialist in clinical genetics. If relevant germline alterations were detected, genetic counseling by a clinical geneticist was offered. The MTB consists of an interdisciplinary team coordinated by the Tuebingen Center for Personalized Medicine and includes experts in clinical and translational oncology, pathology, bioinformatics, molecular biology, radiology, and human genetics. An electronic Web-based platform (MTB platform) was established to introduce patients to the MTB team with all necessary information to prepare and follow up the weekly face-to-face meetings and subsequently document treatment outcomes (Data Supplement). Best response assessment was based on radiologic imaging in line with RECIST 1.1, for checkpoint inhibitor therapy, iRECIST, or for hepatocellular carcinomas (HCC), mRECIST criteria.8-10
Genetic Tumor/Normal Characterization
Tumor and normal tissues were genetically characterized either by NGS panel sequencing of full coding sequences or by WES (more information in Data Supplement). After the identification of relevant molecular alterations in the MTB, available clinical studies or in-label, off-label, or matched experimental treatments were recommended. Off-label use refers to the administration of an FDA/European Medicines Agency-approved drug outside its approved indication. For recommended off-label therapies, an application for reimbursement was submitted to the patient’s health insurance. Experimental individual treatment describes an individualized therapy (Heilversuch) in patients with exhausted standard therapeutic options according to §34 Arzneimittelgesetz (German Pharmacy Law). Medications used within the scope of a Heilversuch do not need to be FDA/EMA approved. Patients treated within a Heilversuch have been registered at the local authority, in this case the Regierungspräsidium Tuebingen.
Statistical Analysis
Progression-free survival (PFS) and overall survival (OS) were analyzed with the Kaplan-Meier method dependent on the treatment response (SD and PR v progressive disease [PD]) compared by log-rank testing. Of the 25 patients who were treated according to MTB recommendations, 5 could not be included in this analysis because they did not have any follow-up imaging for response monitoring.
RESULTS
Diagnostic Procedure and Clinical Work-Up
Ninety-six patients received NGS of advanced GI tumors and were presented to the MTB. This cohort included 32 colorectal cancer (CRC), 22 PC, 19 biliary tract cancer (BTC), 11 HCC, 9 upper GI tract cancer (UGC), and 3 neuroendocrine tumors (NET; Fig 1A). The mean number of systemic anticancer pretreatments was 2.8 (Fig 1B). Ninety-one patients received a gene panel analysis that examined between 337 and 710 genes, and 5 patients had WES (Fig 1C). Genes represented in the different panels are summarized in the Data Supplement, together with the total size and the average and median coverage of the 649 and 710 gene panels.
FIG 1.
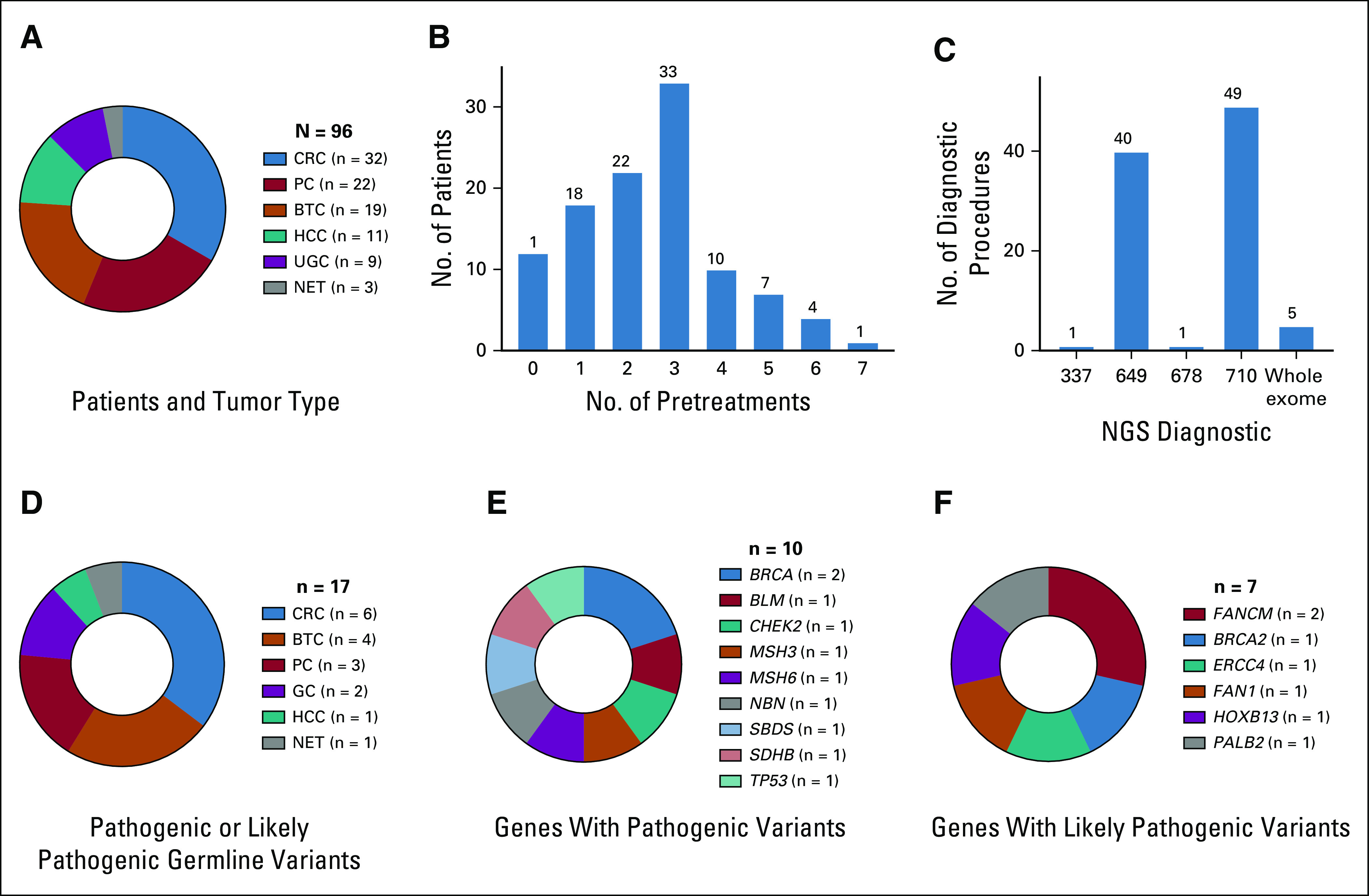
Next-generation sequencing (NGS) of GI tumors. (A) No. of patients with different tumor types. (B) No. of pretreatments before presentation at the molecular tumor board. (C) Performed diagnostic test. (D) No. of identified pathogenic or likely pathogenic germline variants per tumor type. (E) Genes that show pathogenic germline variants. (F) Genes that show likely pathogenic germline variants. BTC, biliary tract cancer; CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; NET, neuroendocrine tumor; PC, pancreatic cancer; UGC, upper GI tract cancer.
Germline Variants in GI Cancers
Germline findings were ranked according to a 5-tiered classification.11,12 Sixteen patients (17%) had germline variants classified as “pathogenic” or “likely pathogenic” (Figs 1D-1F), and in 1 patient with gastric cancer, 2 likely pathogenic variants were detected (PALB2 and FANCM). Five patients (5%) had pathogenic or likely pathogenic germline variants in one of the BRCA2, MSH6, or SDHB genes. These alterations belong to a list of secondary findings that should be reported because of the possibility for interventions to significantly reduce morbidity and mortality.13 In addition, 3 patients showed germline variants in pharmacologically relevant genes, 2 in DPYD and 1 in G6PD (Data Supplement) without showing unusual adverse reactions during chemotherapy. Seventeen additional patients had a variant of unknown significance (Data Supplement), of which the CHEK2 variant p.Ile157Thr was found in 3 different patients. This variant has been reported with classifications ranging from variant of unknown significance to pathogenic.14
A germline alteration was directly responsible for MTB recommendations for 5 patients (5%): BRCA2 for poly(ADP-ribose)-polymerase (PARP) inhibition (3 PC), CHEK2 combined with 2 somatic ATM truncations for ATR-inhibition and carboplatin (1 CRC), and MSH6 and therefore a resulting high tumor mutational burden (TMB) for pembrolizumab (1 CRC). Regarding patient histories, one had a microsatellite instability (MSI)–high CRC; the 3 patients with BRCA mutations had 1 first-degree relative with breast, pharyngeal, or bladder cancer; and the patient with a CHEK2 mutation had a first-degree relative with ovarian cancer. Taken together, for 19 out of 96 patients (20%), germline variants classified as pathogenic, likely pathogenic, or pharmacogenomic could be identified (Fig 2A).
FIG 2.
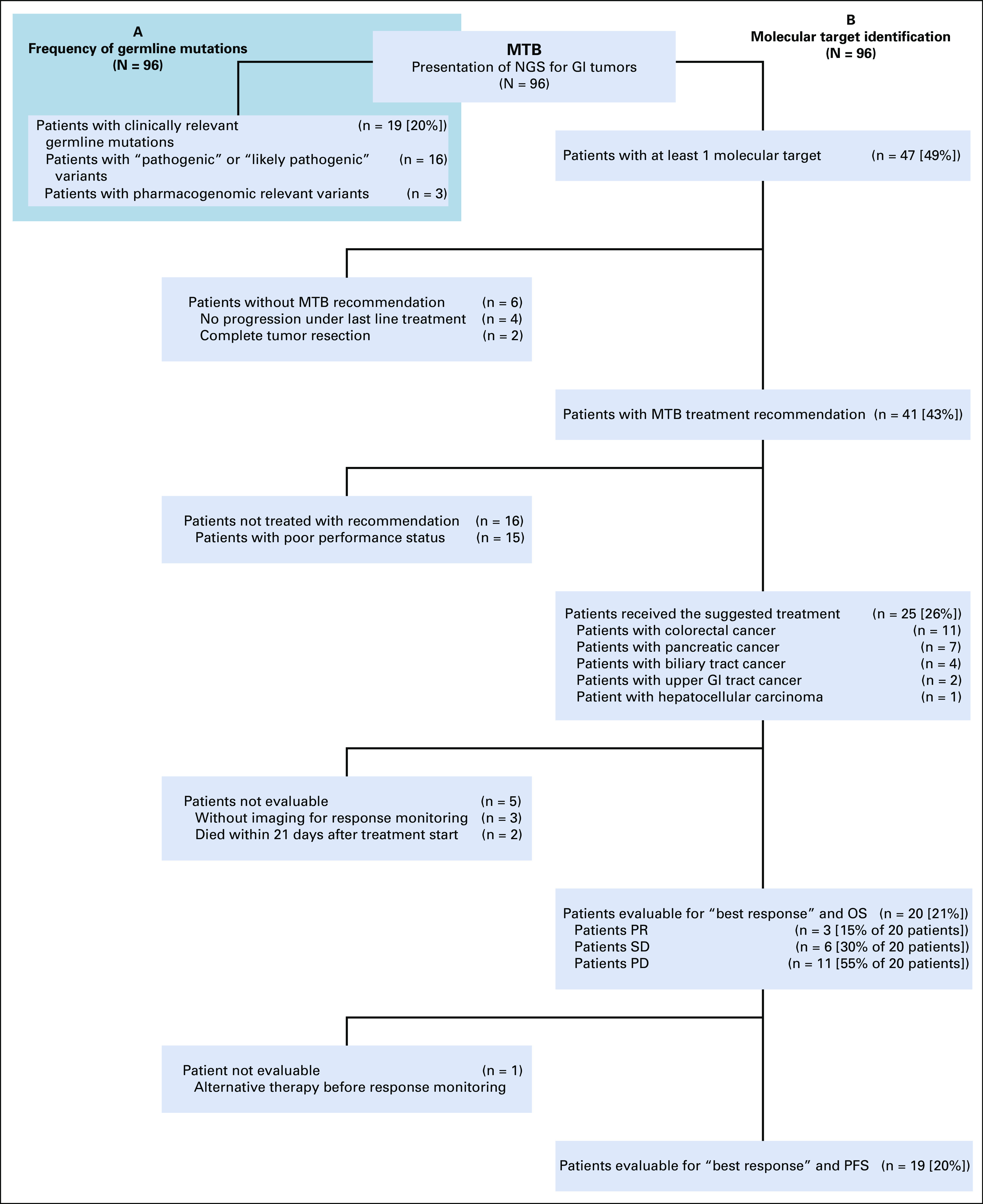
Course of patients after initiating next-generation sequencing (NGS). (A) Frequency of clinically relevant germline variants. (B) Molecular target identification and course of patients. MTB, molecular tumor board; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PR, partial response; SD, stable disease
TMB
TMB is currently regarded as relevant in predicting therapeutic success with checkpoint inhibitors beyond tumors with mismatch repair deficiency.15-18 This has been shown for patients with CRC and UGC,18 but is less clear for hepatobiliary cancer and PC. The median TMB ranged from 6.3 Var/Mbp for CRC to 2.3 Var/Mbp for BTC (Fig 3A). The 3 NET showed low TMB between 0.5 and 1.1 Var/Mbp. For MTB discussions, a high TMB was important for therapy decisions regarding checkpoint inhibitors,17,19-21 defined as ≥ 10 mutations per megabase. For tumors known to have a low TMB in general, those with values between 7.5 and 10 Var/Mb were considered TMB-high. Checkpoint inhibition on the basis of high TMB was recommended for 10 patients (4 CRC, 3 BTC, 1 HCC, 1 gastric cancer, 1 PC; Table 1). Eight of these 10 patients were also tested for MSI, but only 2 patients were found to have a MSI-high tumor.
FIG 3.

Tumor mutational burden and target identification per tumor type. (A) Tumor mutational burden was calculated for 88 patients. Two patients had 3, and 3 patients had 2, different calculations of separate tissue samples over the observation period, which are included in the analysis. The median is shown for each tumor type. (B) Frequency of potential target identification by the molecular tumor board (MTB), patients with target identification but no treatment, and patients with MTB-recommended treatment initiation. (C) Frequency of target identification per tumor type. BTC, biliary tract cancer; CRC, colorectal cancer; HCC, hepatocellular carcinoma; NET, neuroendocrine tumor; PC, pancreatic cancer; UGC, upper GI tract cancer; w/o, without.
TABLE 1.
Patients Treated According to MTB Recommendation

Molecular Target Identification by the MTB
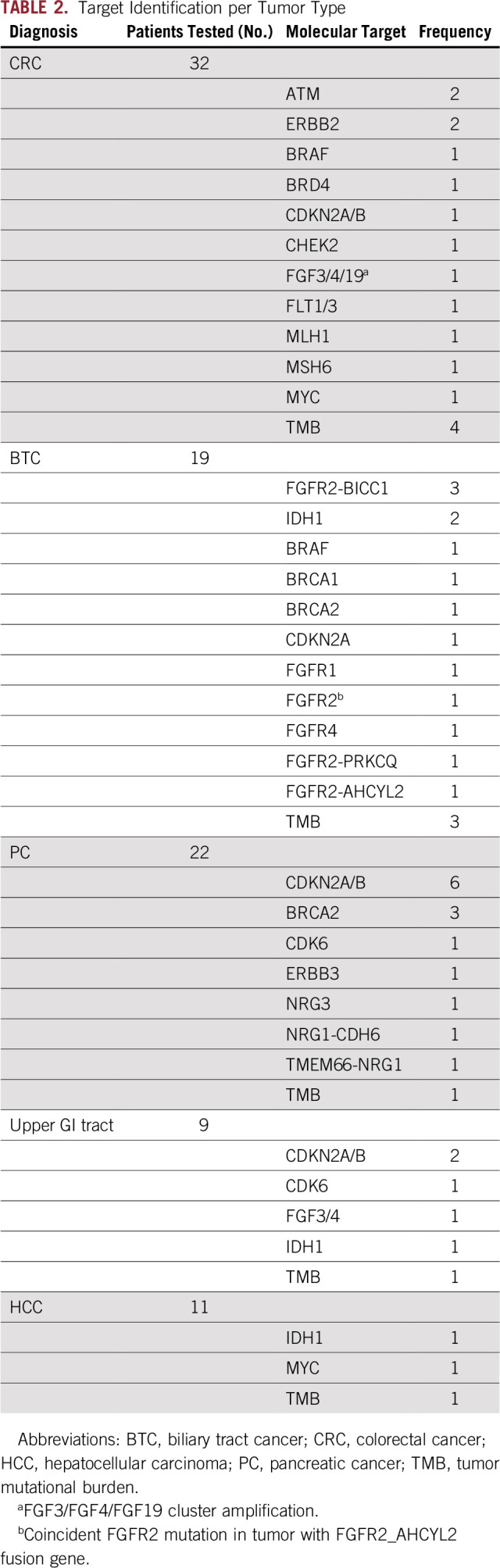
At least 1 potential molecular target was identified in 47 of 96 patients (49%; Figs 2B and 3B). The frequency of target identification varied among tumor types (Fig 3C, Table 2), with 74% for BTC, 56% for upper GI cancers, 50% for PC, 44% for CRC, and 27% for HCC. In 3 cases of NET, no target could be identified. The most frequently altered genes with single-nucleotide variants or copy number variants were CDKN2A/B, BRCA2, IDH1, ERBB2, MYC, FGF3, FGF4, FLT3, FGFR4, CDK6, BRAF, and ATM (Table 2; Data Supplement). Three different fusion genes were detected exclusively with FGFR2 in BTC. Of 10 CDKN2A/B alterations, 6 were detected in PC, 2 in UGC, 1 in BTC, and 1 in CRC. Three out of 4 BRCA2 alterations were detected in PC, and 1 in BTC (Table 2).
TABLE 2.
Target Identification per Tumor Type
Outcome and Clinical Course of Patients
The course of all patients presented to the MTB is shown in Fig 2B. Six patients did not receive a recommendation because of complete tumor resection in 2 patients or sustained disease control during ongoing chemotherapy in 4 patients. Targets in these 6 patients would have been FGFR2 fusions, IDH1 mutation, CDKN2A/B deletion, or high TMB (Data Supplement). MTB recommendations for matched treatments were given for 41 patients (43%). Twenty-five patients with a mean of 3.4 previous anticancer treatments received the suggested medication (61% of patients with MTB recommendation; 26% of the whole cohort). The other 16 patients could not be treated because of poor performance status. The alterations in this subgroup without treatment included CDKN2A/B deletions, BRAF mutations, BRCA deletions, FLT1/3 amplification, FGFR2 fusion, IDH mutation, and high TMB (Data Supplement).
Treatment was initiated in 11 patients with CRC (34% of patients with CRC), 7 patients with PC (32% of patients with PC), 4 patients with BTC (21% of patients with BTC), 2 patients with UGC (22% of patients with UGC), and 1 patient with HCC (9% of patients with HCC). Detailed information for all treated patients is listed in Table 1, and the exact molecular alteration that led to the recommendation is included in the Data Supplement. The applied drugs were either used in-label, used off-label, obtained via a clinical study, or supplied for a matched experimental treatment (Table 1, Drug Availability column). Twenty patients were treated and evaluable for best response analysis. Three patients reached PR (15%) and 6, SD (30%), and 11 had tumor progression regardless of treatment (PD, 55%). The disease control rate was 45%. Patients who showed PD as best response were treated with checkpoint inhibition (high TMB), olaparib (BRCA2 germline variant), BRD4 inhibition (BRD4 mutation), regorafenib (FGFR alteration), aurora A kinase inhibition (Myc amplification), ATR-inhibitor with carboplatin (ATM mutation), or palbociclib (CDKN2A/B deletion; Table 1).
Patients who reached either SD or PR were treated with PD-1 blocking antibodies (high TMB), ATR inhibitor with carboplatin (simultaneous mutations in ATM, CHEK2, and TP53BP1), trastuzumab with lapatinib (ERBB2 amplification), pertuzumab with erlotinib (NRG1 fusion, high-expression ERBB3 and NRG3), IDH1 inhibitor BAY 1436032 (IDH1 mutation), or lenvatinib (FGFR2 fusion gene; Table 1). For each treated patient, PFS and OS are shown in Figs 4A and 4B, respectively. The median PFS for all treated and evaluable patients was 2.8 months (range, 1.0-9.0 months) and the median OS of all treated patients was 5.2 months (range, 0.1 months to not reached).
FIG 4.
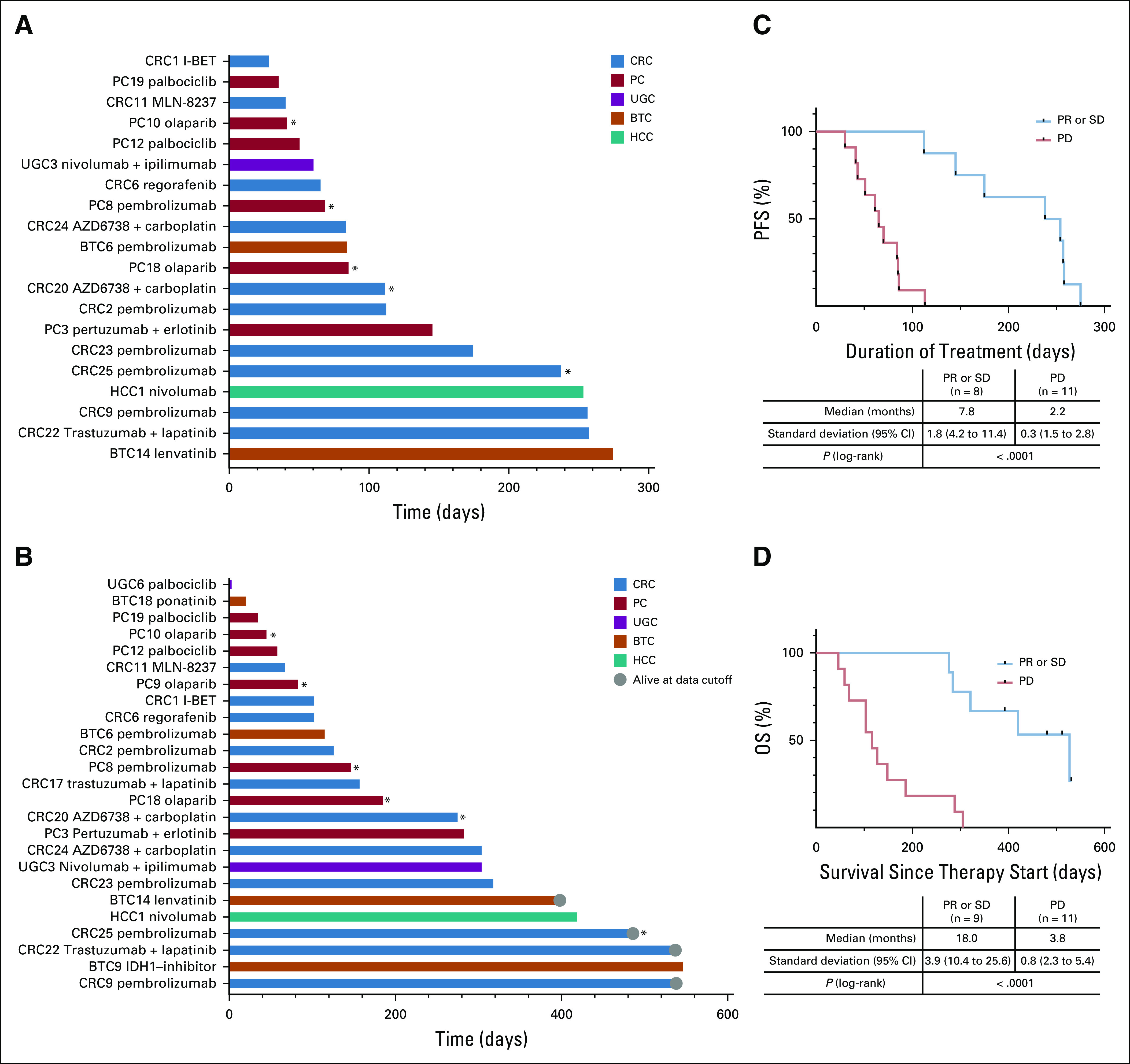
Outcomes of patients treated according to molecular tumor board (MTB) recommendations. (A) Progression-free survival (PFS), defined as the time from start of an MTB-recommended treatment to radiographic progression or death, and (B) overall survival (OS), defined as the time from start of an MTB-recommended treatment to death as a result of any cause, in days. Shown are individual patient identifiers and treatments. (*) Patients who were treated according to identified germline alterations. Kaplan-Meier analysis of (C) PFS and (D) OS in evaluable patients according to best response. The estimation compared patients who reached a partial response (PR) or stable disease (SD) with patients with progressive disease (PD). Tick marks indicate censored data. BTC, biliary tract cancer; CRC, colorectal cancer; HCC, hepatocellular carcinoma; PC, pancreatic cancer; UGC, upper GI cancer.
Of note, all patients who survived > 1 year after treatment initiation reached either SD or PR (Data Supplement). Therefore, as a surrogate end point to estimate whether patients might benefit from the MTB-recommended treatment, the achievement of SD for at least 3 months or even PR as best response was compared with PD in a Kaplan-Meier estimation for PFS and OS. Patients who reached SD or PR showed both a statistically significant longer median PFS of 7.8 months (95% CI, 4.2 to 11.4 months) versus 2.2 months (95% CI, 1.5 to 2.8 months; P < .0001) and median OS of 18.0 months (95% CI, 10.4 to 25.6 months) versus 3.8 months (95% CI, 2.3 to 5.4 months; P < .0001; Figs 4C and 4D).
DISCUSSION
Comprehensive NGS panel or WES of GI tumors and matched normal tissue samples was performed for 96 patients. In several studies, the inclusion of germline findings enabled a more comprehensive characterization of tumor biology, potential resistances, or even treatment opportunities.22,23 In this study, germline variants classified as pathogenic, likely pathogenic, or pharmacogenomic could be identified in 19 patients (20%). These numbers are higher than in a recent investigation that reported pathogenic or likely pathogenic germline predisposition variants in 8.8% of GI tumors,7 and could be mainly a result of the smaller sample size in our study. The importance of germline variants for genetic counseling usually has to be regarded in the context of associated tumor types. Whether this also holds true for therapeutic implications is unclear and is currently under discussion. For example, for BRCA mutations, the tumor lineage indeed seems to determine the therapeutic benefit of PARP inhibition.24 Conversely, emerging data suggest that patients with germline mutations that are not typically associated with a diagnosed cancer might nevertheless benefit from drugs that target this alteration.25 Germline variant reporting and the inclusion of experts in clinical genetics, should therefore be a prerequisite for MTBs.22,23,26
TMB is regarded as relevant in predicting therapeutic outcome with checkpoint inhibitors.15-18 The minimum size of tumor panels to reliably calculate TMB was suggested to include at least 300 genes, or more precisely, 1.5 Mb of the target region,21,27,28 which is met in our approach. Of 8 patients treated with checkpoint inhibitors, 4 reached PR or SD with a duration of at least 4.8 months. One patient with CRC and high TMB (118 Var/Mb) who showed progression under PD-1 inhibition even responded after the addition of CTLA4 inhibition on progression. In the 4 patients who did not show any response to checkpoint inhibition, the molecular analysis did not reveal one of the currently discussed mechanisms of resistance.29 Of note, only a minority of patients with high TMB also had MSI-high tumors. This observation is in line with an investigation in a broad range of different tumor types, showing that only 16% with high TMB were classified as MSI high.21 Another study in 6,004 patients with CRC identified 465 patients with high TMB; however, only 65% of these could be classified as MSI high.30 These observations suggest that a reliable method for TMB estimation should be used if patients with GI tumors are evaluated for personalized treatment options.
Of 96 patients with advanced GI tumors, MTB treatment recommendations were given for 41 (43%), which means an additional therapy option beyond established treatment lines for 4 out of 10 patients. The identification of parameters that reflect the benefit to patients is challenging. Best response analysis revealed disease control in 45% of evaluable patients. However, the achievement of a PR or SD is meaningful only if other relevant outcomes are improved as well. We performed an exploratory analysis of PFS and OS for the patients who received MTB-recommended treatment. The difference in OS of > 14 months in our cohort, compared with patients with PD as best response (Fig 4D), is remarkable; however, it has to be interpreted with caution and should be confirmed in larger patient populations with GI or other cancers. If this observation could be confirmed, 1 goal in the constant improvement of MTB recommendations should be to gradually increase the percentage of patients who reach disease control of at least 3 months in advanced cancers.
Early cohorts with molecular-matched therapies have been reported for CRC31 and BTC.32 In the first study, 68 out of 254 patients with advanced CRC received selected matched therapies to KRAS/BRAF/PIK3CA mutations, PTEN, or phosphorylated MET expression. PR or SD of ≥ 16 weeks were seen for only 11 patients, and median time-to-treatment failure was 7.9 weeks.31 In our cohort with an extended molecular profiling technique, actionable alterations in CRC were identified for 44%, and 4 out of 10 evaluable patients showed either PR or SD of ≥ 16 weeks. A subgroup with BTC from the MOSCATO-01 trial reported molecular targets in 23 of 34 patients, with 18 receiving matched therapies (53%).32 The overall response rate and PFS of ≥ 6 months in that study were 33% and 37%, respectively. In line with this observation, we identified targets in 74% of patients with BTC, which further emphasizes that BTC might be the most promising GI cancer subgroup for MTB-guided therapies.33
To constantly improve the quality of future recommendations, several issues must be considered. First, the diagnostic era of characterizing tumors has just begun with panel or exome sequencing. Several techniques, such as transcriptome34,35 and epigenome36 analysis or whole-genome37 and single-cell sequencing, are expected to improve tumor characterization up front.38 Such a constant evolution will require more specialists in these methods to join academic MTBs, which means a new era of interdisciplinary patient care to bridge the gap between a growing complexity in diagnostic procedures and clinicians at the bedside. Second, recent data suggest that the administration of customized multidrug regimens that target multiple identified molecular alterations39 or a sequential treatment of standard chemotherapy followed by matched therapies40 could further improve therapeutic success. Recent examples in CRC are PD-L1 and CTLA-4 inhibition in MSI-high cancers41 or the combination of a BRAF inhibitor, MEK inhibitor, and anti–epidermal growth factor receptor antibody in BRAF V600E mutated cancers.42 Third, a reduction in dropout rates might be achieved with more focused and earlier patient selection for NGS-based diagnostic procedures and a shortening of time intervals to get access to suggested drugs. Fourth, each patient who is treated according to an MTB recommendation outside clinical trials should be regarded as an N-of-1 trial.43 This implies a thorough documentation of adverse events and a reliable response assessment along a predefined routine.
In conclusion, GI tumors are an important group in the field of personalized medicine. We were able to show that integrating NGS into everyday practice allowed us to identify an unexpectedly high number of germline variants and unique treatment options. Patients with advanced GI cancer who reach disease control with a matched treatment seem to benefit substantially with respect to PFS and OS. The inclusion of additional complex diagnostic procedures and the integration of individualized tumor characterization at an earlier disease stage might substantively enhance the treatment opportunities in everyday clinical practice for these patients in the near future.
ACKNOWLEDGMENT
We thank Prof. Dr. rer. nat. Peter Martus for his statistical advice.
AUTHOR CONTRIBUTIONS
Conception and design: Michael Bitzer, Olaf Riess, Daniel Zips, Lars Zender, Ghazaleh Tabatabai, Nisar P. Malek
Financial support: Ghazaleh Tabatabai, Nisar P. Malek
Administrative support: Janina Beha, Nisar P. Malek
Provision of study material or patients: Michael Bitzer, Falko Fend
Collection and assembly of data: Michael Bitzer, Leonie Ostermann, Marius Horger, Saskia Biskup, Kristina Ruhm, Öznur Öner, Christopher Schroeder, Olaf Riess, Falko Fend, Daniel Zips, Martina Hinterleitner, Lars Zender, Janina Beha
Data analysis and interpretation: Michael Bitzer, Leonie Ostermann, Marius Horger, Saskia Biskup, Martin Schulze, Franz Hilke, Konstantin Nikolaou, Christopher Schroeder, Olaf Riess, Daniel Zips, Martina Hinterleitner, Lars Zender, Janina Beha, Nisar P. Malek
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Michael Bitzer
Consulting or Advisory Role: Bristol-Myers Squibb, Bayer Vital GmbH, EISAI, Ipsen, Lilly
Travel, Accommodations, Expenses: Ipsen, Celgene
Saskia Biskup
Employment: CeGaT
Stock and Other Ownership Interests: CeGaT
Martin Schulze
Employment: Praxis für Humangenetik Tuebingen
Franz Hilke
Honoraria: Agilent Technologies
Research Funding: Novartis (Inst)
Travel, Accommodations, Expenses: Agilent Technologies
Konstantin Nikolaou
Honoraria: Siemens Healthineers, Bayer Schering Pharma
Consulting or Advisory Role: Siemens Healthineers
Speakers' Bureau: Siemens Healthineers
Research Funding: Siemens Healthineers (Inst), Bayer Schering Pharma (Inst)
Travel, Accommodations, Expenses: Siemens Healthineers, Bayer Schering Pharma
Christopher Schroeder
Research Funding: Illumina (Inst), Novartis (Inst)
Olaf Riess
Honoraria: AstraZeneca, Takeda/Shire
Consulting or Advisory Role: Illumina
Research Funding: Illumina (Inst)
Falko Fend
Consulting or Advisory Role: Roche, EUSA Pharma
Daniel Zips
Research Funding: Elekta (Inst), Siemens (Inst), Sennewald (Inst)
Travel, Accommodations, Expenses: Elekta (Inst)
Martina Hinterleitner
Travel, Accommodations, Expenses: Novartis/Ipsen
Lars Zender
Leadership: HeparegeniX GmBH
Consulting or Advisory Role: Boehringer Ingelheim
Research Funding: HeparegeniX GmBH
Patents, Royalties, Other Intellectual Property: Patent on MKK4 Inhibition for the treatment of acute and chronic liver diseases
Travel, Accommodations, Expenses: Ipsen
Ghazaleh Tabatabai
Honoraria: AbbVie, Bayer, Medac, Novocure (Inst)
Consulting or Advisory Role: AbbVie, Bayer
Travel, Accommodations, Expenses: Novocure (Inst)
Nisar P. Malek
Honoraria: Spring Bank
Travel, Accommodations, Expenses: Falk Foundation
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hoefflin R, Geißler A-L, Fritsch R, et al. Personalized clinical decision making through implementation of a molecular tumor board: A German single-center experience. JCO Precision Oncol. 2018 doi: 10.1200/PO.18.00105. 10.1200/PO.18.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33:2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryce AH, Egan JB, Borad MJ, et al. Experience with precision genomics and tumor board, indicates frequent target identification, but barriers to delivery. Oncotarget. 2017;8:27145–27154. doi: 10.18632/oncotarget.16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. Erratum: Nat Med, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7:283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng DT, Prasad M, Chekaluk Y, et al. Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med Genomics. 2017;10:33. doi: 10.1186/s12920-017-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang KL, Mashl RJ, Wu Y, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. 2018;173:355–370.e14. doi: 10.1016/j.cell.2018.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e152. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lencioni R, Montal R, Torres F, et al. Objective response by mRECIST as a predictor and potential surrogate end-point of overall survival in advanced HCC. J Hepatol. 2017;66:1166–1172. doi: 10.1016/j.jhep.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. doi: 10.1038/gim.2016.190. Erratum: Genet Med, 2017. [DOI] [PubMed] [Google Scholar]
- 14.Schrader KA, Cheng DT, Joseph V, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104–111. doi: 10.1001/jamaoncol.2015.5208. Erratum: JAMA Oncol, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkins RW, Thummalapalli R, Carter J, et al. Molecular and genomic determinants of response to immune checkpoint inhibition in cancer. Annu Rev Med. 2018;69:333–347. doi: 10.1146/annurev-med-060116-022926. [DOI] [PubMed] [Google Scholar]
- 17.Hellmann MD, Callahan MK, Awad MM, et al. : Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 33:853-861.E4, 2018. [Erratum: Cancer Cell 35:329, 2019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao D, Margolis CA, Vokes NI, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet. 2018;50:1271–1281. doi: 10.1038/s41588-018-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.AlDubayan SH. Leveraging clinical tumor-profiling programs to achieve comprehensive germline-inclusive precision cancer medicine. JCO Precis Oncol. 2019 doi: 10.1200/PO.19.00108. 10.1200/PO.19.00108. [DOI] [PubMed] [Google Scholar]
- 23.Dumbrava EI, Brusco L, Daniels M, et al. Expanded analysis of secondary germline findings from matched tumor/normal sequencing identifies additional clinically significant mutations. JCO Precis Oncol . 2019 doi: 10.1200/PO.18.00143. 10.1200/PO.18.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571:576–579. doi: 10.1038/s41586-019-1382-1. Erratum: Nature, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thavaneswaran S, Rath E, Tucker K, et al. Therapeutic implications of germline genetic findings in cancer. Nat Rev Clin Oncol. 2019;16:386–396. doi: 10.1038/s41571-019-0179-3. Erratum: Nat Rev Clin Oncol, 2019. [DOI] [PubMed] [Google Scholar]
- 26.Ngeow J, Eng C. Precision medicine in heritable cancer: When somatic tumour testing and germline mutations meet. NPJ Genomic Med. 2016;1:15006. doi: 10.1038/npjgenmed.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garofalo A, Sholl L, Reardon B, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8:79. doi: 10.1186/s13073-016-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchhalter I, Rempel E, Endris V, et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int J Cancer. 2019;144:848–858. doi: 10.1002/ijc.31878. [DOI] [PubMed] [Google Scholar]
- 29.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fabrizio DA, George TJ, Jr, Dunne RF, et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol. 2018;9:610–617. doi: 10.21037/jgo.2018.05.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dienstmann R, Serpico D, Rodon J, et al. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther. 2012;11:2062–2071. doi: 10.1158/1535-7163.MCT-12-0290. [DOI] [PubMed] [Google Scholar]
- 32.Verlingue L, Malka D, Allorant A, et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial. Eur J Cancer. 2017;87:122–130. doi: 10.1016/j.ejca.2017.10.013. Erratum: Eur J Cancer 87:122-130, 2017. [DOI] [PubMed] [Google Scholar]
- 33.Valle JW, Lamarca A, Goyal L, et al. New horizons for precision medicine in biliary tract cancers. Cancer Discov. 2017;7:943–962. doi: 10.1158/2159-8290.CD-17-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roychowdhury S, Chinnaiyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin. 2016;66:75–88. doi: 10.3322/caac.21329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schütte M, Ogilvie LA, Rieke DT, et al. Cancer precision medicine: Why more is more and DNA is not Enough. Public Health Genomics. 2017;20:70–80. doi: 10.1159/000477157. [DOI] [PubMed] [Google Scholar]
- 36.Bitzer M, Malek NP. Personalized epigenetics. Med Epigenet. 2016:843–858. [Google Scholar]
- 37.Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109:513–522. doi: 10.1111/cas.13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17:557–569. doi: 10.1038/nrc.2017.58. [DOI] [PubMed] [Google Scholar]
- 39.Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat Med. 2019;25:744–750. doi: 10.1038/s41591-019-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 42.Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381:1632–1643. doi: 10.1056/NEJMoa1908075. [DOI] [PubMed] [Google Scholar]
- 43.Schork NJ. Personalized medicine: Time for one-person trials. Nature. 2015;520:609–611. doi: 10.1038/520609a. [DOI] [PubMed] [Google Scholar]