

Abstract
PURPOSE
Germline mutations in DNA repair (DR) genes and susceptibility genes CDKN2A and HOXB13 have previously been associated with prostate cancer (PC) incidence and/or progression. However, the role and prevalence of this class of mutations in metastatic PC (mPC) are not fully understood.
PATIENTS AND METHODS
To evaluate the frequency of pathogenic/likely pathogenic germline variants (PVs/LPVs) in men with mPC, this study sequenced 38 DR genes, CDKN2A, and HOXB13 in a predominantly white cohort of 317 patients with mPC. A PC registry at the University of Utah was used for patient sample acquisition and retrospective clinical data collection. Deep target sequencing allowed for germline and copy number variant analyses. Validated PVs/LPVs were integrated with clinical and demographic data for statistical correlation analyses.
RESULTS
All pathogenic variants were found in men self-reported as white, with a carrier frequency of 8.5% (DR genes, 7.3%; CDKN2A/HOXB13, 1.2%). Consistent with previous reports, mutations were most frequently identified in the breast cancer susceptibility gene BRCA2. It was also found that 50% of identified PVs/LPVs were categorized as founder mutations with European origins. Correlation analyses did not support a trend toward more advanced or earlier-onset disease in comparisons between carriers and noncarriers of deleterious DR or HOXB13 G84E mutations.
CONCLUSION
These findings demonstrate a lower prevalence of germline PVs/LPVs in an unselected, predominantly white mPC cohort than previously reported, which may have implications for the design of clinical trials testing targeted therapies. Larger studies in broad and diverse populations are needed to more accurately define the prevalence of germline mutations in men with mPC.
INTRODUCTION
Despite improvements in the diagnosis and treatment of prostate cancer (PC), it remains the most common cancer diagnosed in US men and is the second leading cause of cancer-related death.1 Major recognized risk factors for PC include increasing age, African heritage,2,3 and family history of PC.4 It was previously determined that PC risk is elevated in families with hereditary breast and ovarian cancer (HBOC) because of deleterious mutations in BRCA1 and BRCA2.5,6 Similarly, there is evidence of elevated PC risk in families with hereditary nonpolyposis colorectal cancer or Lynch syndrome.7,8
Although there is strong evidence for heritability in PC, few moderate to highly penetrant gene mutations that contribute to PC susceptibility have been identified. Linkage analysis and candidate gene studies led to the identification of a recurrent mutation (G84E) in the HOXB13 gene that increases risk of PC9,10 and contributes to approximately 5% of hereditary PCs worldwide.11 In a meta-analysis of US PC cases (n = 9,461) and controls (n = 5,039), HOXB13 G84E carriers had a statistically significantly increased risk of developing PC compared with noncarriers (odds ratio, 5.10; 95% CI, 3.21 to 8.10; P < .00001).12 Recent studies of men with PC have also frequently found a correlation between mutations in DNA repair (DR) genes and an elevated risk of development of metastatic PC (mPC) and/or lethal PC.13-15 In a study of 7 castration-resistant mPC (mCRPC) cohorts, presumed pathogenic germline variants were found highly enriched in DR genes, occurring at a frequency of 11.8%.14
In this study, we evaluated the prevalence and possible associated clinical phenotypes of pathogenic/likely pathogenic variants (PVs/LPVs) in DR genes, CDKN2A, and HOXB13 in 317 men treated for mPC. Metastatic disease characteristics in this cohort included both de novo mPC and mPC progression after diagnosed biochemical recurrence.
PATIENTS AND METHODS
Patient Cohort
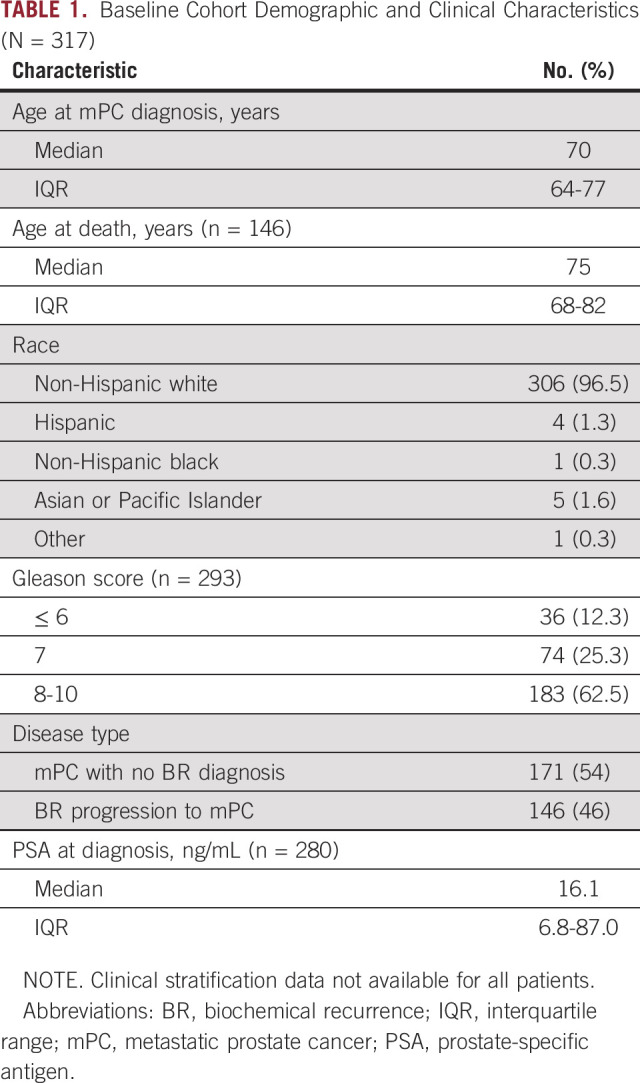
The study cohort comprised 317 men who were currently being treated for mPC at the University of Utah Huntsman Cancer Institute (n = 166) or who had been treated but had since died (n = 151). Cohort demographic and baseline clinical characteristics are summarized in Table 1. Patient selection was based on mPC disease state and was agnostic to age at diagnosis, ethnic background, and known familial predisposition to cancer. Clinical data for all patients in the cohort were extracted from the electronic medical record and included age at diagnosis, Gleason score, and baseline and follow-up laboratory results, including prostate-specific antigen (PSA) level. All aspects of the study were approved by the University of Utah Institutional Review Board. Because family history of cancer was not included in the patient clinical data, patients with mPC were identified in the Utah Population Database (UPDB) for available genealogy data.16
TABLE 1.
Baseline Cohort Demographic and Clinical Characteristics (N = 317)
Sequencing and Bioinformatic Processing
Genomic DNA extracted was from peripheral whole blood samples obtained from 317 patients with mPC. A custom QIAseq Targeted DNA Panel (QIAseq V3; Qiagen, Hilden, Germany) was designed for full exon and splice site coverage with or without 2 bp of 40 cancer-associated genes, including 38 DR genes, CDKN2A, and HOXB13 (Appendix Table A1). Next-generation sequencing libraries were built using the Qiagen GeneRead DNA I Amp Kit in accordance with manufacturer instructions. Targeted sequencing was performed on an Illumina HiSEquation 2500 (San Diego, CA) in accordance with Illumina standard protocol.
Sequencing results were aligned to GRCh37 and postprocessed following recommendations from GATK Best Practices.17 Fastq reads and alignments were processed with custom scripts18 for extracting and deduplicating alignments based on the QIAseq unique molecular indexes. Variants were called with GATK HaplotypeCaller and annotated using Ensembl Variant Effect Predictor.19 Mean target read depth coverage across all targets was approximately 350×, with the exception of ATR exons 31 to 34, which failed to amplify across all samples and were excluded from analysis.
To identify putative large indels (> 40 bp), per-amplicon mean read depth data were generated for all passing samples via alignment against amplicon bed file targets. Cross-sample, cross-target normalizations were performed, and normalized read depth data were reviewed for targets with ≥ 200× coverage for significant differences from mean target depth of coverage (standard deviation, ± 4). The copy number variation detection process was developed and executed internally.
Variant Classification and Validation
Preliminary selection of candidate PVs/LPVs included the following classification resources: American College of Medical Genetics and Genomics (ACMG) PVS1 null variants (nonsense, frameshift, splice site ± 2bp of exon boundaries, initiation codon, and single- or multiexon deletion),20 ClinVar classifications,21 and REVEL missense scores ≥ 0.5 (sensitivity, 76%; specificity, 89%).22 High- and moderate-penetrance variants with preliminary classification as PVs/LPVs were submitted to genetic counselors (GCs; W.K. and S.E.G.), who evaluated conflicting interpretations and identified the classification with the strongest clinical relevance. All verified PVs/LPVs occurred in genes included on the ACMG list and/or in the National Comprehensive Cancer Network guidelines on HBOC and colorectal cancer (version 1.2018; Table 2). Whether carrier status of these PVs/LPVs currently influences cancer preventative care and/or treatment is indicated in the first column of Table 2 (data provided by GCs). The Non-Finnish European (NFE) Genome Aggregation Database (gnomAD; groups contributing exome and genome variant data to this resource are provided online23) cohort24 was used for variant minor allele frequency (MAF) comparisons. Additional variants of interest (VOIs) were identified based on the classification pipeline used for the mPC PVs/LPVs and GC feedback. To assist with classification, VOIs were additionally annotated and stratified with scores generated through wAnnovar25 and BayesDel26 (if available). VOI retention required a deleterious LRT and/or FATHM score, an average GERP++/SiPhy conservation rank score > 0.75, and a BayesDel predicted deleterious score. The reported maximum allele frequency reflected the highest MAF found in gnomAD germline populations.24 Passing VOIs are listed in Appendix Table A2.
TABLE 2.
PVs/LPVs (n = 28) in Patients With mPC (n = 27)a

All reported PVs/LPVs were validated with Sanger sequencing using custom primers and Invitrogen Platinum Hot Start PCR Master Mix (Carlsbad, CA) for standard targets and Thermo Scientific Phusion Green HS High Fidelity PCR Master Mix (Waltham, MA) for high GC content (amplicon GC base content > 65%) and long-range amplicons > 5 kb.
Statistical Analysis
Prevalence and distribution of PVs/LPVs across the mPC cohort were analyzed for possible correlation between disease characteristics and treatment outcomes in general and in response to androgen-deprivation therapy (ADT). PV/LPV-carrier disease characteristics and clinical outcomes were compared against those of noncarrier patients to determine if underlying genotypes correlated with discrete phenotypes. Comparisons between the carrier and noncarrier groups included serum PSA levels at initial diagnosis using an unequal variance Welch t test, age at PC progression landmarks using an unpaired t test, and stratified Gleason score burden comparisons using a 2-sided Fisher’s exact test. A Pearson’s χ2 test was used to calculate Gleason score percentage overall P value. MAF P values between PV/LPV carriers and noncarriers were calculated using a 2-sided Fisher’s exact test. A P value < .05 (2 sided) was considered statistically significant. mPCa MAF significance evaluations were based in part based on The Cancer Genome Atlas Research Network control data.27
RESULTS
Identification of Deleterious Variants
To determine the frequency of PVs/LPVs in DR genes and susceptibility genes CDKN2A and HOXB13, we analyzed the complete coding and with or without 2 splice sites of these regions of interest in 317 patients with mPC. Overall, we identified 28 PVs/LPVs in 27 carriers (1 patient carried 2 PVs/LPVs), resulting in a carrier frequency of 8.5% (DR genes, 7.3%; CDKN2A/HOXB13 G84E, 1.2%; Table 2). HOXB13 G84E carrier frequency of 0.95% was not significantly enriched in the mPC cohort compared with the NFE genomAD carrier frequency of 0.49% (n = 64,053; P = .202).
All identified PVs/LPVs occurred in patients self-reported as white. The functional consequences of these mutations included 22 truncating alterations and 5 single-nucleotide polymorphisms. No copy number variants were validated in qualifying patients. Eighteen (64%) of the 28 total PVs/LPVs currently influenced PC preventative care and/or treatment. Of the genes that harbored deleterious mutations, BRCA2 and CHEK2 were the most represented, with overall patient carrier frequencies of 1.9% in BRCA2 and 1.6% in CHEK2 (Fig 1).
FIG 1.

Frequency distribution of genes containing pathogenic/likely pathogenic germline variants (PVs/LPVs). The other category represents genes for which number of PVs/LPVs is n = 1.
Additionally, there were 52 VOIs found across 23 sites that did not meet PV/LPV classification requirements but are provided in Appendix Table A2 for reference as possible risk factors. Appendix Table A2 includes a truncating nonsense BRCA2 variant (p.Lys3326Ter) with a carrier frequency of 2.5%.
Statistical Correlation Analyses
No statistically significant differences in baseline characteristics between PV/LPV carrier and noncarrier patients were observed (Appendix Table A3). Additionally, no significant correlation was found between DR PV/LPV status and response to ADT. In this retrospective mPC case study, in which men received ADT standard care that did not recommend quarterly scans, disease progression during ADT was determined based on PSA progression per Prostate Cancer Working Group 2 criteria, unless radiographic or clinical progression occurred before PSA progression (data not shown). Because of the inherent treatment heterogeneity within a retrospective disease state–based cohort, it is important to note that several patients in our mPC cohort were treated with PARP inhibitors during clinical trials and that 1 patient was treated with olaparib outside of a protocol and experienced clinically meaningful benefit.28 A cross-study comparison of PV/LPV carrier frequencies in DR genes ATM, BRCA1, BRCA2, MLH1, MSH2, MSH6, PALB2, and RAD51C in mPC cohorts is reviewed in Appendix Table A4, showing an overall PV/LPV rate of 6.3% (107 of 1691) in men with mPC.
Characterization of Founder Mutations
PVs/LPVs were searched against published founder mutations to further characterize the at-risk carrier patient population through the origins of the mutations. No founder mutations originating outside of Europe were identified, a finding reflective of the self-reported white ethnic background of 97% of the mPC cohort. Of the 28 total PVs/LPVs listed in Table 2, 50% are previously identified and published European founder mutations and include the pathogenic Ashkenazi Jewish founder mutation BRCA1 n.5382insC (Table 3).
TABLE 3.
Founder Mutations

DISCUSSION
In a cohort of men with mPC, predominantly of European ancestry, we observed that 23 (7.2%) of the 317 men harbored ≥ 1 pathogenic variants in DR genes. A direct comparison of the frequency of PVs/LPVs found in the 20 DR genes reported in the Pritchard et al14 mPC trial against the same 20 genes in our mPC cohort showed a lower occurrence in the mPC cohort (11.8% v 5.7%). The DR frequency observed in our mPC cohort was somewhat lower than frequencies reported by 4 comparable PC studies for the 8 DR genes in common across all of the groups (Appendix Table A4). However, the enrichment of rare DR germline mutations within our cohort was consistent with the hypothesis that mutations in DR genes increase mPC risk.
Two factors likely contributing to the lower germline DR mutation frequency in our study are the absence of enrichment for both known family history of cancer and Ashkenazi Jewish founder mutations in BRCA1 and BRCA2. Regarding family history of PC, prior studies have suggested that men with ≥ 1 first-degree relatives who have been diagnosed with a noncutaneous cancer are more likely to harbor deleterious DR mutations.14,29 UPDB genealogy data were not available for a majority of men in our cohort (189 [60%] of 317 patients), and just 63% of the men with available UPDB family history were identified as having ≥ 1 first-degree relatives with cancer; therefore, we suspect that our cohort was not highly enriched for this important risk factor. Additionally, although BRCA1 and BRCA2 Ashkenazi Jewish founder mutations are important risk factors for oncogenesis, the patient demographics in our mPC cohort reflect the primarily European ancestry of the local aging population, which makes the single Ashkenazi Jewish BRCA1 founder mutation occurrence in this cohort incidental to our findings. Examples of Ashkenazi Jewish elevated BRCA1 and BRCA2 founder mutation carrier frequencies across multiple types of cancer (eg, HBOC, PC, and pancreatic cancer) can be found in numerous publications, including elevated carrier frequencies identified in breast cancer cohorts both with (14.2%30) and without (10.3%31 and 11.7%32) a family history of cancer. BRCA1/BRCA2 founder mutation carrier frequencies in unselected Ashkenazi Jewish men with PC have been reported at 3.2% (n = 940) for BRCA2 only33 and a combined frequency of 5.2% (n = 251) for BRCA1 and BRCA2,34 a higher overall BRCA1/BRCA2 carrier frequency than identified in our mPC cohort (2.8% v 5.2%).
Known deleterious founder mutations were highly represented in our mPC cohort, representing 50% of the identified PVs/LPVs in the predominantly white mPC cohort (97%). The identified founder mutations were all of European origin. Knowledge of the commonality of DR and HOXB13 founder mutations in men with mPC has clinical utility, because coupling patient ethnic data with relevant founder mutations and locally enriched population risk factors may increase the success of preliminary germline testing.35 Importantly, clinically relevant penetrance, risk, and treatment efficacy data are likely to be enriched for founder mutations, because they have been the focus of multiple previous studies.
A particular Northern European founder mutation, HOXB13 G84E, has a strong correlation with increased lifetime risk of PC36 and poor prognosis in some studies.37 The HOXB13 gene is expressed in the prostate from early developmental stages into adulthood and has been found to affect prostate cell proliferation and differentiation,38,39 as well as androgen receptor regulation.40 To quantify family history impact on expected carrier rates, a meta-analysis of HOXB13 G84E frequencies in European ancestry PC cases was reviewed.12 It was found that the mutation occurred at a rate of 5.11% (n = 2,272) in PC cohorts with a family history10,11,41 compared with 0.95% (n = 8,917) in family history–nonselective PC cohorts (> 70% without family history10,42-44; P < .00001). The observed HOXB13 G84E carrier rate in this family history–unselective study was found to be in alignment with comparable PC studies. The prevalence of HOXB13 G84E in this mPC cohort is a reflection of the Northern European genetic background of many historical Utah settlers and may be an important PC risk factor in the local community. No trend toward early-onset disease in age at initial diagnosis or age at metastasis in G84E carriers was observed (data not shown).
It is also well known that deleterious germline mutations in DNA repair gene BRCA2 increase the risk of prostate oncogenesis. A particular BRCA2 C-terminus truncating mutation (p.Lys3326Ter) was validated in 8 patients with mPC (carrier frequency, 2.5%; MAF, 0.013), functionally resulting in the loss of the second of 2 nuclear localization signal (NLS) sequences within the C-terminal region. Although p.Lys3326Ter is a truncating mutation in BRCA2, it was not included as a PV/LPV in our report because of the current ClinVar classification for hereditary cancer as benign with 1 conflicting PV interpretation.45 One of the 4 ClinVar benign interpretations concluded that p.Lys3326Ter is a significant but low-risk allele for HBOC (Int. Genetics / Lab. Corp. of America). However, this mutation has been reported in recent publications to be a low- to moderate-risk variant that alters BRCA2 activity in multiple types of cancer.45-52 The role of this mutation as a risk modifier when coupled with a BRCA1 LPV/PV has also been previously explored; Raffaele et al53 classified this mutation as a powerful modifier in early-onset neoplasia when coupled with a BRCA1 PV, reporting increased pathogenicity in heterogeneous carriers. Interestingly, 1 of the BRCA2 p.Lys3326Ter mutations in the mPC cohort cooccurred with Ashkenazi Jewish founder mutation BRCA1 p.Gln1777ProfsTer74. Overall, the 2.5% carrier frequency of BRCA2 p.Lys3326Ter found in the mPC cohort is elevated compared with the genomAD NFE carrier frequency of 1.7% (n = 64,427). However, it is important to note that although BRCA2 p.Lys3326Ter was enriched in the mPC cohort, the estimated maximum MAF of individual PVs/LPVs occurring in BRCA2 is approximately 0.003,54 and the approximately 0.009 NFE gnomAD MAF is above that threshold. A consideration of these data, including an elevated mPC MAF and possible loss of functional efficiency via a diminishment of the NLS, supports a mild-penetrance risk classification for BRCA2 p.Lys3326Ter for hereditary cancer-predisposing syndrome.
This study has some limitations. First, because the cohort in this study was predominantly of European descent, complementary studies of non-European mPC cohorts would provide more comprehensive DR gene and HOXB13 distribution and prevalence estimates. Second, statistical comparisons of the clinical data of mPC PV/LPV carriers versus mPC noncarriers were confounded in part by the low number of PV/LPV carriers present (n = 27) and the high degree of heterogeneity of patient disease characteristics and treatment courses. The underlying genotypic and phenotypic diversity in this disease highlights the value of large patient cohorts and more expansive genomic interrogations. Larger-scale analyses generate more comprehensive statistics that translate to more robust mPC preventative care and treatment.
In conclusion, our study provides supporting evidence that deleterious germline mutations in DR genes and HOXB13 predispose men with European ancestry to mPC. Although the DR mutation frequency was lower than the initial report (11.8% by Pritchard et al14), it was consistent with other more recent studies (Appendix Table A4).14,29,55,56 The combined carrier frequency in the 8 genes shared across these comparable studies (ATM, BRCA1, BRCA2, MLH1, MSH2, MSH6, PALB2, and RAD51C) is 6.3% (107 of 1,691), decreased from the initial report (8.7%). These findings have implications for the design of clinical trials in men with mPC. The TOPARP trial, a phase II clinical trial evaluating olaparib in men with mCRPC, showed that men with deleterious mutations in a DR gene had high response rates (14 [88%] of 16) to PARP inhibitors.57 Currently, 4 separate phase II or III clinical trials are evaluating PARP inhibitors in men with mCRPC (ClinicalTrials.gov identifiers: NCT02854436, NCT02975934, NCT02952534, and NCT02952534). Most of the trials evaluating PARP inhibitors in mCRPC are designed using the frequency of DR PVs/LPVs published as observed in a hormone-sensitive mPC/mCRPC cohort.14 In contrast, our cohort of predominately European descent had a lower frequency of PVs/LPVs at 5.7% in the same 20 genes evaluated by Pritchard et al.14 If investigators designing a trial overestimate the frequency of deleterious DR mutations, it could lead to methodologic issues that may unexpectedly influence the results of the trial. Overestimation of enrollment could result in an underpowered study at time of closure or a trial that is unable to enroll a sufficient number of patients. Moving forward, our results suggest that investigators evaluating novel therapies in men with deleterious DR mutations should consider the population structure of study sites when designing clinical trials.
ACKNOWLEDGMENT
We thank the Genome Aggregation Database and all groups that provided exome and genome variant data to this resource; the TCGA Research Network, control data from which was used for metastatic prostate cancer minor allele frequency significance evaluations; Scott Tomlins, MD, PhD, for assisting with gene target selection; the University of Utah Center for Clinical and Translational Science (funded by National Institutes of Health Clinical and Translational Science Awards); the Pedigree and Population Resource; and the University of Utah Information Technology Services and Biomedical Informatics Core for establishing the Master Subject Index between the Utah Population Database, the University of Utah Health Sciences Center, and Intermountain Health Care.
APPENDIX
TABLE A1.
QiaSeq V3 Custom Gene Panel With 38 DR Genes, CDKN2A, and HOXB13
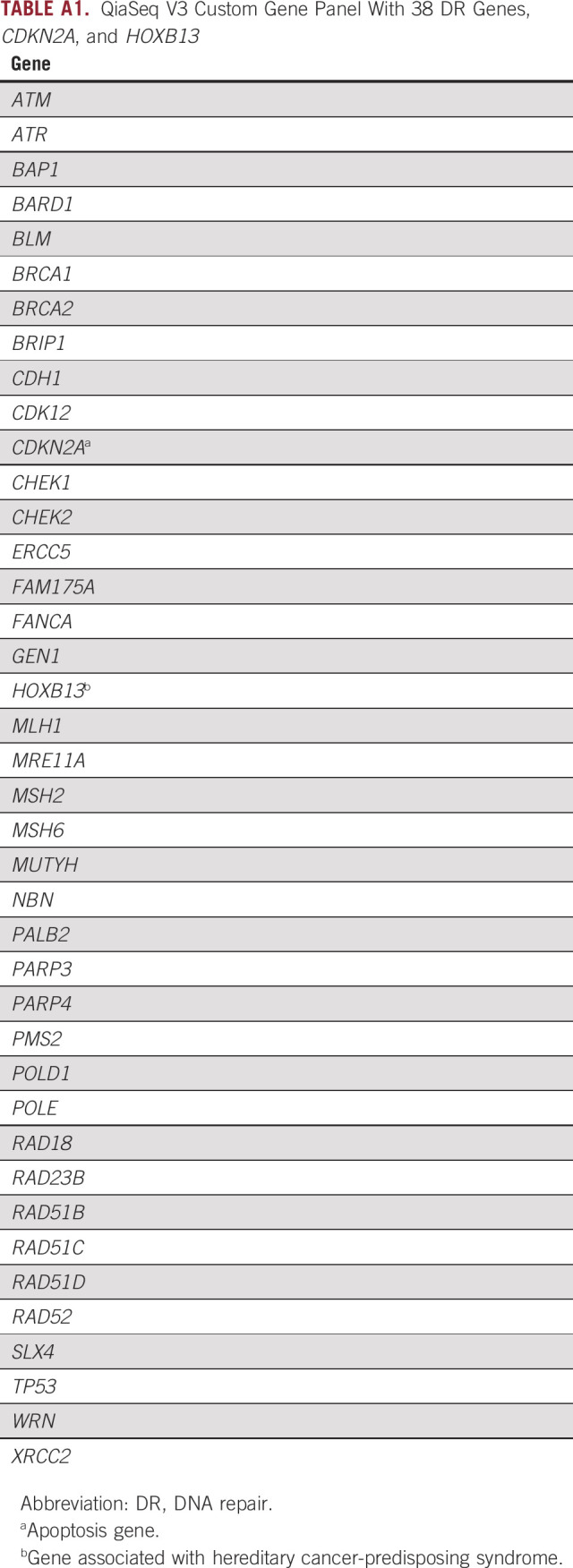
TABLE A2.
Rare VOIs (n = 23) in Patients With mPC (n = 49)

TABLE A3.
Comparison of Baseline Characteristics Between Clinically Actionable PV/LPV Carriers and Noncarrier Patients (N = 317)
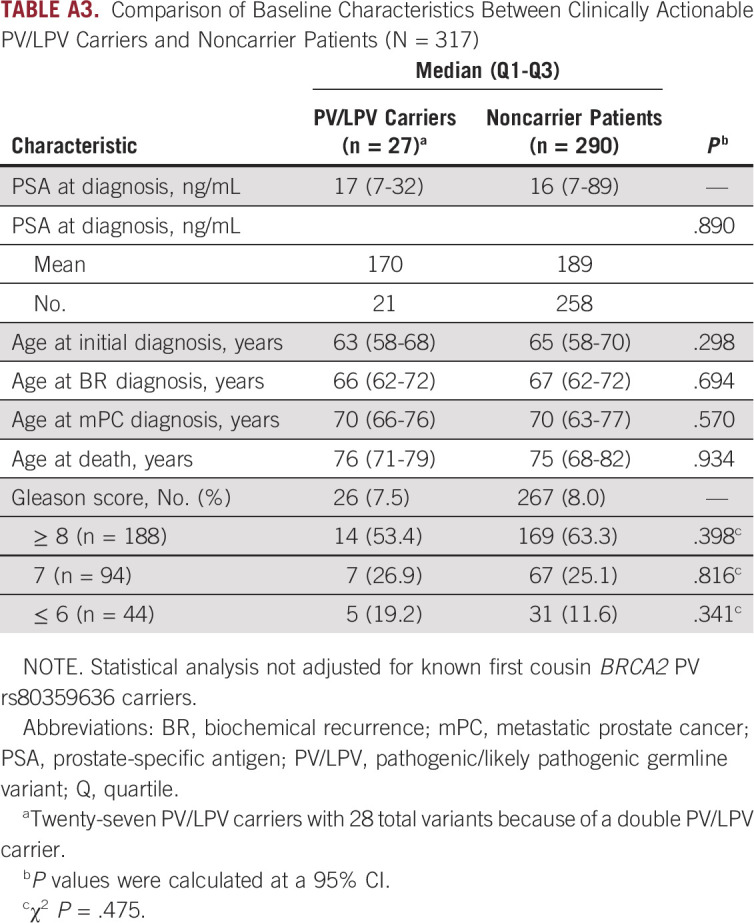
TABLE A4.
Comparison of DR Gene Mutations Among Studies With Comparable Cohort Demographics

Footnotes
Supported by National Institutes of Health Grant No. P30CA042014 awarded to the Huntsman Cancer Institute for the Genetic Counseling and High-Throughput Genomics and Bioinformatics Analysis Shared Resource and generous donors; by the Huntsman Cancer Foundation and Department of Defense Grant No. PC170413 (L.C.-A.); by the University of Utah, the Huntsman Cancer Institute, and Huntsman Cancer Institute Cancer Center Support Grant No. P30 CA42014 from the National Cancer Institute in partial support for all data sets within the Utah Population Database; and in part by AstraZeneca.
AUTHOR CONTRIBUTIONS
Conception and design: Neeraj Agarwal, Kathleen A. Cooney
Financial support: Neeraj Agarwal, Kathleen A. Cooney
Administrative support: Ashley L. Kapron, Kathleen A. Cooney
Provision of study materials or patients: Andrew W. Hahn, Benjamin L. Maughan, Lisa Cannon-Albright, Neeraj Agarwal
Collection and assembly of data: Julie L. Boyle, Andrew W. Hahn, Ashley L. Kapron, Wendy Kohlman, Samantha E. Greenberg, Timothy J. Parnell, Benjamin L. Maughan, Lisa Cannon-Albright, Neeraj Agarwal, Kathleen A. Cooney
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Wendy Kohlmann
Employment: BioFire Diagnostics (I)
Benjamin L. Maughan
Consulting or Advisory Role: Janssen Oncology, Exelixis, Tempus, Peleton, Bristol-Myers Squibb, Astellas Medivation, Bayer
Research Funding: Clovis Oncology (Inst), Bristol-Myers Squibb (Inst), Bavarian Nordic (Inst)
Travel, Accommodations, Expenses: Exelixis
Bing-Jian Feng
Research Funding: Pfizer (Inst), Regeneron (Inst)
Patents, Royalties, Other Intellectual Property: Inventor of PERCH software, which has been nonexclusively licensed to Ambry Genetics for clinical genetic testing service and research.
Neeraj Agarwal
Consulting or Advisory Role: Pfizer, Astellas Medivation, Bristol-Myers Squibb, AstraZeneca, Nektar, Eli Lilly, Bayer, Foundation One, Pharmacyclics, Foundation Medicine, Astellas Pharma, Exelixis, Janssen Oncology, Merck, Novartis
Research Funding: Bayer (Inst), Bristol-Myers Squibb (Inst), GlaxoSmithKline (Inst), Takeda Pharmaceutical (Inst), Novartis (Inst), Pfizer (Inst), BN ImmunoTherapeutics (Inst), Exelixis (Inst), TRACON Pharma (Inst), Rexahn Pharmaceuticals (Inst), Amgen (Inst), AstraZeneca (Inst), Active Biotech (Inst), Bavarian Nordic (Inst), Calithera Biosciences (Inst), Celldex (Inst), Eisai (Inst), Genentech (Inst), Immunomedics (Inst), Janssen (Inst), Merck (Inst), Newlink Genetics (Inst), Prometheus (Inst), Sanofi (Inst)
Kathleen A. Cooney
Patents, Royalties, Other Intellectual Property: Patent awarded for discovery of HOXB13 as prostate cancer susceptibility gene (Inst).
Travel, Accommodations, Expenses: Boston Scientific (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Altekruse SF, Huang L, Cucinelli JE, et al. Spatial patterns of localized-stage prostate cancer incidence among white and black men in the southeastern United States, 1999-2001. Cancer Epidemiol Biomarkers Prev. 2010;19:1460–1467. doi: 10.1158/1055-9965.EPI-09-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeSantis CE, Siegel RL, Sauer AG, et al. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin. 2016;66:290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 4.Albright F, Stephenson RA, Agarwal N, et al. Prostate cancer risk prediction based on complete prostate cancer family history. Prostate. 2015;75:390–398. doi: 10.1002/pros.22925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemminki K, Chen B. Familial association of prostate cancer with other cancers in the Swedish Family-Cancer Database. Prostate. 2005;65:188–194. doi: 10.1002/pros.20284. [DOI] [PubMed] [Google Scholar]
- 6.Leongamornlert D, Mahmud N, Tymrakiewicz M, et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer. 2012;106:1697–1701. doi: 10.1038/bjc.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raymond VM, Mukherjee B, Wang F, et al. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013;31:1713–1718. doi: 10.1200/JCO.2012.44.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haraldsdottir S, Hampel H, Wei L, et al. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16:553–557. doi: 10.1038/gim.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlsson R, Aly M, Clements M, et al. A population-based assessment of germline HOXB13 G84E mutation and prostate cancer risk. Eur Urol. 2014;65:169–176. doi: 10.1016/j.eururo.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 10.Ewing CM, Ray AM, Lange EM, et al. Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med. 2012;366:141–149. doi: 10.1056/NEJMoa1110000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Lange EM, Lu L, et al. HOXB13 is a susceptibility gene for prostate cancer: Results from the International Consortium for Prostate Cancer Genetics (ICPCG) Hum Genet. 2013;132:5–14. doi: 10.1007/s00439-012-1229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shang Z, Zhu S, Zhang H, et al. Germline homeobox B13 (HOXB13) G84E mutation and prostate cancer risk in European descendants: A meta-analysis of 24,213 cases and 73, 631 controls. Eur Urol. 2013;64:173–176. doi: 10.1016/j.eururo.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Dong X, Wang L, Taniguchi K, et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003;72:270–280. doi: 10.1086/346094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. doi: 10.1038/s41391-018-0086-1. Marshall CH, Fu W, Wang H, et al: Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: Association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis 22:59-65, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. University of Utah Health, Huntsman Cancer Institute: Huntsman Cancer Institute research. http://www.huntsmancancer.org/research/shared-resources/utah-population-database/data.
- 17. Van der Auwera GA, Carneiro MO, Hartl C, et al: From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 43:11.10.1-11.10.33, 2013. [DOI] [PMC free article] [PubMed]
- 18. Huntsman Cancer Institute: Unique molecular index scripts. https://github.com/HuntsmanCancerInstitute/UMIScripts.
- 19.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landrum MJ, Lee JM, Benson M, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Genome Aggregation Database: About gnomAD. http://gnomad.broadinstitute.org/about.
- 24.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang X, Wang K. wANNOVAR: Annotating genetic variants for personal genomes via the Web. J Med Genet. 2012;49:433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng BJ. PERCH: A unified framework for disease gene prioritization. Hum Mutat. 2017;38:243–251. doi: 10.1002/humu.23158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. National Cancer Institute: The Cancer Genome Atlas program. http://cancergenome.nih.gov.
- 28.Rathi N, Anderson N, Greenberg S, et al. DNA damage repair (DDR) mutations and the utility of high-risk genetics clinics in metastatic castration-refractory prostate cancer (mCRPC) World J Oncol. 2018;9:119–122. doi: 10.14740/wjon1144w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leongamornlert D, Saunders E, Dadaev T, et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. Br J Cancer. 2014;110:1663–1672. doi: 10.1038/bjc.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stadler ZK, Salo-Mullen E, Patil SM, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012;118:493–499. doi: 10.1002/cncr.26191. [DOI] [PubMed] [Google Scholar]
- 31.Walsh T, Mandell JB, Norquist BM, et al. Genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi Jewish women. JAMA Oncol. 2017;3:1647–1653. doi: 10.1001/jamaoncol.2017.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warner E, Foulkes W, Goodwin P, et al. Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst. 1999;91:1241–1247. doi: 10.1093/jnci/91.14.1241. [DOI] [PubMed] [Google Scholar]
- 33.Giusti RM, Rutter JL, Duray PH, et al. A twofold increase in BRCA mutation related prostate cancer among Ashkenazi Israelis is not associated with distinctive histopathology. J Med Genet. 2003;40:787–792. doi: 10.1136/jmg.40.10.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchhoff T, Kauff ND, Mitra N, et al. BRCA mutations and risk of prostate cancer in Ashkenazi Jews. Clin Cancer Res. 2004;10:2918–2921. doi: 10.1158/1078-0432.ccr-03-0604. [DOI] [PubMed] [Google Scholar]
- 35.Neuhausen SL. Founder populations and their uses for breast cancer genetics. Breast Cancer Res. 2000;2:77–81. doi: 10.1186/bcr36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. doi: 10.1371/journal.pgen.1004930. Hoffmann TJ, Sakoda LC, Shen L, et al: Imputation of the rare HOXB13 G84E mutation and cancer risk in a large population-based cohort. PLoS Genet 11:e1004930, 2015 [Errata: PLoS Genet 11:e1005114, 2015; PLoS Genet 11:e1005362, 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Xiao L, Qin Z, et al. Association between germline homeobox B13 (HOXB13) G84E allele and prostate cancer susceptibility: A meta-analysis and trial sequential analysis. Oncotarget. 2016;7:67101–67110. doi: 10.18632/oncotarget.11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooney KA, Beebe-Dimmer JL. HOXB13 mutations and prostate cancer risk. BJU Int. 2016;118:496–497. doi: 10.1111/bju.13477. [DOI] [PubMed] [Google Scholar]
- 39.Beebe-Dimmer JL, Hathcock M, Yee C, et al. The HOXB13 G84E mutation is associated with an increased risk for prostate cancer and other malignancies. Cancer Epidemiol Biomarkers Prev. 2015;24:1366–1372. doi: 10.1158/1055-9965.EPI-15-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10:361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 41.Breyer JP, Avritt TG, McReynolds KM, et al. Confirmation of the HOXB13 G84E germline mutation in familial prostate cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:1348–1353. doi: 10.1158/1055-9965.EPI-12-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gudmundsson J, Sulem P, Gudbjartsson DF, et al. A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat Genet. 2012;44:1326–1329. doi: 10.1038/ng.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stott-Miller M, Karyadi DM, Smith T, et al. HOXB13 mutations in a population-based, case-control study of prostate cancer. Prostate. 2013;73:634–641. doi: 10.1002/pros.22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akbari MR, Trachtenberg J, Lee J, et al. Association between germline HOXB13 G84E mutation and risk of prostate cancer. J Natl Cancer Inst. 2012;104:1260–1262. doi: 10.1093/jnci/djs288. [DOI] [PubMed] [Google Scholar]
- 45.Meeks HD, Song H, Michailidou K, et al. BRCA2 polymorphic stop codon K3326X and the risk of breast, prostate, and ovarian cancers. J Natl Cancer Inst. 2015;108:djv315. doi: 10.1093/jnci/djv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson ER, Gorringe KL, Rowley SM, et al. Reevaluation of the BRCA2 truncating allele c.9976A > T (p.Lys3326Ter) in a familial breast cancer context. Sci Rep. 2015;5:14800. doi: 10.1038/srep14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimelis H, Mesman RLS, Von Nicolai C, et al. BRCA2 hypomorphic missense variants confer moderate risks of breast cancer. Cancer Res. 2017;77:2789–2799. doi: 10.1158/0008-5472.CAN-16-2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge Y, Wang Y, Shao W, et al. Rare variants in BRCA2 and CHEK2 are associated with the risk of urinary tract cancers. Sci Rep. 2016;6:33542. doi: 10.1038/srep33542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Michailidou K, Hall P, Gonzalez-Neira A, et al: Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 45:353-361, 361e351-361e352, 2013. [DOI] [PMC free article] [PubMed]
- 50. doi: 10.1038/ng.3002. Wang Y, McKay JD, Rafnar T, et al: Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet 46:736-741, 2014 [Erratum: Nat Genet 49:651, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akbari MR, Malekzadeh R, Nasrollahzadeh D, et al. Germline BRCA2 mutations and the risk of esophageal squamous cell carcinoma. Oncogene. 2008;27:1290–1296. doi: 10.1038/sj.onc.1210739. [DOI] [PubMed] [Google Scholar]
- 52.Martin ST, Matsubayashi H, Rogers CD, et al. Increased prevalence of the BRCA2 polymorphic stop codon K3326X among individuals with familial pancreatic cancer. Oncogene. 2005;24:3652–3656. doi: 10.1038/sj.onc.1208411. [DOI] [PubMed] [Google Scholar]
- 53.Palmirotta R, Lovero D, Stucci LS, et al. Double heterozygosity for BRCA1 pathogenic variant and BRCA2 polymorphic stop codon K3326X: A case report in a southern Italian family. Int J Mol Sci. 2018;19:E285. doi: 10.3390/ijms19010285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song W, Gardner SA, Hovhannisyan H, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: Insights of relevance to variant classification. Genet Med. 2016;18:850–854. doi: 10.1038/gim.2015.180. [DOI] [PubMed] [Google Scholar]
- 55.Annala M, Struss WJ, Warner EW, et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur Urol. 2017;72:34–42. doi: 10.1016/j.eururo.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 56.Antonarakis ES, Lu C, Luber B, et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur Urol. 2018;74:218–225. doi: 10.1016/j.eururo.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hauke RJ, Jr, Sissung TM, Figg WD. Discussing the predictive, prognostic, and therapeutic value of germline DNA-repair gene mutations in metastatic prostate cancer patients. Cancer Biol Ther. 2017;18:545–546. doi: 10.1080/15384047.2017.1345398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sokolenko AP, Iyevleva AG, Preobrazhenskaya EV, et al. High prevalence and breast cancer predisposing role of the BLM c.1642 C>T (Q548X) mutation in Russia. Int J Cancer. 2012;130:2867–2873. doi: 10.1002/ijc.26342. [DOI] [PubMed] [Google Scholar]
- 59.Suspitsin EN, Sibgatullina FI, Lyazina LV, et al. First two cases of Bloom syndrome in Russia: Lack of skin manifestations in a BLM c.1642C>T (p.Q548X) homozygote as a likely cause of underdiagnosis. Mol Syndromol. 2017;8:103–106. doi: 10.1159/000454820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamel N, Feng BJ, Foretova L, et al. On the origin and diffusion of BRCA1 c.5266dupC (5382insC) in European populations. Eur J Hum Genet. 2011;19:300–306. doi: 10.1038/ejhg.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dørum A, Møller P, Kamsteeg EJ, et al. A BRCA1 founder mutation, identified with haplotype analysis, allowing genotype/phenotype determination and predictive testing. Eur J Cancer. 1997;33:2390–2392. doi: 10.1016/s0959-8049(97)00328-6. [DOI] [PubMed] [Google Scholar]
- 62.Cybulski C, Huzarski T, Górski B, et al. A novel founder CHEK2 mutation is associated with increased prostate cancer risk. Cancer Res. 2004;64:2677–2679. doi: 10.1158/0008-5472.can-04-0341. [DOI] [PubMed] [Google Scholar]
- 63.Chen Z, Greenwood C, Isaacs WB, et al. The G84E mutation of HOXB13 is associated with increased risk for prostate cancer: Results from the REDUCE trial. Carcinogenesis. 2013;34:1260–1264. doi: 10.1093/carcin/bgt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aretz S, Tricarico R, Papi L, et al. MUTYH-associated polyposis (MAP): Evidence for the origin of the common European mutations p.Tyr179Cys and p.Gly396Asp by founder events. Eur J Hum Genet. 2014;22:923–929. doi: 10.1038/ejhg.2012.309. [DOI] [PMC free article] [PubMed] [Google Scholar]