

Graphical abstract
Abbreviations: ACE2, Angiotensin-converting enzyme 2; AD, Alzheimer’s disease; ADP, Adenosine diphosphate; aIIb/b3, Glycoprotein IIb/IIIa; ARDS, Acute respiratory distress syndrome; ARE, Antioxidant response elements; ASC, Apoptosis-associated speck-like protein containing a CARD; ATP, Adenosine triphosphate; BALF, Bronchoalveolar lavage; bZIP, Basic leucine zipper; CATs, Catalase; CBP, CREB binding protein; COVID-19, Coronavirus disease 19; CREB, cAMP response element-binding protein; Cul3, Cullin 3; DAMP, Death associated molecular pattern; G-CSF, Granulocyte colony stimulating factor; GPx, Glutathione peroxidase; GSH, Intracellular glutathione; Gsk-3, Glycogen synthase kinase-3; HCV, Hepatitis C virus; HDAC3, Histone deacetylase 3; HO-1, Heme Oxygenase-1; IFN-γ, Interferon-gamma; IKK, IkB Kinase; IL-6, Interleukin -6; IRAKs, Interleukin (IL)- 1R-associated kinase; IκB, Inhibitor of kappa B; Keap1, Kelch-like ECH associated protein 1; LiCl, Lithium chloride; MCP-1, Monocyte chemoattractant peptide; MIP1α, Macrophage inflammatory protein 1α; Myd88, Myeloid differentiation primary response 88; NAD(P)H, Nicotinamide adenine dinucleotide phosphate hydrogen; NF-κB, Nuclear factor-κB; NLRP3, NOD-like receptors protein 3; NLRP3, Nucleotide-binding domain (NOD)-like receptor protein 3; NOX, NADPH oxidase; N-protein, Nucleocapsid protein; Nrf2, Nuclear factor erythroid 2-related factor; NTD, N-terminal domain; O.−2, Superoxide anion; O2, Oxygen molecule; OxPLs, Oxidized phospholipids; PAMP, Pathogen associated molecular pattern; PAR, Protease-activated receptors; PD, Parkinson’s disease; PEDV, Porcine epidemic diarrhea virus; PPRPattern, recognition receptor; PSGL, P-Selectin glycoprotein ligand-1; RIG-I, Retinoic acid-inducible gene I; ROS, Reactive oxygen species; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; sgmRNA, Sub genomic messenger RNA; SODs, Superoxide dismutase; TAK1, Transforming growth factor-β (TGF-β)-activated kinase 1; TF, Tissue factor; TIRAP, TIR-domain-containing adaptor protein; TLR-3, Toll like receptor-3; TNF, Tumor necrosis factor; TNF-R, Tumor necrosis factor receptor; TNFα, Tumour necrosis factor-alpha; TRAF6, Tumour necrosis factor receptor associated factor 6; TRS, Transcription regulating sequence; vWF, von Willebrand factor; XO, Xanthine oxidase; XOR, Xanthine oxidoreductase
Keywords: COVID-19, Gsk-3, NF-κB, Nucleocapsid protein, Oxidative stress, SARS-CoV-2
Abstract
The coronavirus disease 19 (COVID-19) outbreak caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) had turned out to be highly pathogenic and transmittable. Researchers throughout the globe are still struggling to understand this strain's aggressiveness in search of putative therapies for its control. Crosstalk between oxidative stress and systemic inflammation seems to support the progression of the infection. Glycogen synthase kinase-3 (Gsk-3) is a conserved serine/threonine kinase that mainly participates in cell proliferation, development, stress, and inflammation in humans. Nucleocapsid protein of SARS-CoV-2 is an important structural protein responsible for viral replication and interferes with the host defence mechanism by the help of Gsk-3 protein. The viral infected cells show activated Gsk-3 protein that degrades the Nuclear factor erythroid 2-related factor (Nrf2) protein, resulting in excessive oxidative stress. Activated Gsk-3 also modulates CREB-DNA activity, phosphorylates NF-κB, and degrades β-catenin, thus provokes systemic inflammation. Interaction between these two pathophysiological events, oxidative stress, and inflammation enhance mucous secretion, coagulation cascade, and hypoxia, which ultimately leads to multiple organs failure, resulting in the death of the infected patient. The present review aims to highlight the pathogenic role of Gsk-3 in viral replication, initiation of oxidative stress, and inflammation during SARS-CoV-2 infection. The review also summarizes the potential Gsk-3 pathway modulators as putative therapeutic interventions in combating the COVID-19 pandemic.
1. Introduction
In late December 2019, Wuhan, China, got attention worldwide after getting several patients diagnosed with pneumonia, following a viral infection. On 11th February 2020, the pathogenic strain of the virus was taxonomically designated as "Severe Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)", by the International Committee on Taxonomy of Viruses (ICTV). The associated diseased condition was termed COVID-19 by the World Health Organization (WHO) [1]. The WHO announced SARS-CoV-2 virus infection a pandemic as it infected nearly 9 million persons, and engulfed more than 460,000 worldwide [2]. SARS-CoV-2 is a member of β coronaviruses, consisting of ∼30 kb single-stranded positive-sense RNA as genetic material [3]. It shows 79 % genetic similarity between another human coronavirus, i.e., SARS-CoV, while 98 % similarity with bat coronavirus RaTG1 [4] and shares a high similarity index with pangolin coronavirus [5]. Respiratory droplets are the primary source of viral transmission either through nasopharyngeal or oral route. Dry cough and high fever are the significant symptoms observed in patients within 11–15 days following viral infection. The disease pathophysiology of COVID-19 also shows a close resemblance with previous reported SARS-CoV-associated respiratory disease.
The majority of respiratory viral infections are associated with the recruitment of immune cells, the release of pro-inflammatory cytokines, oxidative stress, and finally, phagocytosis of the infected cells [6]. However, in the case of SARS-CoV-2 infection, aggressive inflammation and oxidative stress help in viral replication and damage the airway epithelial cell that results in acute respiratory distress syndrome (ARDS), which makes the condition worst. Glycogen synthase kinase-3 (Gsk-3) is a serine/threonine evolutionary conserved central molecule that mainly participates in cell proliferation, migration, development, apoptosis, and immune regulation (acquired and innate). Activation of Gsk-3 is associated with suppression of host immunity and inhibition of antioxidant response [7]. It is also supports viral genome replication within the host cell in disease progression [8]. The present review provides an in-depth knowledge of oxidative stress, inflammation, and viral replication related to Gsk-3 during SARS-CoV-2 infection. Further, the review highlights the Gsk-3 pathway modulators' putative role as therapeutic interventions in combating the COVID-19 pandemic.
2. Gsk-3 structure
Gsk-3 is a versatile serine/threonine kinase that regulates glycogen metabolism. It consists of two isoforms, Gsk-3α, and Gsk-3β, encoded by two separate genes [9]. Both the isoforms share 98 % sequence similarity between kinase domains, despite they never compensate for each other's loss of function. Gsk-3 has two prime functional domains, a substrate-binding domain, which acquires substrates to Gsk-3, while the other kinase domain is responsible for phosphorylation of the substrate [10]. The N-terminal region of Gsk-3 contains ATP binding domain, whereas the C-terminal region consists of a large conserved "activation loop" responsible for the enzyme's full activation [7,11]. Activation of Gsk-3 depends on the site-specific phosphorylation that is controlled by various kinases [7]. Gsk-3 prefers pre-phosphorylate substrate by recognizing consequence sequences S/T-X-X-X-phospho-S/T on substrate [12]. Gsk-3 is also involved in Wnt/β-catenin and Sonic hedgehog cell signalling pathways mediating in cell proliferation, differentiation, maturation, and cell adhesion [13]. Transcription factors (c-Jun, CREB, STAT3, C/EBPα, NFAT, myc, NF-κB, and p53) are the major substrate of Gsk-3 that can manipulate the expression of several other genes [14,10]. Impaired activity of Gsk-3 has recognized in several clinical conditions, such as Metabolic disorders, Cancers, Alzheimer's disease (AD), Parkinson's Disease (PD), Bipolar disorders, and various other neurodegenerative diseases [13,15].
3. SARS-CoV-2 infection and inflammation
The severity of symptoms and death in SARS-CoV-2 infected patients depends on the viral infection and is greatly affected by the aggressive behaviour of the host immune system. COVID-19 patients' systemic cytokine profile shows a close resemblance with cytokine release syndrome, characterized by macrophage activation, an elevated level of cytokines like tumour necrosis factor-alpha (TNFα), interleukin-6 (IL-6) and interferon-gamma (IFN-γ) [16]. Further, elevated levels of these cytokines trigger ARDS, characterized by a low level of oxygen in blood and difficulty in breathing, leading to the death of the infected patients [17]. Previous data on SARS-CoV demonstrated that the virus predominately affects the endothelial cells of the airway, alveoli, vascular system, and macrophages in the pulmonary organ. SARS-CoV-2 employs the same host receptor angiotensin-converting enzyme 2 (ACE2) for infection, like SARS-CoV, indicating that both the viruses target the same set of cells for infection [18]. The expression of the ACE2 receptor is reduced in the lungs following SARS-CoV infection [19], disrupting the renin-angiotensin system that affects fluid/electrolyte balance, blood pressure, increases the vascular permeability and inflammation in the airway.
In counter-defence, the virus encodes numerous immune-suppressive proteins that help it to evade from host immune response [20] and helps in replication. Similarly, to counter such problem, SARS-CoV-2 evolves with numerous structural and non-structural proteins that act as an antagonist of interferon signalling. Interruptions in interferon signalling happened at various stages, (1) preventing the recognition of viral RNA through pattern recognition receptor (PPR), (2) inhibiting the synthesis of type I interferon protein via interrupting the toll-like receptor-1 (TLR-1) and retinoic acid-inducible gene I (RIG-I) signalling, (3) disturbing STAT-1 signalling [21] and (4) initiating the host mRNA degradation and interrupting host translation machinery [22] (Fig. 1 ).
Fig. 1.

Mechanism of inflammation during SARS-CoV-2 infection.
SARS-CoV-2 invade host cells with angiotensin-converting enzyme2 (ACE2) receptors present on the cell surface membrane. Virus nucleocapsid (N) protein hijack the host interferon type-1 signalling for the successful replication, transcription, and new viral progeny. Infected host cells undergo pyroptosis and release numerous proinflammatory cytokines (IL-6, IL-2, and TNFα) DAMPs and PAMPs. These free contents recognized by the neighboring cells and proceed with the same process of pyroptosis by using the NLRP3 receptor and also trigger the proinflammatory cytokines synthesis by TLR4-Nf-κB pathway. Monocytes (transform later into macrophage) and T-lymphocytes are attracted to the infected site and promote further pro-inflammation events, resulting in cytokine storm damage to the lung and other organs in the body.
TLR4: Toll like receptor 4; Myd88: Myeloid differentiation primary response 88; IL-1β: Interleukin 1 beta; IL-18: Interleukin 18; DAMPs: Death- associated molecular pattern; PAMPs: Pathogen-associated molecular pattern; NLRP3: Nucleotide-binding domain (NOD)-like receptor protein 3; ASC: Apoptosis-associated speck-like protein containing a CARD; IL-6: Interleukin 6; IL-2: Interleukin 2; TNFα: Tumor necrosis factor alpha; STAT: Signal transducer and activator of transcription; JAK: Janus kinase.
At the time of replication, cytopathic viruses, including SARS-CoV-2, show a massive death and injury of the infected epithelial and endothelial cells, triggering the excessive release of cytokines and chemokines [18]. In addition to this, inflammation-induced cell death-pyroptosis also observed in SARS-CoV-2 patients that further provoke the systemic inflammatory response. Pyroptosis signalling proceeds via NOD-like receptors protein 3 (NLRP3) present on the cell membrane, activate caspase-1 through ASC (Apoptosis-associated speck-like protein containing a caspase recruitment domain) adaptor protein. Activated caspase-1 further triggers the synthesis of proinflammatory cytokines such as IL-1β and IL-6 (Fig. 1) [23]. These cytokines further attract the other immune cells, mostly T-lymphocytes and monocytes, at the site of infection [24,25]. Bronchoalveolar lavage (BALF) fluid from the sever COVID-19 patients showed CCL2 and CCL7 chemokines, which require the recruitment of CCR2+ monocytes [26]. Further, BALF analysis also revealed a highly inflammatory population of monocyte-derived FCN1+ macrophage [27]. In addition to these responses, sever cases of SARS-CoV-2 infection also disclose a significant expansion in the population of proinflammatory monocytes CD14+ and CD16+ in the peripheral blood, as compared to mild cases [28,29].
Around 80 % of SARS-CoV-2 infected patients show lymphopenia-infiltration of lymphocyte and immune cells' requirement at the site of infection [30,31]. In most of the patients, these recruited cells clear the infection recedes the inflammatory response, and leads to recovery. However, some patients show cytokine storms because of an imbalance in the immune cascade that further inflames the lungs. SARS-CoV-2 infected patients also show alleviation in the T cell population, which is more noticeable in severe cases. The level of helper T cell (CD4+), cytotoxic T cell (CD8+), and regulatory T cell were below the average level in severe cases of COVID-19, as compared to mild cases [31]. CD8+ T cells directly attack and kill the virus-infected cells, while CD4+ participates in the production of cytokines to recruit other immune cells. At the same time, regulatory T cell maintains the normal immune homeostasis, along with inhibition of proliferation, the proinflammatory activity of maximum lymphocytes, natural killer cells, and B-cells (Fig. 1).
Severe hospitalized COVID-19 patients' blood plasma exhibits a higher level of granulocyte colony-stimulating factor (G-CSF), IL-2, IL-6, IL-10, monocyte chemoattractant peptide (MCP)-1), macrophage inflammatory protein 1α (MIP1α) and TNFα [32]. The blood plasma of the infected patients shows a significantly higher level of IL-6 in severe cases compared to mild or non-severe cases, which further contributes to macrophage activation syndrome [31]. Pulmonary infiltration-based assessment in ARDS patients also revealed that a more significant portion of lung injury is associated with a higher level of IL-6 in peripheral blood [33]. All of these evidences suggest that SARS-CoV-2 infection is responsible for dysregulation of the host immune system with the abnormal synthesis of cytokines, chemokines, and a decrease in the level of lymphocytes that ultimately leads to cytokine storm responsible for multi-organ failure.
3.1. Role of Nuclear factor-κB in disease progression
Nuclear factor-κB (NF-κB) is the leading player that responds immediately following the pathogen's invasion by promoting inflammation, controlling cell proliferation, and survival. NF-κB is a heterodimeric transcription factor that belongs to the Rel protein family. There are 05Rel proteins present in mammalian cells that further divided into two classes. RelA (p65), RelB, and c-Rel are grouped in first-class, characterized by the presence of transactivation domain, while NF-kB1 (p50) and NF-kB2 (p52) belongs to the second group that is devoid of transcriptional-modulation activity. So, both the classes of proteins need to be heterodimerized with each other to perform their functions. Under normal physiological conditions, RelA and p50, the heterodimer's predominant form is inactivated in the cytoplasm by IkB protein [34]. A pathogenic stimulus provoked by a bacteria or a virus invasion, exposure of mitogen, proinflammatory cytokines, growth factors, and stress, activates IkB Kinase (IKK), which further phosphorylates and degrades IkB protein via ubiquitination process [7].
Excessive cytokine release (especially IL-6) plays a crucial role in SARS-CoV-2 infection and further progression of pathogenic conditions [35,36]. NF-κB is a transcription factor that controls the expression of proinflammatory genes responsible for the cytokine storm. A study suggested that the nucleocapsid protein of SARS-CoV directly interacts with NF-κB, translocate it to the nucleus, and finally upregulates IL-6 gene expression [37]. Several studies indicated that SARS-CoV directly or indirectly activates NF-κB protein, following infection [[38], [39], [40]]. NF-κB also activated by receptors present on the cell surface membrane-like, Toll-like receptor 4 (TLR 4). Pathogen associated molecular pattern (PAMP) and Death associated molecular pattern (DAMP) are inflammatory, stimulating molecules released by virus-infected cells, which act as ligands for TLR 4, subsequently activating NF-κB protein via MyD88-dependent pathway [7]. Oxidative stress is another important factor responsible for cytokine storm generation via cross-talk between Nuclear factor erythroid 2-related factor (Nrf2) and NF-κB pathway. NF-κB suggested as a negative regulator of Nrf2 driven genes, either by recruiting histone deacetylase 3 (HDAC3), which promote local histone hypoacetylation or deprive the CBP (CREB binding protein) (Fig. 1) [7,41].
4. SARS-CoV-2 infection and oxidative stress
Oxygen is a crucial molecule in the aerobic system to maintain normal life processes. Under normal cellular conditions, the oxygen molecule utilized to generate chemical energy in the form of ATP in a very tight and controlled manner. The oxygen molecule combustion generates a small number of reactive oxygen species (ROS), which utilized for usual cell signalling cascades. ROS are oxygen molecules with an unpaired electron that behaves as free radicals and reactive metabolites. Several ROS forms were discovered so far, such as peroxidase, oxygen-free radicals, nitrogen oxide, and singlet oxygen molecules [7]. Generally, ROS associated cellular damage is processed via sophisticated antioxidant machinery, involving both enzymatic (Catalase (CATs), Superoxide dismutase (SODs), and glutathione peroxidase (GPx) and non-enzymatic (Glutathione and nicotinamide adenine dinucleotide phosphate hydrogen [NAD(P)H] mechanisms [7]. In normal physiological conditions, the antioxidant systems can work simultaneously to combat the exceeded levels of ROS. However, in a pathological state, ROS overwhelmed the antioxidant mechanism and generated “oxidative stress” in cells. All the crucial cellular components such as proteins, lipids, nuclear, and mitochondrial DNA get degraded under the influence of oxidative stress, subsequently triggering the process of cell death.
The available literature of clinical and preclinical experiments proposed that oxidative burst is another prompting factor for mortality following SARS-CoV infection [42,43]. SARS-CoV-2 infection activates the host airway epithelium and alveolar macrophage, further releasing cytokines to attract another immune cell from the blood (Neutrophils and monocyte that further differentiate into macrophage) at the site of injury. Recruitment of these cells ensures the clearance of the virus, but due to imbalanced host immune system, they also start to release excessive cytokines that further aggravate to cytokine storm. The recruited phagocytic cell participates in ROS generation, along with inflammatory response [43]. Nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidase) and xanthine oxidase (XO) are the two well-known enzymes responsible for oxidative stress in respiratory viral infections [44]. NADPH oxidase (NOX) is an evolutionary conserved, membrane-bounded enzyme complex that catalyzes the molecular oxygen into superoxide [44]. Human’s NADPH oxidase family consists of 7 members, NOX1−5, DUOX1, and DUOX2 [45]. Its C-terminal region comprises NADPH binding site, flavin adenine dinucleotide binding domain, while the N-terminal region consists of 6 transmembrane α helical domains, with four conserved heme-binding sites [45]. NOX2 is predominantly expressed in the recruited phagocytes (neutrophils and macrophages) at the viral infection site and contributes to oxidative stress [46]. A study reported that alveolar macrophage depended oxidative stress is responsible for acute lung injury progression following H5N1 viral infection in mice, mostly via oxidized phospholipid and superoxide. However, the same pathological events reduced following the suppression of p47phox, a regulatory subunit of NOX2 [19]. In a study, Influenza A virus-infected NOX2-/y mice showed reduced oxidative stress, improved alveoli epithelium condition, less production of superoxide, and reduced airway inflammation compared to wild type mice (Fig. 2 ) [46].
Fig. 2.

Possible mechanism of oxidative stress in COVID-19 patients following SARS-CoV-2 infection.
Proinflammatory cytokines and chemokines are released by the SARS-CoV-2 infected cells, to attract the phagocytes. Airway epithelium and phagocytic cells (Neutrophils and Monocyte derive macrophage) carry different isoforms of NOXs, which trigger the generation of superoxide ions during cellular stress condition and directly participate in oxidative stress. Xanthine oxidoreductase is also transformed into xanthine oxidase -during stressed cellular environments and triggers ROS generation by metabolizing hypoxanthine. Tumour necrosis factor (TNF) also enhances oxidative stress by initiating the NF-κB pathway and inhibiting the complex II of the electron transport chain.
TNF: Tumor necrosis factor; TNF-R: Tumor necrosis factor receptor; ATP: Adenosine triphosphate; XOR: Xanthine oxidoreductase; XO: Xanthine oxidase; ARE: Antioxidant Response Element; Nf-κB: Nuclear Factor kappa-light-chain-enhancer of activated B cells; O2: Oxygen molecule; O.−2: Superoxide anion.
Similarly, ex-vivo influenza A virus-infected alveolar macrophage exhibited an increase in superoxide synthesis via NOX2 enzyme complex [47]. Xanthine oxidase (XO) is another ROS generating enzyme that participates in oxidative stress, following respiratory viral infection. In the mammalian system, this enzyme is existing in interchangeable form between XO to Xanthine oxidoreductase (XOR) [7]. XOR is predominantly distributed in healthy tissues and reduces NAD+ to NADH by utilizing electron form substrate. While during inflammation, XOR is converted into XO by oxidation of cysteine amino acid or calcium-dependent proteolysis. XO shows more affinity toward molecular oxygen, resulting in the transfer of a univalent and divalent electron to oxygen that further generates superoxide and hydrogen peroxide, respectively (Fig. 2) [48]. In-vitro rhino virus’s infection in primary bronchial and A549 respiratory epithelial cell lines decreased the intracellular glutathione (GSH) level, leading to oxidative stress via enhanced superoxide production. Serine protease inhibitor or XO inhibitor, oxypurinol treatment enhanced the intracellular levels of GSH, and decreased superoxide generation, thus revealed that XO also participates in oxidative stress during infection [49].In-vivo analysis also revealed that XO is the main contributor to superoxide synthesis during a respiratory viral infection. Mice infected with influenza virus showed a higher mortality rate, which found to be associated with XO and superoxide in BALF and serum analysis. However, allopurinol and chemical modified superoxide dismutase decreased the oxidative stress and mortality rate [50]. These evidences revealed that XO also participates in the viral associated disease progression via oxidative stress. A part of these activated phagocyte releases pro-oxidant mediators such as TNF and IL-1, which further enhances the oxidative stress in host cells during viral infection. TNF binds with the complex II of the mitochondrial respiratory chain, hampering oxidative phosphorylation via restricting electrons transport. As a result, the electron transport chain becomes leakier, and lastly, it enhances superoxide production [51]. TNF also helps in detachment of Nf-κB protein from IKB complex, resulting in suppression of antioxidant gene expression via binding to their promoter region following translocation from the cytoplasm to the nucleus [52] (Fig. 2). During stress condition, neutrophils also release lactoferrin along with lysosomal proteins under the influence of IL-1, which further binds to iron and start to accumulate in the reticuloendothelial system. When an iron-binding threshold reached, superoxide ions combine with free iron to generate hydroxyl radicals via Fenton reaction and enhances oxidative stress [53,54].
4.1. Nrf2 a key regulator of antioxidant genes
Nrf2 is the main transcription factor that plays an important role to overcome oxidative stress. It is a basic leucine zipper (bZIP) protein that belongs to the cap ‘n’ collar family of transcription factors. Nrf2 consist of 6 highly conserved functional domain termed as Neh 1–6 (Nrf2-ECH homologies 1–6). Neh1 is a leucine zipper domain through which Nrf2 interact with other transcription factors, whereas Neh2 is the Kelch-like ECH associated protein 1(Keap1) binding domain [55]. Keap1 is a cystine rich and cytoplasmic protein, whose N-terminal domain binds with Cul3-dependent E3 ubiquitin ligase complex, while C-terminal domain binds with Nrf2 protein. Under normal physiological conditions, Keap1 protein ubiquitinates the Nrf2 resulting in its proteasomal degradation [55].
However, during stress conditions Nrf2 detached from the Keap1 protein, translocate to the nucleus, heterodimerize with small musculoaponeurotic fibrosarcoma (MAFs) proteins, and finally initiates or suppresses the transcription of genes that consists of electrophile response elements (ERE) or antioxidant response elements (ARE) in their promoters [7]. Nrf2 regulates more than 500 genes expression belonging to oxidative stress, inflammation, autophagy, metabolism and excretion [55]. The pulmonary system is more exposed to oxidative stress because of its highly vascular nature and indirect contact with environmental oxidant, which had already proven in numerous respiratory diseases. It was found that, lung-specific Nrf2 conditional knockout rodents showed pulmonary protective behaviour in respiratory disorders [56].
5. Systemic oxidative stress and inflammation linked thrombus formation in SARS-CoV-2 infection
Abnormal coagulation, a higher level of D-dimers, and low platelet count are the signs of poor prognosis and significant reasons for multiple organ failure and death in severe cases of COVID-19 [57,58]. Microthrombus had reported in the lungs, the heart, the kidneys, and the brain of COVID-19 patients [59,60]. Cytokine storm induces aberrant coagulation by expressing the tissue factor (TF) pathway [61,62]. TF is a member of cytokine receptor superfamily and type I integral membrane glycoprotein, which is highly abundant in the vasculature sub-endothelium, especially in the brain, lungs, gut, skin, as well as in the monocytes. In response to proinflammatory cytokines, especially IL-6, the expression of TF is upregulated in the monocytes and the perivascular cells, resulting in TF exposure to circulation [63,64]. The exposed portion of TF forms a complex with circulating factor VII, thus enhance its catalytic activity that further activates downstream circulating factors, such as IX and X. Activated factor X participates in the transformation of prothrombin into thrombin, that finally leads to the formation of blood clots by conversion of fibrinogen into fibrin [65,66] (Fig. 3 ).
Fig. 3.

Mechanism of hypercoagulation in COVID-19 patients following SARS-CoV-2 infection.
During systemic inflammation, circulating pro-inflammatory cytokines can activate and triggers the expression of tissue factor (TF) on the circulating monocytes and endothelial cells. Exposed TF form complex with circulating factor VII (TF-VII complex) and initiate the coagulation pathways by triggering the downstream process. TF-VII complex also activates the platelets via the PAR signalling pathway and helps in binding on the endothelial cells via von Willebrand factor-aIIb/b3receptors interaction, resulting in platelets aggregation. Neutrophils also bind with the activated endothelial and release the neutrophil extracellular trap, which activates the coagulation cascade, binds with the platelets, and further enhances the coagulation pathway. Activated endothelial cells also synthesize and express monocyte chemoattractant and cell adhesion molecules. Activated monocyte recruited on the endothelial, release pro-inflammatory cytokines and monocyte–derived microvesicles, which further provoke the coagulation process.
DAMPs: Death- associated molecular pattern; PAMPs: Pathogen-associated molecular pattern; TLR4: Toll like receptor 4; Nf-κB: Nuclear Factor kappa-light-chain-enhancer of activated B cells; IL-6: Interleukin 6; PAR: Protease-activated receptor; PSGL: P-Selectin glycoprotein ligand-1; ADP: Adenosine diphosphate; TF: Tissue factor; vWF: von Willebrand factor; aIIb/b3: Glycoprotein IIb/IIIa.
Platelet activation, different proinflammatory events, and fibrin clot formation are the main consequences of the cytokine storm that also provokes thrombin production via protease-activated receptors (PAR) signalling pathway. PAR is a unique G-protein coupled cell surface receptor that carries its ligand and remains inactive until unmasked by proteolytic cleavage by the TF-factor-VIIa complex [67]. Thrombin mediated PAR activated platelets undergo a morphological transformation, release platelet activators such as serotonin, adenosine diphosphate (ADP), thromboxane A2, translocate cell adhesion molecule P-selectin and CD40 ligand on the surface of platelet along with activation of the integrin aIIb/b3 receptor [67]. The released thromboxane A2 and ADP trigger activation of neighbouring platelets via thromboxane receptor and P2Y12, respectively [68]. Activated aIIb/b3 on platelets' surface binds with von Willebrand factor (vWF) and fibrinogen that contributes to platelet aggregation [67]. Activated endothelial cells following PAR signalling also exposes cell adhesion molecules (E-selectin, P-selectin, ICAM-1 and VCAM-1) and expresses monocyte chemo attractant protein-I, that facilitates recruitment, adhesion, and migration of leukocytes and platelets during viral infection [69]. Adherent leukocyte-platelets interaction provides positive feedback to amplify the overall inflammatory response and pro-coagulation events [69]. These events are prothrombotic, which further contributes to blood clot formation (Fig. 3).
Along with cytokine storm, oxidized phospholipids (OxPLs) also participate in the coagulation cascade via the TLR-4 receptors present on the monocytes and endothelial cells. OxPLs concerned as PAMs patterns that are recognized by numerous conserved pattern-recognition receptors. In an experimental model of acute lung injury, OxPLs triggered cytokine storm release via TLR4-TRIF-TRAF6-NF-κB pathway. IL-6 further promoted TF expression on monocytes and activated the endothelial cell to express monocyte adherent protein for their requirement, which finally participated in inflammatory events [19]. Thrombotic complications can be reduced in pre-existing metabolic and cardiovascular disorders in COVID-19 patients by interfering with the OxPLs activated monocyte or endothelial cell [36]. Additionally, during inflammation, the natural anticoagulant pathways such as TF pathway inhibitors or antithrombin are nearly diminished, subsequently facilitating coagulation cascade [69].
6. Gsk-3 and SARS-CoV-2 infection
The virus has to undergo many complex processes that are tightly regulated to infect a host cell. It begins with viral genomic RNA entry into the host cytosol, transcription, and finally budding off as viral progeny. These viral progenies are similar to their parent in morphology and function that consists of 4 structural proteins, spike (S) protein, envelop (E) protein, matrix (M) protein and nucleocapsid (N) protein [70]. The N protein of severe acute respiratory syndrome coronavirus is the most abundant protein existing in an infected host cell among all other proteins.
Protein sequencing also revealed that N protein is highly conserved among the species, which consists of 3 domains, including the N-terminal domain (NTD/domain 1), C-terminal domain (CTD/domain 3) and linker region (LKR/domain 2). N-terminal domain enriched with positive charge amino acids, which is responsible for binding with viral RNA. Whereas, C-terminal domain mapped between 373 and 390 amino acids is enriched with lysin, thought to be responsible for nuclear localization signal [71]. The C-terminal domain of the N protein is also responsible for protein dimerization. Both the domains of N protein i.e., domain 1 and domain 3 are linked to each other through linker region (domain 2) that consists of serine-arginine motif, responsible for the multimerization of the N-protein [72] and predicted as a hot spot region for phosphorylation. In brief, N-protein divided into three main domains that play diverse functions during different stages of the virus life cycle. N-protein is a type of capsid protein whose primary function is to pack the virus's genomic RNA into the protective covering.
Interestingly, to perform such activity, N-protein should recognize the viral genomic RNA, associate with it, and finally, oligomerize by self-association to form capsid or nucleocapsid. Protection of viral genome, timely replication, and proper transmission is the capsid's primary function [71]. N-protein also inhibits host cell proliferation and cytokines by interacting with elongation factors 1α, resulting in a halt of translation mechanism [73]. Moreover, the N-protein of SARS-CoV also hijacks the host innate immune system for the progress of new viral progeny and associated disease development. Post-translational phosphorylation of the virus N-protein is essential for their activity, which results in an increased affinity of N-protein toward virus RNA rather than non-viral RNA [74]. Gsk-3 found to be an essential kinase responsible for the phosphorylation of N-protein on the serine residue in linker-region [74] (Fig. 4 ). SARS-CoV infected VeroE6 cells showed an 85 % reduction in viral titer and cytopathic effects following the treatment of Gsk-3 inhibitor kenpaullone and lithium chloride, thus suggested phosphorylation by this kinase be strongly linked with the viral replication [74]. Several sub-genomic mRNAs synthesized due to discontinued transcription mechanisms during the coronavirus replication, which encodes major structural proteins [75]. Transcription regulating sequence (TRS) responsible for the discontinuous transcription process exists in front of each gene (body TRS) and after the leader sequence (leader TRS) [76]. Template-switching events happen via base pairing between the body TRS and leader TRS to synthesize the discontinuous minus-stranded RNAs. Discontinuous nested plus-strand sub-genomic mRNA transcribed from the previously generated minus-stranded RNAs [77]. This discontinuous transcription mechanism tightly controlled for the successful compilation of the virus life cycle. Among all the structural proteins, N-protein tightly regulates the discontinued transcription mechanism as the synthesis of sub-genomic mRNA is reduced following the N segment's deletion from the replicon [78]. The studies showed that phosphorylated N-protein at ser197 via Gsk-3 interacts with the DDX1 RNA helicase to participate in discontinues to a continuous process of virus replication [8]. Moreover, phosphorylation of N-protein at the serine-arginine motif also inhibits the translational machinery of the host cell [79] and interferes with antiviral response signalling. All this evidence suggested that Gsk-3 protein plays a crucial role in the virus life cycle and interrupts host-defence mechanisms for the infection progression.
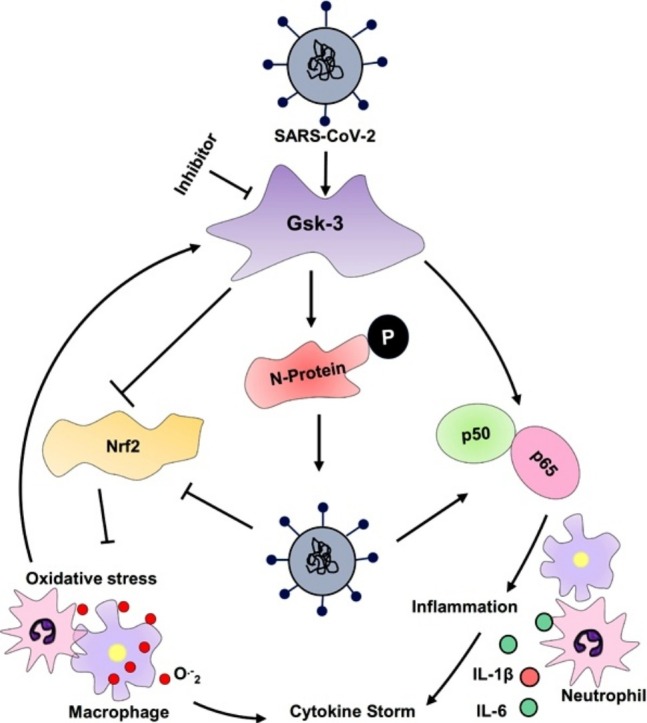
Fig. 4.

Putative role of Gsk-3 in the SARS-CoV-2 replication and COVID-19 pathogenesis.
Activated Gsk-3 phosphorylate the nucleocapsid protein of SARS-CoV-2 and support viral genome replication and transcription. Simultaneously, activated Gsk-3 also phosphorylate and degrade the significant oxidative stress combating transcription factor Nrf2, resulting outburst of different reactive oxygen species. Circulating DAMPs and PAMPs bind with TLR4 and stimulate the pro-inflammatory cytokine synthesis via Myd88 dependent pathway. β-catenin is a downstream protein of Gsk-3, phosphorylate the p50 sub-unit of NfκB, altered their DNA binding ability, and suppresses inflammatory events. CREB is another transcription factor that initiates the expression of anti-inflammatory genes to overcome the disease condition. During virus infection, activate Gsk-3 degrade the β-catenin protein and phosphorylate the CREB, consequential, lost CBP binding site, and enhances pro-inflammatory cytokine synthesis, especially IL-6. LiCl and Tideglusib are the two well-known non-selective metal cation and non - ATP competitive inhibitors of Gsk-3.
SARS-CoV-2: Severe acute respiratory syndrome coronavirus 2; N-protein: Nucleocapsid protein; sgmRNA: Subgenomic messenger RNA; DAMPs: Death- associated molecular pattern; oxPls: Oxidized phospholipids; TLR: Toll like receptor; Gsk-3: Glycogen synthase kinase; Keap1: Kelch-like ECH-associated protein 1; Nrf2: nuclear factor erythroid 2–related factor 2; Cul3: Cullin 3; CREB: cAMP response element-binding protein; CBP: CREB binding proteins; ARE: Antioxidant Response Element References; Myd88: Myeloid differentiation factor 88; IRAKs: Interleukin (IL)- 1R-associated kinase; TRAF6: Tumour necrosis factor receptor associated factor 6; TAK1: Transforming growth factor-β (TGF-β)-activated kinase 1; IκB: Inhibitor of kappa B; TIRAP: TIR-domain-containing adaptor protein; LiCl: Lithium chloride.
As described, ROS's overproduction and alteration in antioxidant mechanisms are mainly responsible factors for the viral replication and development of associated conditions [6]. Nrf2 is a transcription factor responsible for combating the oxidative stress by upregulating antioxidant genes that consist of ARE in their promoter [7]. The respiratory syncytial virus-infected Nrf2 knockout mice showed severe infection and increased viral titer, enhanced mucous secretion, inflammation, and epithelium injury compared to the wild type mice [80]. Besides, influenza-infected Nrf2 knockout mice, following cigarette smoking, could not combat oxidative stress and showed higher mortality compared to wild-type strain due to higher secretion of mucous and peri bronchial inflammation [80]. Similarly, the direct correlation of oxidative stress markers with SARS-CoV infection is remarkably observed in various animal model systems [80]. Gsk-3β, a direct upstream regulator of Nrf2 found to be active in numerous virus-infected cells. It also participates in disease progression by supporting the replication process or inhibiting the host defence mechanisms [81]. Active Gsk-3β phosphorylates Nrf2 on serine reside of Neh6 domain that finally gets degraded by cullin-1/Rbx1-mediated ubiquitination process, and provokes oxidative stress [7]. HO-1 expression was also significantly upregulated following Gsk-3β inhibition in Huh 7.5.1 hepatocytes infected with JFH1 hepatitis C virus (HCV) [82]. These literature findings strongly support the role of Gsk-3 in oxidative stress following viral infection (Fig. 4).
Gsk-3 involvement is not only restricted up to oxidative stress, but it also provokes systemic inflammation for disease progression by synthesizing the pro-inflammatory molecules such as IL-6, IL-1β, IL-18, IFN-γ and TNF-α [81]. Gsk-3β mainly participates in inflammatory reactions via TLRs [83]. TLRs are a type of pattern recognition receptors that responds to PAMs released by the virus-infected cell. Corona virus-infected organism shows a significant upregulation of OxPLs, which act as PAMs. OxPLs induces the production of pro-inflammatory cytokines by translocation of NF-κB from the cytosol to the nucleus via TLR4–TRIF–TRAF6 pathway [6]. β-catenin protein, a direct downstream target of Gsk-3β, forms a complex with both the units of NF-κB, alters its DNA binding activity that ultimately inhibits inflammatory cascade. Activated Gsk-3β phosphorylates β-catenin protein for proteasomal degradation that directly promotes the inflammatory events [84]. In a study, it was found that Gsk-3β directly phosphorylated P65 unit of NF-κB at ser276, enhanced CBP-P65 interaction, followed by transcription of pro-inflammatory gene expression [85]. Activated Gsk-3β also reduced CREB-DNA interaction via phosphorylation at Ser129 residue on CREB protein that resulted in NF-κB binding with CBP, which enhanced the expression of IL-1β and TNF-α and reduced IL-10 [7] (Fig. 4).
7. Gsk-3 pathway modulators and SARS-CoV-2 infection
Gsk-3 shows a remarkable impact on numerous physiological processes and plays a crucial role in the pathogenesis of various clinical conditions. Lithium is a non-selective metal cation most frequently used as a Gsk-3 inhibitor to treat patients diagnosed with mood and bipolar disorders (Fig. 4) [81]. A study found that lithium chloride (LiCl) treatment helped overcome the transmissible gastroenteritis coronavirus infection in porcine [86]. Similarly, replication and entry of porcine epidemic diarrhea virus (PEDV) in the Vero cells also reduce following the LiCl treatment [87]. LiCl treatment also protects from the coronavirus infectious bronchitis virus in avian by suppressing the synthesis of genomic RNA and sub genomic mRNA without affecting the host translation mechanisms [88]. To date, the interaction of lithium carbonate with the recently exercised antiviral drugs (viz. ribavirin, ritonavir, and lopinavir) employed against SARS-CoV-2 has not been revealed [89]. Tideglusib is another non-ATP competitive inhibitor belonging to the thiadiazolidinone class of drugs, characterized by irreversible Gsk-3 inhibition, which initially designed to treat Alzheimer's disease (AD) (Fig. 4). Treatment of tideglusib improved the process of adult neurogenesis via enhancing the cascade of proliferation in the stem cell pool, differentiation, migration, and maturation both in in-vitro and in-vivo studies [90].
Moreover, thiadiazolidinone treatment in the double transgenic mouse model of AD showed improved memory formation via preventing hyperphosphorylation of tau protein, reactive astrogliosis, and reduced Aβ plaque disposition via modulating the CREB signalling [91,92]. A study also showed potent antiviral activity of thiadiazolidinone against COVID-19 in cell-based assay [93]. Safety and efficacy of tideglusib already proved in phase II clinical trial in AD patients [94,95]. In a randomized trial, tideglusib treatment also presented an improved condition in the patients who have progressive supranuclear palsy via reducing the atrophy in the brain's different regions [96]. Clinical trial II/III for tideglusib announced in 2020 after evaluating the pharmacokinetic activity, safety, and efficacy in the myotonic dystrophy affected patients in 2018. Although these indirect and direct targeted effects could prove to be the potential success of GSK-3 inhibition in reference for the treatment of COVID-19, yet all the risks must be keenly and precisely understood before the administration of any treatment.
8. Conclusion
This review provided a direct link of Gsk-3 with SARS-CoV-2 infection, involving regulation of SARS-CoV-2 genome transcription and defeating host defence mechanisms. Hyperinflammation and oxidative stress are the two significant factors responsible for the development of ARDS in COVID-19 patients. Activated Gsk-3 directly phosphorylates nucleocapsid protein of the virus that further helps in viral progeny formation. Simultaneously, activated Gsk-3 supports NF-κB signalling, resulting in oxidative burst and cytokine storm. In the present scenario, the strategies employed for the management of COVID-19 targets either pathogen or host, but has low succeeding rate as both the events are transpiring synchronously.
Moreover, the virus has a very high mutation rate, while vaccines are designed only against specific strains, increasing the chances of failure. Alternately, targeting Gsk-3 may be a better option as it is an evolutionarily conserved protein, and is involved in host-pathogen interaction. Its many indirect inhibitors used to cure other pathological conditions, while direct inhibitors are in clinical trials. In order to overcome the difficult circumstances of the COVID-19 outbreak, Gsk-3 apprehended as a striking target. In the process of curbing the pandemic, Gsk-3 can act as a potent therapeutic attribute. Thus necessary efforts must be taken to devise comprehensive and in-depth research towards this target.
Acknowledgements
The authors are thankful to the Director, CSIR-IHBT, Palampur (HP), for his support. The financial support of CSIR, New Delhi, in the form of project MLP0204 is highly acknowledged. Mr. Anil Kumar Rana is grateful to the Department of Science and Technology for providing DST-INSPIRE fellowship vide letter no: DST/INSPIRE fellowship/[IF160224]. Mr. Shubham Nilkanth Rahmatkar is thankful to the CSIR for providing Junior Research Fellowship [31/GATE/54(06)/2019-EMR-I]. The institute communication number for this manuscript is 4636.
Biographies

Anil Kumar Rana obtained his master degree in Molecular and Human Genetics from Centre for Genomics, Jiwaji University, India. In his Master’s he studied the epigenetic alteration in Transgelin gene in patients with gall bladder cancer. Mr. Rana is awarded with INSPIRE fellowship by Department of Science and Technology, New Delhi for outstanding performance in Master program. Currently, he is perusing his doctorate from AcSIR, CSIR-IHBT in the area neuroscience. He is currently working on understanding the role of Gsk-3 in neurobehavior impairment following cerebral ischemia-reperfusion injury.

Shubham Nilkanth Rahmatkar is a Junior Research Fellow (JRF) at Pharmacology and Toxicology Laboratory in CSIR-IHBT, Palampur, India. He completed his master’s degree in Pharmacology from S.K.B. College of Pharmacy, Maharashtra, India in 2018. In his Master’s degree he has explored the role of hippocampal agmatine in β amyloid 1–42 induced memory impairment, neuroinflammation and BDNF signalling disruption in mice. He is awarded with GATE Fellowship by CSIR, India. He is pursuing his Ph.D. from AcSIR, CSIR-IHBT in the area of Pharmacology. He is currently working on role of natural products in cognitive dysfunction.

Amit Kumar earned his M. Pharmacy degree in Pharmacology from University Institute of Pharmaceutical Sciences, Panjab University, Chandigarh, India. Mr. Kumar has worked with various animal models and treatment strategies of schizophrenia, especially in glutamate hypothesis during his masters. He is currently working as a Ph.D. scholar at CSIR-Institute of Himalayan Bioresource Technology, Palampur, India and working with different animal models of epilepsy to find potential leads at preclinical level.

Damanpreet Singh did his M. Pharmacy from Guru Nanak Dev University, India. Thereafter, he obtained his Ph.D. in the area of Pharmacology from Punjabi University, India. He then joined the Department of Pharmaceutical Sciences and Drug Research, Punjabi University, India as an Assistant Professor and worked in the area of neuropharmacology to find out potential targets and ligands of natural origin for comprehensive management of epilepsy. Then he joined the CSIR-Institute of Himalayan Bioresource Technology, India as a Scientist and continued to explore the molecular basis of natural products in management of neurological disorders. Presently, he is working as a Senior Scientist in the Pharmacology and Toxicology Laboratory of the same institute. His research group is focused on identification of mTOR and Gsk-3 pathway inhibitors of natural origin to explore their therapeutic potential for the management of chronic neurological conditions.
References
- 1.Baig A.M., Khaleeq A., Ali U., Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020;11:995–998. doi: 10.1021/acschemneuro.0c00181. [DOI] [PubMed] [Google Scholar]
- 2.Arico E., Bracci L., Castiello L., Gessani S., Belardelli F. Are we fully exploiting type I Interferons in today’s fight against COVID-19 pandemic? Cytokine Growth Factor Rev. 2020 doi: 10.1016/j.cytogfr.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shereen M.A., Khan S., Kazmi A., Bashir N., Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020;24:91–98. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delgado-Roche L., Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020;51:384–387. doi: 10.1016/j.arcmed.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rana A.K., Singh D. Targeting glycogen synthase kinase-3 for oxidative stress and neuroinflammation: opportunities, challenges and future directions for cerebral stroke management. Neuropharmacology. 2018;139:124–136. doi: 10.1016/j.neuropharm.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Wu C.H., Chen P.J., Yeh S.H. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host Microbe. 2014;16:462–472. doi: 10.1016/j.chom.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., Yu T. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beurel E., Grieco S.F., Jope R.S. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen P., Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 12.Forde J.A., Dale T.C. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell. Mol. Life Sci. 2007;64:1930–1944. doi: 10.1007/s00018-007-7045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong H., Zou H., Semenov M.V., Moshinsky D., He X., Huang H., Li S., Quan J., Yang Z., Lin S. Characterization and development of novel small-molecules inhibiting GSK3 and activating Wnt signalling. Mol. Biosyst. 2009;5:1356–1360. doi: 10.1039/b905752h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen P., Frame S. The renaissance of GSK3 nat. Rev. Mol. Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 15.Lei P., Ayton S., Bush A.I., Adlard P.A. GSK-3 in neurodegenerative diseases. Int. J. Alzheimers Dis. 2011;2011 doi: 10.4061/2011/189246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B., Zhou X., Qiu Y., Feng F., Feng J., Jia Y., Zhu H., Hu K., Liu J., Liu Z., Wang S. Clinical characteristics of 82 death cases with COVID-19. Gut. 2020;69:1002–1009. doi: 10.1136/gutjnl-2020-320926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu Y., Cheng Y., Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol. Sin. 2020;35:1–6. doi: 10.1007/s12250-020-00207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai Y., Kuba K., Neely G.G., Yaghubian-Malhami R., Perkmann T., van Loo G., Ermolaeva M., Veldhuizen R., Leung Y.C., Wang H., Liu H. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siu K.L., Chan C.P., Kok K.H., Woo P.C., Jin D.Y. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus M protein is mediated through the first transmembrane domain. Cell. Mol. Immunol. 2014;11:141–149. doi: 10.1038/cmi.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narayanan K., Huang C., Lokugamage K., Kamitani W., Ikegami T., Tseng C.T., Makino S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon. in infected cells J. Virol. 2008;82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang M. 2020. Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-nCoV Infection. Available at SSRN 3527420. [DOI] [Google Scholar]
- 24.Tian S., Hu W., Niu L., Liu H., Xu H., Xiao S.Y. Pulmonary pathology of early phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer [e-pub ahead of print] J. Thorac. Oncol. 2020 doi: 10.1016/j.jtho.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., Tai Y. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Resp. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., Yang D. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27:883–890. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Chen L., Li J., Wang X., Wang F., Liu L. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang D., Guo R., Lei L., Liu H., Wang Y., Wang Y., Dai T., Zhang T., Lai Y., Wang J., Liu Z. COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome. MedRxiv. 2020 doi: 10.1101/2020.03.24.20042655. [DOI] [Google Scholar]
- 29.Zhou Y., Fu B., Zheng X., Wang D., Zhao C., qi Y., Sun R., Tian Z., Xu X., Wei H. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl. Sci. Rev. 2020;7:998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S., Du B. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., Tian D.S. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W., He J., Wu S. The definition and risks of cytokine release syndrome-like in 11 COVID-19-infected pneumonia critically ill patients: disease characteristics and retrospective analysis. Medrxiv. 2020 doi: 10.1093/infdis/jiaa387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q., Verma I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 35.Hsueh P.R., Chen P.J., Hsiao C.H., Yeh S.H., Cheng W.C., Wang J.L., Chiang B.L., Chang S.C., Chang F.Y., Wong W.W., Kao C.L. Patient data, early SARS epidemic. Taiwan Emerg. Infect. Dis. 2004;10:489–493. doi: 10.3201/eid1003.030571. [DOI] [PubMed] [Google Scholar]
- 36.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., Guan L. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X., Wu K., Wang D., Yue X., Song D., Zhu Y., Wu J. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-κB. Virology. 2007;365:324–335. doi: 10.1016/j.virol.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao Q.J., Ye L.B., Timani K.A., Zeng Y.C., She Y.L., Ye L., Wu Z.H. Activation of NF‐κB by the full‐length nucleocapsid protein of the SARS coronavirus. Acta. Biochim. Biophys. Sin. 2005;37:607–612. doi: 10.1111/j.1745-7270.2005.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeDiego M.L., Nieto-Torres J.L., Regla-Nava J.A., Jimenez-Guardeño J.M., Fernandez-Delgado R., Fett C., Castaño-Rodriguez C., Perlman S., Enjuanes L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014;88:913–924. doi: 10.1128/JVI.02576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siu K.L., Yuen K.S., Castaño-Rodriguez C., Ye Z.W., Yeung M.L., Fung S.Y., Yuan S., Chan C.P., Yuen K.Y., Enjuanes L., Jin D.Y. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delgado-Roche L., Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020;51:384–387. doi: 10.1016/j.arcmed.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smits S.L., de Lang A., van den Brand J.M., Leijten L.M., van IJcken W.F., Eijkemans M.J., van Amerongen G., Kuiken T., Andeweg A.C., Osterhaus A.D., Haagmans B.L. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J.Z., Zhang R.Y., Bai J. An anti-oxidative therapy for ameliorating cardiac injuries of critically ill COVID-19-infected patients. Int. J. Cardiol. 2020;312:137–138. doi: 10.1016/j.ijcard.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernard K., Hecker L., Luckhardt T.R., Cheng G., Thannickal V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014;20:2838–2853. doi: 10.1089/ars.2013.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panday A., Sahoo M.K., Osorio D., Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015;12:5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vlahos R., Selemidis S. NADPH oxidases as novel pharmacologic targets against influenza A virus infection. Mol. Pharmacol. 2014;86:747–759. doi: 10.1124/mol.114.095216. [DOI] [PubMed] [Google Scholar]
- 47.To E.E., Broughton B.R., Hendricks K.S., Vlahos R., Selemidis S. Influenza Avirus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production inmacrophages. Free Radic. Res. 2014;48:940–947. doi: 10.3109/10715762.2014.927579. [DOI] [PubMed] [Google Scholar]
- 48.Cantu-Medellin N., Kelley E.E. Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol. 2013;1:353–358. doi: 10.1016/j.redox.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Papi A., Contoli M., Gasparini P., Bristot L., Edwards M.R., Chicca M., Leis M., Ciaccia A., Caramori G., Johnston S.L., Pinamonti S. Role of xanthine oxidase activation and reduced glutathione depletion in rhinovirus induction of inflammation in respiratory epithelial cells. J. Biol. Chem. 2008;283:28595–28606. doi: 10.1074/jbc.M805766200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akaike T., Ando M., Oda T., Doi T., Ijiri S., Araki S., Maeda H. Dependence on O2-generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Invest. 1990;85:739–745. doi: 10.1172/JCI114499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulze-Osthoff K., Bakker A.C., Vanhaesebroeck B., Beyaert R., Jacob W.A., Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions Evidence for the involvement of mitochondrial radical generation. J. Biol. 1992;267:5317–5323. [PubMed] [Google Scholar]
- 52.Schreck R., Rieber P., Baeuerle P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF‐kappa B transcription factor and HIV‐1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klempner M.S., Dinarello C.A., Gallin J.I. Human leukocyticpyrogen induces release of specific granule contents from human neutrophils. J. Clin. Invest. 1978;61:1330–1336. doi: 10.1172/JCI109050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tarifeno-Saldivia E., Aguilar A., Contreras D., Mercado L., Morales-Lange B., Márquez K., Henríquez A., Riquelme-Vidal C., Boltana S. Iron overload is associated with oxidative stress and nutritional immunity during viral infection in fish. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Q., Gao Y., Ci X. Role of Nrf2 and its activators in respiratory diseases. Oxid. Med. Cell. Longev. 2019;2019 doi: 10.1155/2019/7090534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cho H.Y., Kleeberger S.R. Noblesse oblige: NRF2 functions in the airways. Am. J. Respir. Cell Mol. Biol. 2014;50 doi: 10.1165/rcmb.2014-0116PS. pp. 844-710.1165/rcmb.2014-0116PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiang-hua Y., Le-min W., Ai-bin L., Zhu G., Riquan L., Xu-you Z., Wei-wei R., Ye-nan W. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am. J. Respir. Crit. Care Med. 2010;182:436–437. doi: 10.1164/ajrccm.182.3.436. [DOI] [PubMed] [Google Scholar]
- 58.Tang N., Bai H., Chen X., Gong J., Li D., Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020;18:1094–1099. doi: 10.1111/jth.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu P.P., Blet A., Smyth D., Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. 2020;142:68–78. doi: 10.1161/CIRCULATIONAHA.120.047549. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y., Xiao M., Zhang S., Xia P., Cao W., Jiang W., Chen H., Ding X., Zhao H., Zhang H., Wang C. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N. Engl. J. Med. 2020;382:e38. doi: 10.1056/NEJMc2007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simmons J., Pittet J.F. The coagulopathy of acute sepsis. Curr. Opin. Anaesthesiol. 2015;28:227–236. doi: 10.1097/ACO.0000000000000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iba T., Levy J.H., Raj A., Warkentin T.E. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Clin. Med. 2019;8:728. doi: 10.3390/jcm8050728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osterud B. Cellular interactions in tissue factor expression by blood monocytes. Blood coagulation & fibrinolysis: an international journal in haemostasis and thrombosis. Blood Coagul. Fibrinolysis. 1995;6:20–25. [PubMed] [Google Scholar]
- 64.Pawlinski R., Mackman N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb. Res. 2010;125:70–73. doi: 10.1016/j.thromres.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morrissey J.H. Tissue factor interactions with factor VII: measurement and clinical significance of factor VIIa in plasma. Blood. Coagul. Fibrin. 1995;6:14–19. [PubMed] [Google Scholar]
- 66.Danckwardt S., Hentze M.W., Kulozik A.E. Pathologies at the nexus of blood coagulation and inflammation: thrombin in hemostasis, cancer, and beyond. J. Mol. Med. 2013;91:1257–1271. doi: 10.1007/s00109-013-1074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coughlin S.R. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 68.Foley J.H., Conway E.M. Cross talk pathways between coagulation and inflammation. Circ. Res. 2016;118:1392–1408. doi: 10.1161/CIRCRESAHA.116.306853. [DOI] [PubMed] [Google Scholar]
- 69.Yang Y., Tang H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016;13:432–442. doi: 10.1038/cmi.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saikatendu K.S., Joseph J.S., Subramanian V., Neuman B.W., Buchmeier M.J., Stevens R.C., Kuhn P. Ribonucleo capsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 2007;81:3913–3921. doi: 10.1128/JVI.02236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Surjit M., Lal S.K. The SARS-CoV nucleocapsid protein: a protein with multifarious activities. Infect. Genet. Evol. 2008;8:397–405. doi: 10.1016/j.meegid.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fung T.S., Liu D.X. Post-translational modifications of coronavirus proteins: roles and function. Future Virol. 2018;13:405–430. doi: 10.2217/fvl-2018-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou B., Liu J., Wang Q., Liu X., Li X., Li P., Ma Q., Cao C. The nucleocapsid protein of severe acute respiratory syndrome coronavirus inhibits cell cytokinesis and proliferation by interacting with translation elongation factor 1α. J. Virol. 2008;82:6962–6971. doi: 10.1128/JVI.00133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu C.H., Yeh S.H., Tsay Y.G., Shieh Y.H., Kao C.L., Chen Y.S., Wang S.H., Kuo T.J., Chen D.S., Chen P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 2009;284:5229–5239. doi: 10.1074/jbc.M805747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sawicki S.G., Sawicki D.L., Siddell S.G. A contemporary view of coronavirus transcription. J. Virol. 2007;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pasternak A.O., Spaan W.J., Snijder E.J. Nidovirus transcription: how to make sense…? J. Gen. Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- 77.Pasternak A.O., Spaan W.J., Snijder E.J. Regulation of relative abundance of arterivirus subgenomic mRNAs. J. Virol. 2004;78:8102–8113. doi: 10.1128/JVI.78.15.8102-8113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuniga S., Cruz J.L., Sola I., Mateos-Gómez P.A., Palacio L., Enjuanes L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010;84:2169–2175. doi: 10.1128/JVI.02011-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peng T.Y., Lee K.R., Tarn W.Y. Phosphorylation of the arginine/serine dipeptide‐rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2008;275:4152–4163. doi: 10.1111/j.1742-4658.2008.06564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Komaravelli N., Casola A. Respiratory viral infections and subversion of cellular antioxidant defenses. J. Pharmacogenomics Pharmacoproteomics. 2014;5 doi: 10.4172/2153-0645.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoffmeister L., Diekmann M., Brand K., Huber R. GSK3: a kinase balancing promotion and resolution of inflammation. Cells. 2020;9:820. doi: 10.3390/cells9040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang Y., Bao H., Ge Y., Tang W., Cheng D., Luo K., Gong G., Gong R. Therapeutic targeting of GSK3β enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis. C Gut. 2015;64:168–179. doi: 10.1136/gutjnl-2013-306043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martin M., Rehani K., Jope R.S., Michalek S.M. Toll-like receptor–mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deng J., Miller S.A., Wang H.Y., Xia W., Wen Y., Zhou B.P., Li Y., Lin S.Y., Hung M.C. β-catenin interacts with and inhibits NF-κB in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- 85.Agarwal D., Dange R.B., Raizada M.K., Francis J. Angiotensin II causes imbalance between pro‐and anti‐inflammatory cytokines by modulating GSK‐3β in neuronal cultur. Br. J. Pharmacol. 2013;169:860–874. doi: 10.1111/bph.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ren X., Meng F., Yin J., Li G., Li X., Wang C., Herrler G. Action mechanisms of lithium chloride on cell infection by transmissible gastroenteritis coronavirus. PLoS One. 2011;6 doi: 10.1371/journal.pone.0018669. pp. e18669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H.J., Gao D.S., Li Y.T., Wang Y.S., Liu H.Y., Zhao J. Antiviral effect of lithium chloride on porcine epidemic diarrhea virus in vitro. Res. Vet. Sci. 2018;118:288–294. doi: 10.1016/j.rvsc.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khalil R.B. Lithium chloride combination with rapamycin for the treatment of COVID-19 pneumonia. Med. Hypotheses. 2020;142:109798. doi: 10.1016/j.mehy.2020.109798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nowak J.K., Walkowiak J. Is lithium a potential treatment for the novel Wuhan (2019-nCoV) coronavirus? A scoping review. F1000Research. 2020;9 doi: 10.12688/f1000research.22299.2. [DOI] [Google Scholar]
- 90.Morales-Garcia J.A., Luna-Medina R., Alonso-Gil S., Sanz-SanCristobal M., Palomo V., Gil C., Santos A., Martinez A., Perez-Castillo A. Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem. Neurosci. 2012;3:963–971. doi: 10.1021/cn300110c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sereno L., Coma M., Rodriguez M., Sanchez-Ferrer P., Sanchez M.B., Gich I., Agullo J.M., Perez M., Avila J., Guardia-Laguarta C., Clarimon J. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 92.DaRocha-Souto B., Coma M., Perez-Nievas B.G., Scotton T.C., Siao M., Sánchez-Ferrer P., Hashimoto T., Fan Z., Hudry E., Barroeta I., Serenó L. Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol. Dis. 2012;45:425–437. doi: 10.1016/j.nbd.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 94.del Ser T., Steinwachs K.C., Gertz H.J., Andres M.V., Gomez-Carrillo B., Medina M., Vericat J.A., Redondo P., Fleet D., Leon T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimers Dis. 2013;33:205–215. doi: 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- 95.Lovestone S., Boada M., Dubois B., Hüll M., Rinne J.O., Huppertz H.J., Calero M., Andres M.V., Gomez-Carrillo B., Leon T., Del Ser T. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015;45:75–88. doi: 10.3233/JAD-141959. [DOI] [PubMed] [Google Scholar]
- 96.Hoglinger G.U., Huppertz H.J., Wagenpfeil S., Andrés M.V., Belloch V., León T., Del Ser T., TAUROS MRI Investigators, Gmez J.C., Tijero B., Villoria R. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov. Disord. 2014;29:479–487. doi: 10.1002/mds.25815. [DOI] [PubMed] [Google Scholar]