

Abstract
The extent to which sequence variation impacts plant fitness is poorly understood. High-resolution maps detailing the constraint acting on regulatory sites would be beneficial, as functional annotation of noncoding sequences remains sparse. Here we present a fitness consequence map for rice (Oryza sativa). We obtained fitness consequence scores (ρ) for 246 inferred genome classes derived from nine functional genomic and epigenomic datasets, including chromatin accessibility, mRNA/sRNA transcription, DNA methylation, histone modifications, and engaged RNA polymerase activity. These were integrated with genome-wide polymorphism and divergence data from 1,477 rice accessions and 11 reference genome sequences in the Oryzeae. We found ρ to be multimodal, with ≈9% of the rice genome falling into classes where more than half of the bases would likely have a fitness consequence if mutated. Around 2% of the rice genome showed evidence of weak negative selection, frequently at candidate regulatory sites, including a novel set of 1,000 potentially active enhancer elements. This fitness consequence map provides perspective on the evolutionary forces associated with genome diversity, aids in genome annotation, and can guide crop breeding programs.
Keywords: selection, sequence constraint, neutral, epigenome, enhancer, promoter, noncoding DNA, histone mark, methylation, polymerase activity, genome annotation, crop genomics, evolutionary genomics
INTRODUCTION
Determining the likely impact of sequence variation at noncoding sites continues to be problematic, in part because functional annotation is generally sparse. Nonetheless, high-resolution maps of sequence constraint that reveal both functional coding and regulatory sites would benefit crop breeding and genetic engineering interventions. Several approaches to estimating selective constraint from sequence conservation across species have been developed, and more recently complemented by approaches that utilize large population genomic datasets1,2. While conservation-based approaches frequently provide high spatial resolution, and population-based approaches can be used to infer recent changes in constraint, neither class of approaches has until recently been able to simultaneously provide both perspectives.
One contemporary approach to determining recent selection on genome sequences at high resolution is INSIGHT3 (Inference of Natural Selection from Interspersed Genomically coHerent elemenTs), which infers the fraction of nucleotide sites under selection. This is accomplished by comparing patterns of within-species sequence polymorphism with between-species divergence across dispersed genomic sites, relative to nearby neutrally evolving sites3. The shorter evolutionary timescales associated with intra-species variation make this approach more robust to evolutionary turnover, and its applicability to short sequence domains (e.g., regulatory sites) makes it particularly powerful for surveying the fitness consequences of point mutations in noncoding DNA4–7. The use of locally-matched rather than global neutral models make the INSIGHT approach similar to the McDonald-Kreitman test4 in its robustness to confounding factors such as non-equilibrium demography, mutation rate variation, and background selection.
The influence of natural selection at each site is summarized by ρ, the probability that a mutation at that site will affect fitness. Values of ρ closer to 1 suggest that a larger proportion of sites in a sequence class are under selection3 compared to neutral regions (for which ρ is closer to 0). INSIGHT additionally quantifies other parameters, including the number of segregating polymorphic sites per kilobase pair (kbp) under weak negative selection3 (Pw).
Integrating INSIGHT with functional genomic data and aggregating the genome by joint patterns across these functional genomic tracks allows the development of genomic fitness consequence (fitCons) maps that permit selection to be inferred at a high resolution across the genome8,9. By leveraging patterns of polymorphism within species, these maps measure natural selection at shorter time scales than traditional evolutionary conservation methods. Because polymorphic sites are sparse across many genomes, the fitCons approach uses functional genomic data to pool information across putatively functionally similar genomic sites, and therefore define discrete genomic classes for which we can infer ρ and other selection parameters (e.g., Pw).
The fitCons approach, with INSIGHT at its core, was first developed for the human genome. Here we report the first fitness consequence map in a plant genome, using rice (Oryza sativa) as a model system. Rice is one of the most important domesticated food crops in the world, and is the target of intense effort for crop improvement to advance food security and sustainable agriculture10. The rice fitness consequence map will contribute to a better understanding of selection in this key crop species, and potentially guide the identification of functional components of genome structure, for future breeding efforts.
RESULTS
GreenINSIGHT
To produce a rice fitness consequence map, we adapted the INSIGHT model for plants (greenINSIGHT) by modifying elements that were human-specific and accounting for some aspects of plant genome organization and recent introgression (see Methods). In essence the greenINSIGHT pipeline (Supplementary Fig. 11) combines genomic alignments, from which an ancestral base probability distribution is generated, with polymorphism data from the focal species, to infer selection acting at sites of interest relative to a set of matched local neutral sites. Polymorphism data was aggregated from 1,477 O. sativa accessions in the 3k Rice Genome Project panel11 (Supplementary Table 1) and alignments were generated from O. sativa to the genomes of 10 species in the Oryza genus, with the closest outgroup (O. rufipogon) having ~0.3 million years of divergence from O. sativa12 (Supplementary Fig. 1, also see Methods). We also developed an INSIGHT model for Arabidopsis thaliana using an 80-way sub-population alignment of the 1,001 Arabidopsis genome dataset13 (see Methods) and alignments generated previously14. The robustness of INSIGHT to complex demography is particularly relevant in rice because assortative mating in the different O. sativa variety groups (e.g. japonica and indica) has led to relatively differentiated populations3,15,16.
We explored the distribution of ρ reported by greenINSIGHT at a set of well-characterized genomic locations (Fig. 1a). As expected, intergenic regions depleted of open chromatin and distal to genes and conserved non-coding regions (CNSs) (see Methods) had the lowest ρ (<0.03)17. Genomic regions annotated as coding sequences (CDSs) had a significantly higher median ρ, albeit with a broad distribution likely reflective of the considerable variation in constraint across protein domains.
Figure 1. greenINSIGHT across different genomic annotations in rice.
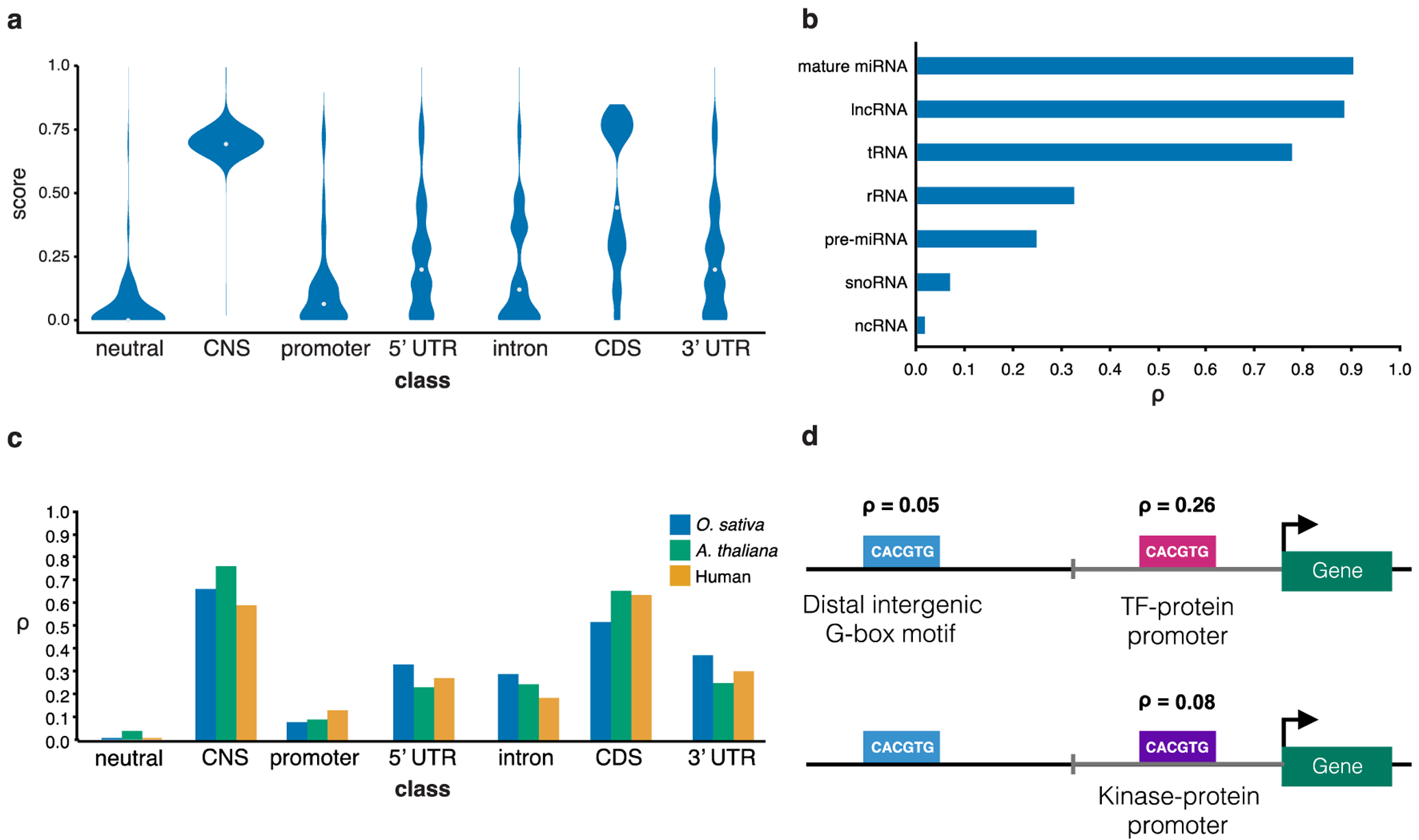
a, Violin plots of the ρ distribution within annotated gene and intergenic site classes. Annotations for neutral and CNS refer to distal neutral and distal CNS. White dots indicate the median ρ for each class. b, Mean ρ across seven types of annotated noncoding RNAs. c, Mean ρ across annotated gene and intergenic site-classes in humans, Arabidopsis, and rice. d) Distribution of rho values at different intergenic locations for the plant G-box motif (CACGTG).
The selection profiles on 5’ and 3’ UTRs were similarly complex, again suggestive of a broad range of functional sites. Promoter regions have ρ distributions similar to neutral sites overlaid with a small number of selected sites. Introns may experience low levels of direct selection, but possibly higher levels of intron-length dependent background selection18,19 due to their linkage with CDSs (Fig. 1a, see also Fig. 3h, Supplementary Fig. 6). Distal CNSs had highest ρ values, similar to those of the more constrained CDSs14.
Figure 3. Distribution of ρ across the rice genome.
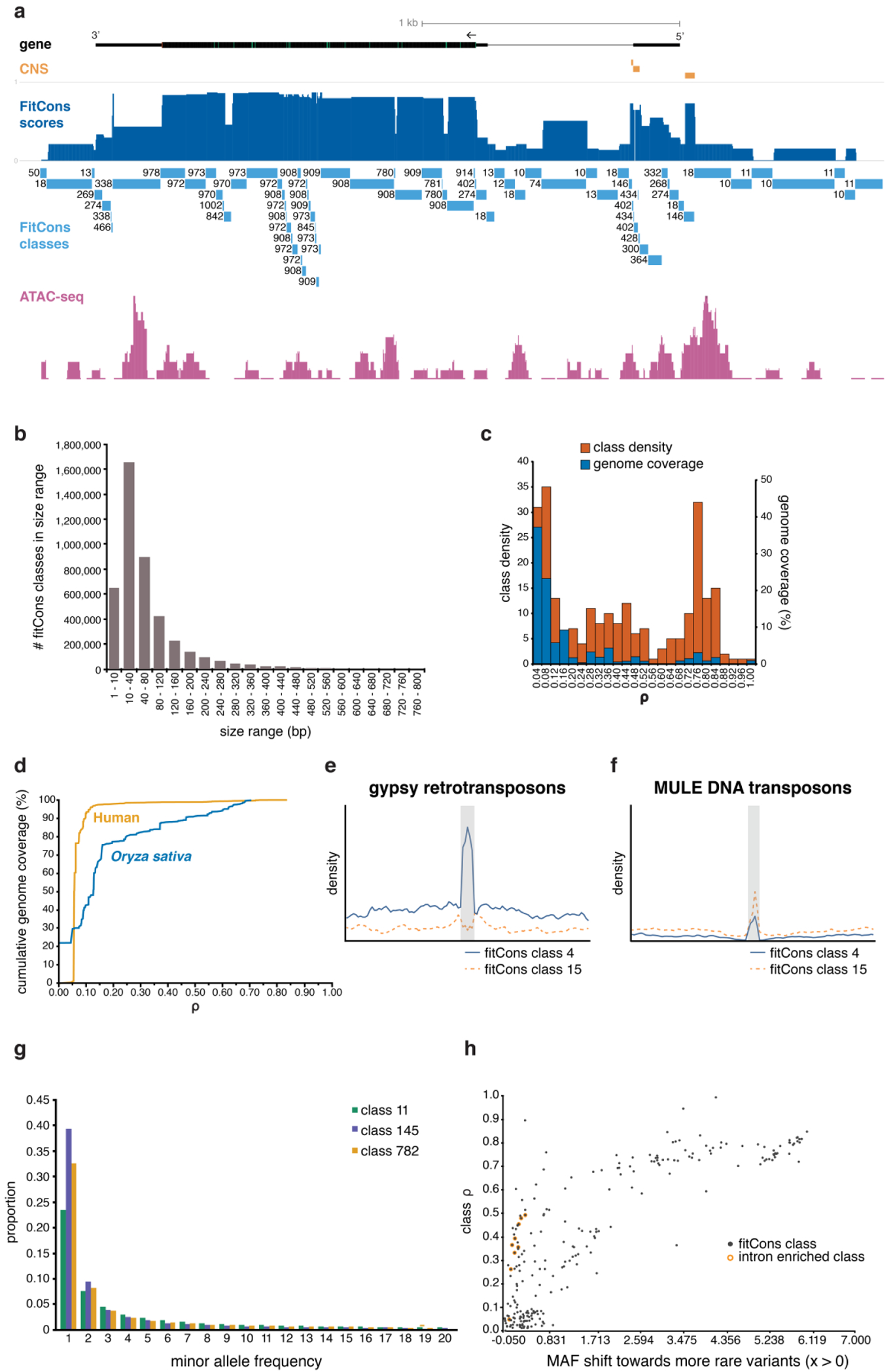
a, Rice locus Os07g0153400 illustrating the partitioning of a locus by genomic class and the associated ρ. A high-ρ class immediately upstream of the transcription start site coincides both with open chromatin and sequence conservation, whilst the short 3’ UTR peak lies immediately downstream of two canonical polyA signals, coinciding with paused-polymerases. b, The size distribution of blocks that make up the 246 genomic classes used to partition the genome. c, Genome-wide distribution of class ρ: most of the genome (right axis, percentage genome coverage) is contained in large low-ρ classes (left axis, class density), contrasting with the much smaller high-ρ classes. d, Cumulative distribution of ρ| in the 3.2-Gbase human genome relative to the 0.4-Gbase rice genome. e, f, Class densities around Type I and Type II transposable elements. g, Standardized minor allele frequencies of rare SNPs (subset of f:1–20) for three fitCons classes; those with higher ρ [class 145 (0.69) > class 782 (0.27) > class 11 (0.001)] show an expected bias towards rare SNPs (Supplementary Table 9). The MAF shift (x) towards rarer variants in each class relative to a control neutral class (class 11) illustrates h, an expected increase with class ρ, with the exception of classes that experience high levels of indirect rather than direct selection (such a subset of intron-enriched classes are highlighted).
Sites that generate noncoding RNAs (ncRNAs) displayed a range of ρ, with sites generating long noncoding RNAs (lncRNAs), mature microRNAs (miRNAs), and transfer/ribosomal RNAs (tRNAs/rRNAs) showing evidence of more selection than sites generating pre-miRNAs, small nucleolar RNAs (snoRNAs) and other unclassified ncRNAs (frequently small interfering RNAs (siRNAs)) (Fig. 1b). Differences between ρ in humans and plants (A. thaliana and O. sativa) were mostly subtle, except in introns (Fig. 1c) where the much longer human20,21 introns likely make background selection less of a confounding factor.
We sought to determine the sensitivity of greenINSIGHT to constraint in regulatory regions by estimating constraint on a well-documented plant transcription factor binding motif, the G-box motif (CACGTG)22,23, in different genomic contexts. This motif is often functional in the promoters of genes targeted by abscisic acid signaling24, and so unsurprisingly ρ was higher in the promoters of transcription factor genes than in the promoters of genes with more enzymatic roles (Fig. 1d). However, the proportion of constrained bases in this motif was, as expected, higher when found in promoters than it was in distal intergenic locations.
Genome partitioning to build a fitCons map of rice
To estimate ρ genome-wide in rice, we used the fitCons approach to partition the genome into a set of classes8 based on their shared functional characteristics. Functional genomic datasets were generated from the leaves of 3-week old Oryza sativa tropical japonica (cultivar Azucena) plants and included total RNA transcription25, small RNA transcription26 (sRNA), ATAC-seq27,28 (Assay for Transposase-Accessible Chromatin using Sequencing), DNA methylation29, Precision nuclear Run-On and sequencing (PRO-seq), which maps transcriptionally engaged polymerase activity at basepair (bp) resolution30,31, and H3K27me3, H3K27ac, H3K18ac and H3K4me3 histone modifications (using ChIP-seq32,33) (Supplementary Table 2).
Epigenomic data broadly supported observations of chromatin states reported elsewhere in plants33–43 (Fig. 2a; Supplementary Table 3). Promoter regions of expressed genes were marked with open chromatin and decreased methylation, while the enhancer mark H3K27ac positively correlated with polymerase activity in distal and proximal gene regions (Fig. 2a). In addition, the distribution of transcriptionally engaged polymerases confirmed 5’ and 3’ polymerase pausing around genes, with the highest signal found in actively expressed genes around transcription start sites (TSSs)36 (Supplementary Fig. 2). Active gene expression was negatively correlated with the presence of H3K27me3, while transposable elements (TEs) showed positive correlations between sRNA transcription and DNA methylation indicative of TE silencing. Most sRNA coverage appeared to arise from broadly distributed siRNAs whilst DNA methylation was mostly found in CpG contexts, with CpG methylation slightly enriched in gene bodies relative to CHG/CHH methylation; however their distribution was similar in TEs as previously reported44 (Supplementary Table 4).
Figure 2. Partitioning and scoring the rice genome for selection (ρ).

a, Pearson’s correlation matrices for high-confidence uniquely mapped reads for histone modification, DNA methylation, open chromatin and transcriptomic datasets in five genomic regions (see also Supplementary Table 3). b, A conceptual overview of the analysis pipeline used to generate the fitness consequence map. See Table 1 and Supplementary Table 5 for details about the 246 fitCons scores. c, Emission parameters from the 20-state ChromHMM model for seven covariates of chromatin state.
The rice genome was partitioned into a set of coherent classes in a two step process - first the epigenomic datasets were used to infer a small set of chromatin states, and in a second step these states were intersected with transcriptomic and annotation data (Fig. 2b) to generate a more nuanced global classification.
The first step employed a Hidden-Markov modeler ChromHMM45 to binarize chromatin signals and produce a low-resolution map of chromatin states across the rice genome (Fig 2b,c see also Methods). The selection of the number of states was informed by the ChromHMM option “CompareModels”46 to determine the correlation between the emissions of models having different numbers of states. Selecting a 50-state model as an over-parameterized reference, we observed a rapid convergence towards this model’s outputs after 15-states were incorporated and the mean state correlation with the closest state in the 50-state model exceeded 0.9 once 20 states were included (Supplementary Fig. 3). We therefore selected a 20-state ChromHMM model (Fig. 2b,c). This number is higher than early estimates of the number of chromatin states in plants47,48 but fewer than the 38 states previously inferred in rice from a broader set of histone marks32. The inferred number of chromatin states is anticipated to vary to some degree with the type of chromatin marks used as input and to this end we selected marks based on testing for high levels of intra-replicate correlation but low levels of correlation between marks in A. thaliana public datasets.
In a second step we intersected these chromatin states with further binarized annotation data and evidence for roles in transcription or transcription initiation. These data included reference genome annotation (coding, exon), phastCons scores, and RNA-seq and PRO-seq alignments (Fig. 2b, also see Methods). The intersection of the 20 states with this data generated a more ontologically complete and higher-resolution set of 640 possible genome classes8, of which 246 were identified with an appreciable coverage of the rice genome (>20 kb of total sequence) (Fig 2b, Supplementary Table 5).
With the rice genome partitioned into 246 genomic classes (fitCons classes), we estimated ρ for each class using greenINSIGHT as previously described8 (Table 1, Supplementary Tables 5 and 6). The 246 class ρ scores that were then distributed back to each nucleotide in each class, giving each nucleotide in the genome a fitCons score (Fig. 3a). A simple validation of class coherence was performed to ensure that the distribution of ρ as a function of class size was different to that expected under a random sampling model (Supplementary Fig. 4, Supplementary Table 7, also see Methods).
Table 1.
Properties of a subset of 246 inferred FitCons classes (see Supplementary Table 5).
| Class ID* | Genome Coverage % | CDS | Introns | Promoter 500 | End 500 | CNS | ATAC | ProSeq | TEs | Class ρ |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.851 | 0.3 | 0.19 | 0.31 | 0.26 | 0 | 0.04 | 0.01 | 2.66 | 0.083 |
| 4 | 6.302 | 0.24 | 0.21 | 0.27 | 0.27 | 0 | 0.01 | 0 | 2.61 | 0.085 |
| 10 | 6.312 | 0.16 | 1.3 | 1.08 | 1.46 | 0 | 0.02 | 0.44 | 0.41 | 0.146 |
| 11 | 22.004 | 0.23 | 1.06 | 1.07 | 1.36 | 0 | 0.02 | 0.21 | 0.66 | 0.001 |
| 15 | 2.988 | 0.66 | 0.38 | 0.81 | 0.55 | 0 | 0.08 | 0.05 | 1.78 | 0.065 |
| 44 | 0.1 | 0.47 | 0.48 | 3.23 | 3.08 | 0 | 15.65 | 13.72 | 0.46 | 0.064 |
| 48 | 0.142 | 0.25 | 0.35 | 4.05 | 3.94 | 0 | 15.16 | 14.95 | 0.52 | 0.019 |
| 49 | 0.41 | 0.06 | 0.33 | 3.77 | 3.57 | 0 | 10.47 | 14.93 | 0.71 | 0.023 |
| 50 | 0.182 | 0.16 | 0.41 | 2.75 | 3.96 | 0 | 7.6 | 17.32 | 0.58 | 0.014 |
| 52 | 0.282 | 0.03 | 0.4 | 3.12 | 2.1 | 0 | 2.34 | 8.11 | 2.03 | 0.084 |
| 102 | 0.005 | 0.04 | 3.93 | 0.63 | 1.38 | 0 | 0.61 | 8.64 | 2.5 | 0.061 |
| 108 | 0.031 | 0.4 | 3.08 | 1.95 | 1.31 | 0 | 12.98 | 17.22 | 0.23 | 0.363 |
| 112 | 0.011 | 0.28 | 2.02 | 2.66 | 2.97 | 0 | 16.71 | 18.24 | 0.4 | 0.256 |
| 138 | 0.268 | 0.8 | 1.63 | 0.94 | 1.21 | 36.36 | 0.01 | 0.44 | 0.04 | 0.693 |
| 144 | 0.12 | 0.81 | 0.52 | 3.62 | 1.26 | 40.82 | 13.72 | 1.14 | 0.04 | 0.738 |
| 145 | 0.158 | 0.54 | 0.82 | 2.26 | 1.5 | 42.34 | 10.45 | 1.19 | 0.02 | 0.684 |
| 174 | 0.005 | 1.29 | 0.48 | 1.47 | 6.19 | 36.38 | 0 | 16.28 | 0.07 | 0.700 |
| 200 | 0.017 | 2.69 | 0.33 | 3.5 | 4.37 | 3.77 | 0.25 | 0 | 0.09 | 0.994 |
| 201 | 0.014 | 1.88 | 0.42 | 1.99 | 2.87 | 4.96 | 10.56 | 0.08 | 0.14 | 0.946 |
| 234 | 0.005 | 1.43 | 4.65 | 0.74 | 1.02 | 22.67 | 0 | 21.87 | 0.01 | 0.802 |
| 300 | 0.07 | 0.36 | 0.21 | 1.76 | 0.86 | 0 | 21.79 | 8.59 | 0.35 | 0.416 |
| 306 | 0.043 | 0.25 | 0.28 | 1.8 | 1.59 | 0 | 14.12 | 11.96 | 0.41 | 0.237 |
| 466 | 0.016 | 0.99 | 0.93 | 0.5 | 1.34 | 41.39 | 1.56 | 11.97 | 0.02 | 0.806 |
| 782 | 1.205 | 7.02 | 0.18 | 0.27 | 0.25 | 0 | 0.01 | 0.31 | 0.14 | 0.271 |
| 908 | 0.122 | 7.7 | 0.06 | 0.41 | 0.28 | 0 | 1.46 | 3.5 | 0.16 | 0.768 |
| 970 | 0.33 | 7.76 | 0.11 | 0.07 | 0.29 | 0 | 0 | 2.78 | 0.01 | 0.817 |
| 974 | 0.378 | 7.77 | 0.09 | 0.04 | 0.26 | 0 | 0 | 1.8 | 0.02 | 0.807 |
| 977 | 0.096 | 7.76 | 0.1 | 0.05 | 0.31 | 0 | 0.27 | 2.98 | 0.01 | 0.802 |
| 978 | 0.055 | 7.74 | 0.14 | 0.23 | 0.32 | 0 | 0.51 | 8.38 | 0.04 | 0.797 |
| 1003 | 0.011 | 7.75 | 0.11 | 0.08 | 0.8 | 0 | 0 | 17.34 | 0.01 | 0.848 |
| 1010 | 0.023 | 7.71 | 0.17 | 0.31 | 0.56 | 0 | 2.79 | 24.31 | 0.06 | 0.824 |
The class ID numbering is not continuous
Distribution of fitCons scores
The 246 rice fitCons classes we inferred were distributed in ~4.3 million blocks ranging in size from 1–600 bps, with most in blocks of 10–40 bps (Fig. 3a,b). We found a multimodal distribution of ρ over the 246 states, with peaks at ρ≈0.08, ≈0.44, and ≈0.76. Most of the genome is comprised of classes with low/moderate ρ (86.4% of the genome has ρ < 0.4), while higher ρ classes (ρ > 0.5) make up only 8.98% of the genome (Fig. 3c). The cumulative distribution of ρ in rice is consistent with similar number of coding sites in a small genome space relative to human, and more intermediate selection on noncoding functional sites, some of which may arise from background selection (Fig. 3d).
Relative to the genome’s broader annotation, classes with low/moderate selection were primarily located in unannotated intergenic regions, or were enriched for TEs (Fig.3c, Supplementary Table 5). In contrast, genome classes with higher ρ were enriched for CDSs and CNSs, with some overlapping regions with open chromatin, and/or actively transcribed sites. Classes that have intermediate values of ρ are enriched for a mixture of genomic annotations, and it is hard to identify and predominant constituents for many of these classes.
As expected, many classes with higher ρ were enriched for known functional elements. For example, classes 974, 977 and 1003 (median ρ = 0.817) were associated with CDSs and classes 138–146 (median ρ = 0.694) and 170–178 (median ρ = 0.7) with a set of inferred CNSs (Table 1, Supplementary Table 5, also see Methods). Several classes were associated with different TE types; for instance, classes 15 (ρ = 0.065) and 4 (ρ = 0.085) were enriched for Mutator-like transposable elements and gypsy LTR retrotransposons, respectively (Fig. 3e, f).
Several of these high ρ classes appeared to be enriched for facultative regulatory sites, showing small but significant correlations with the expression of downstream genes (e.g., class 17: r = 0.113, two-tailed t-test, P = 5.2 × 10−70, N = 24,296; class 145: r = 0.08, P = 4.5 × 10−32, N = 24,296; Supplementary Table 8). Combining all classes, a multiple regression model trained on chromatin classes upstream (500 bps) of genes on chromosomes 2 to 12 had modest but significant predictive power for gene expression when tested on chromosome 1 genes (r = 0.419 two-tailed t-test, P = 3.6 × 10−148, N = 3,502) (Fig. 4). Predictive power arose primarily from epigenomic states around slightly distal promoter sequences, as masking the 50 bp core promoter did not significantly impact model power. Notable for 3’ to 5’ looping models of gene regulation49 downstream gene regions were enriched for a different set of classes with equally high individual associations with gene expression (e.g., class 49: r = 0.18, two-tailed t-test, P = 1.0 × 10−177, N = 24,296; class 50, r = 0.181, P = 7.2 × 10−179, N = 24,296), but that had less power as a combined model (Supplementary Fig. 5).
Figure 4. Proximal upstream chromatin class distribution correlates with downstream gene expression.
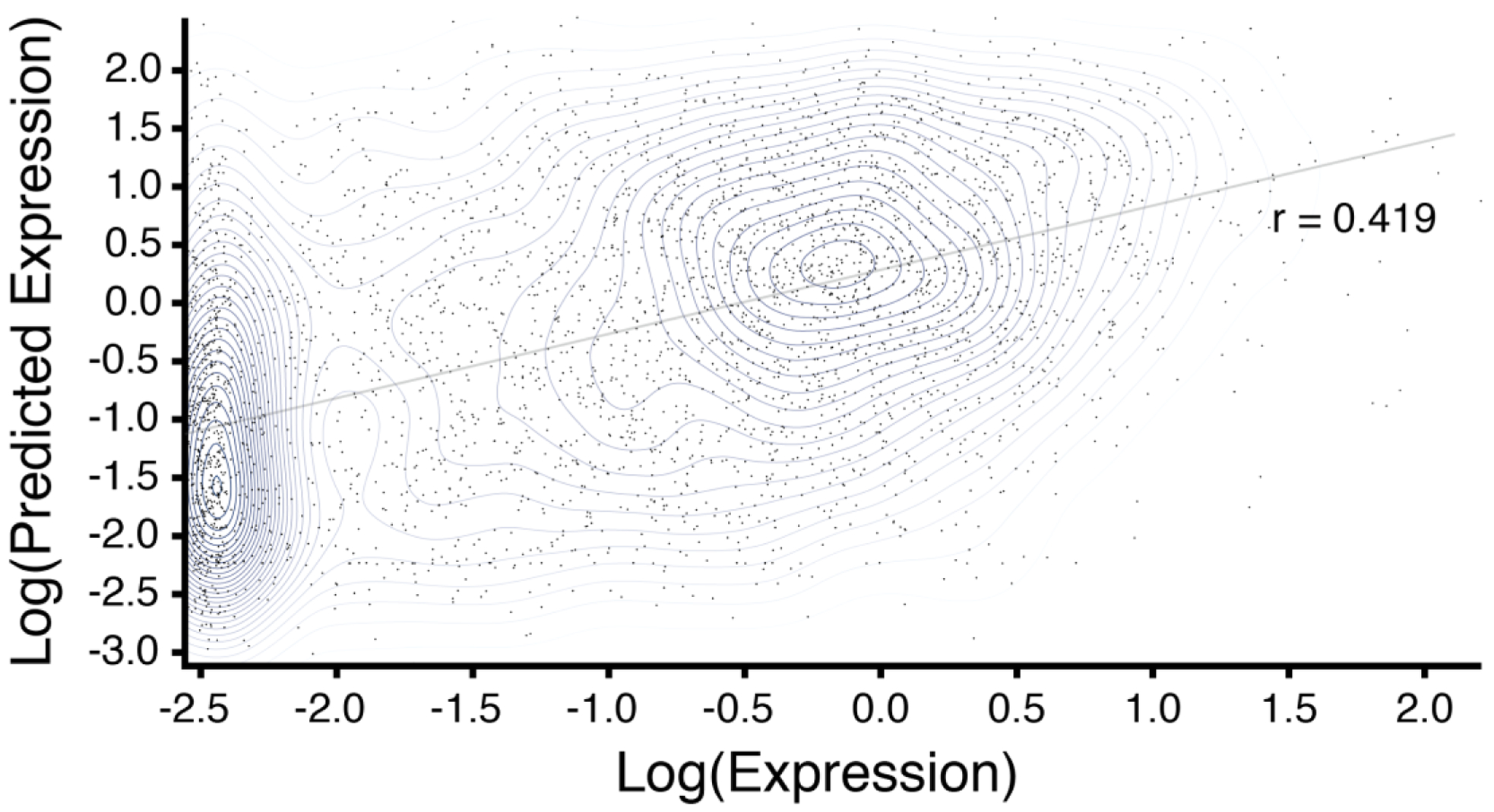
Actual (x-axis) and predicted (y-axis) leaf tissue gene expression per class coverage on chromosome 1. The predicted expression is derived from a multiple linear regression model of upstream chromatin classes used as covariates against gene expression for protein coding genes on rice chromosomes 2–12 [r2 = 0.18, (grey line)].
As expected, the site-frequency spectrum (SFS) of polymorphisms in rice displays a minor allele frequency (MAF) skew towards rare variants in high-ρ classes, such as those enriched in the more conserved promoters (e.g. class 145, ρ = 0.681) and CDSs (e.g. class 782, ρ = 0.294) (Fig. 3g; Supplementary Table 9) relative to low-ρ classes such as the TE-enriched class 11 (ρ = 0.016). This skew was evident across classes in general (r = 0.83, two-tailed t-test, P = 8 × 10−64, N = 246) (Fig. 3h Supplementary Table 9).
Overall, strong purifying selection was the main driver of ρ. However, about 2% of the rice genome appeared to be under weak negative selection. Classes with lower ρ sometimes had notable levels (up to 8%) of sites under weak negative selection that potentially mark recently selected genomic sites (Supplementary Table 5).
Delineating putative noncoding regulatory regions
Among the 246 fitCons classes, we further defined three categories of potentially functional noncoding regions by considering their functional and epigenomic characteristics as well as their ρ scores. The first category, termed “Conserved” classes, includes 46 noncoding element classes with evidence of sequence conservation among Oryza species (phastCons >0.82), (Supplementary Table 5). Conserved classes have high ρ and are prevalent in the promoter regions of a subset of protein-coding genes (within 0–350 bps upstream of TSSs) where they likely act as cis-regulatory elements14,50 (Fig. 5a, Supplementary Fig. 7a). Gene ontology enrichment analysis suggests these are predominantly associated with transcription-factor and developmental genes (FDR = 1.98 × 10−40) (Supplementary Table 10). The density of upstream conserved classes was strongly linked to genes with the highest fold change in expression between tissues (Fig. 5b), again suggesting tissue specific or developmental roles. De novo motif analysis (Homer N:6–8 bps) of these 46 classes revealed generally complex motifs (Fig. 5c, Supplementary Fig. 7b–c,) including well-characterized transcription-factor binding sites (e.g. G-box, RY-repeat motifs, TATA box, etc., see Supplementary Fig. 8)23,51,52.
Figure 5. Characterization of three categories of intergenic fitCons classes.
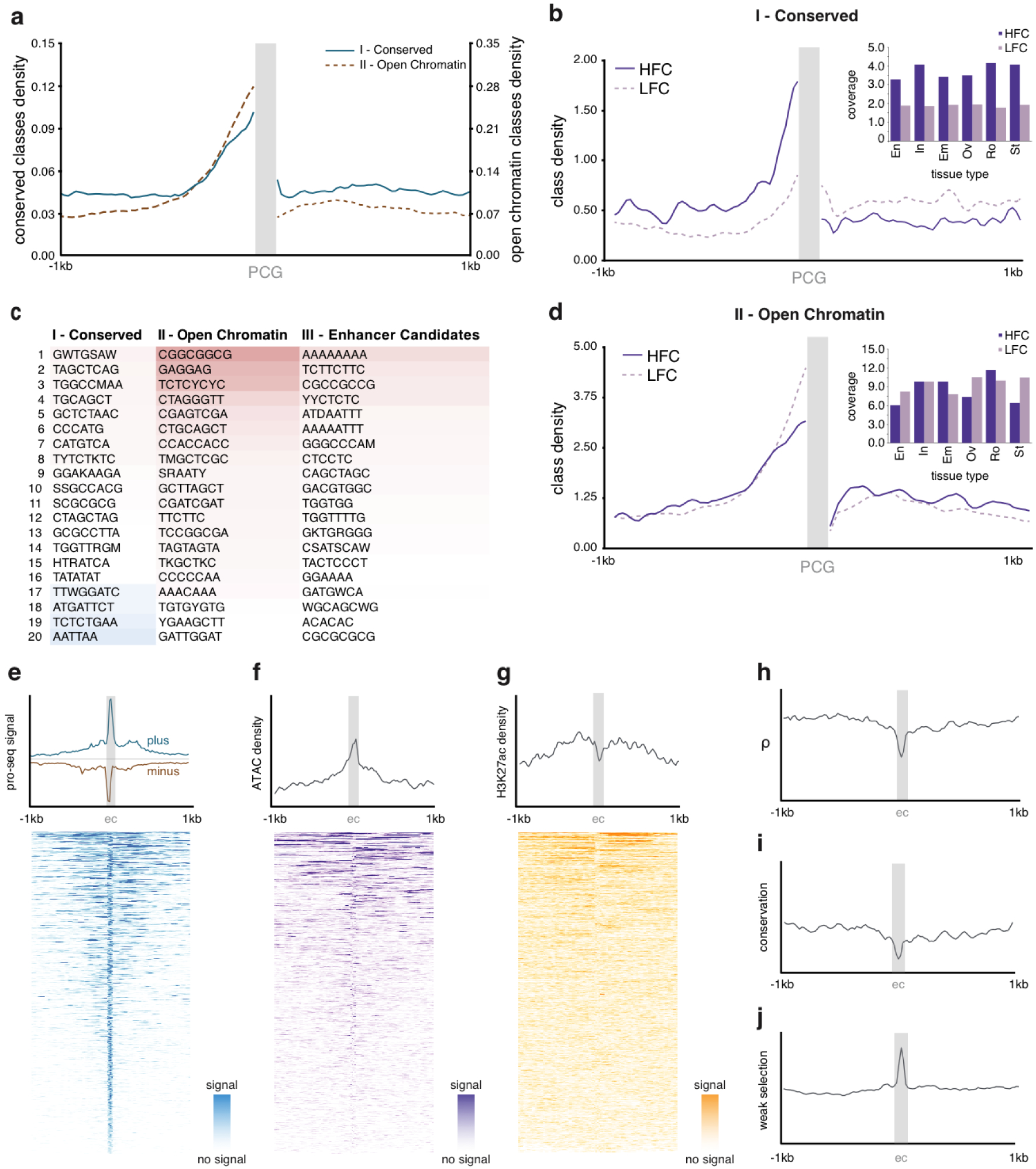
a, The density of higher ρ/conserved classes (category I, blue, solid) and lower ρ/open chromatin classes (category II, red, dotted) around protein-coding genes (PCGs) (grey box) suggest a rice promoter of mean size ≈320 bps b, Regions upstream of genes (grey box) with a high fold-change (HFC – purple line) across tissues are enriched for blocks of conserved classes relative to regions upstream of genes with a stable expression across tissues [low fold-change (LFC - dotted line)]. A breakdown by tissue (inset) suggests that regions upstream of genes with differential expression relative to leaf (En=Endosperm; In=Inflorescence; Em=Embryo; Ov=Ovary; Ro=Root and St=Stamen tissues) consistently show greater promoter coverage with blocks of more conserved classes. c, Motif (6–8 bps) enrichment (blue - red) differs between the three categories of noncoding classes (I - conserved, II- open chromatin and III- enhancer candidate classes). Open chromatin classes are relatively enriched for simple repetitive motifs similar to those found in enhancer candidate regions. d, Genes whose upstream regions are enriched for open chromatin classes rather than conserved classes show a broader activation across tissues, but no similar enrichment for differential expression across tissues. Density plots +/− 1 kb around the 1,000 enhancer candidate (EC) sites show e, a defining bi-directional diverged PRO-seq signal (plus strand – blue, minus strand- red, arbitrary strands) identified by dReg54, f, a marked enrichment for open chromatin, and g, a generally asymmetric H3K27ac location beyond the nucleosome-depleted core (indicated by the dashed rectangle). EC sites are also associated with h, low ρ, i, low conservation, and, j, a two-fold excess of weak negative selection (Pw).
The second category comprises 17 classes, termed “Open Chromatin” classes, that have a broader range of ρ but have ATAC-seq signals that are at ≥10-fold above background (Supplementary Table 5, Supplementary Fig. 7a). While their ρ tends to be similar to ρ for UTRs and promoters (median ρ = 0.256), this ranges from ρ = 0.013 (class 305) to ρ= 0.946 (class 201). These Open Chromatin classes are associated with stable gene expression (lowest fold change: LFC) profiles across multiple tissues (Fig. 5d), and are often enriched for simple tandem repeat motifs (Fig 5c, Supplementary Fig. 7b–c, Supplementary Fig. 9).
The third category includes 11 classes enriched for intergenic bidirectional divergent PRO-seq signals (Fig. 5e). These signals are often characteristic of mammalian enhancer-RNAs53,54, but were also recently suggested in plants36. Using dREG54, to identify enhancer-RNA signals from PRO-seq data, we find 1,000 high-scoring (>1.0) dREG sites in regions >1kb from genes, suggesting that these sites form a set of putative rice enhancer elements (Supplementary Table 11, also see Methods). The dREG locations shared other enhancer-type characteristics, including moderate enrichment for open chromatin (≈7.17 fold) (Fig. 5f), asymmetrically co-located H3K27ac marks (Fig. 5g), and enrichment for motifs similar to those found in open chromatin classes (Fig. 5c, Supplementary Fig. 10). As expected, these 11 classes (e.g. classes 44, 48; Table 1) had a >10 fold enrichment for these high-scoring dREG sites (Supplementary Fig. 7a, Supplementary Table 5) and individual dREG sites were generally found to overlap several of these 11 classes. It was rare for a dREG site, however, to be homogenous across its length with respect to a single fitCons class (Supplementary Table 11).
These 11 classes, termed “Enhancer Candidates”, have weak correlations with the expression of nearby genes (e.g., class 43, r = 0.03; two-tailed t-test, P = 1 × 10−5, N = 24,296) (Supplementary Table 8) consistent with the majority of candidates being in a poised but inactive state. They are also associated with low ρ (< 0.2) (Fig. 5h) and low phastCons conservation (Fig. 5i), but have a > 2-fold excess of sites under weak negative selection (Fig. 5j). The association with weak negative selection was also observed for dREG sites detected in human populations55. Taken together, this may suggest that emergent negative selection, consistent with rapid enhancer turnover, has recently acted on these classes within O. sativa.
DISCUSSION
The INSIGHT and fitCons approaches provide a set of potentially powerful mechanisms for identifying selective constraint on a genome-wide scale. INSIGHT has been used to identify different types of enhancers, such as exon splicing enhancers in human56, shadow enhancers in Drosophila57, and novel motifs like the Coordinator motif found within human cranial neural crest cell specific enhancers58. In the human genome, fitCons maps were revealed to have higher sensitivity than other methods for locating multiple types of functional noncoding elements with putative roles in transcriptional regulation8.
Our fitness consequence map for rice provides a catalogue of putative functional sites that can allow patterns of selection between different genes, genomic regions and genetic pathways to be explored. The map we have developed has limitations; for example, since every base within a fitCons class will receive the same fitCons score (same ρ), we cannot determine which specific bases within a class are under selection. Also, as this is the first instance of inferring a fitCons map in a plant genome, the validation of fitCons scores relative to the actual fitness impacts of mutations remains to be tested. As in every prediction model, subsequent experimental analyses will help shed light on the function of the candidate noncoding regulatory elements that we have described in this study
Nevertheless, some of the broad features of the distribution of ρ in the rice genome confirm what we know about the biology of specific genome elements, suggesting that the inferred ρ values are related to underlying biological features. Integrating evolutionary information with functional genomic and epigenomic data permits identification of important regulatory components of crop genomes10, improving genome annotation, and helping to guide molecular genetic studies. Fitness consequence maps can also help in genetic mapping and crop breeding efforts, including the identification of candidate deleterious mutations59 that can be targeted for removal in next-generation breeding efforts60–62. As more such maps are produced, it will be possible to undertake comparative analyses of genomic selection across species, and help develop more precise genomic breeding programs. To this end, a web interface for greenINSIGHT is available at http://purugganan-genomebrowser.bio.nyu.edu/greenInsight/ and all functional genomic tracks, ρ scores, and fitCons classes for rice can be viewed or downloaded from a custom genome browser (http://purugganan-genomebrowser.bio.nyu.edu/cgi-bin/hgTracks?db=Osaj&position=Osaj.1%3A166356-178595 ).
METHODS
Plant material
Seeds of the cultivated rice Oryza sativa landrace Azucena (IRGC#328; tropical japonica) provided by the International Rice Research Institute (Los Baños, Philippines) were used in this study for functional genomic analyses. Dormancy was broken by incubating seeds for five days at 50°C. Seeds were germinated in water in the dark for 48h at 30°C and were then sown on hydroponic pots suspended in 1x Peters solution and 1.8mM FeSO4 (pH maintained at 5.1–5.8 throughout) (J.R. Peters Inc., Allentown, PA). Plants were grown for 15 days in climate-controlled growth chambers (12h days; 30°C/20°C day/night, 300–500 μmol quanta m−2 s−1; relative humidity 50–70%). Leaf tissue for library construction was collected from 17-day-old, young plants. All freshly collected tissues were wrapped in labeled aluminum foil and immediately immersed in liquid nitrogen, followed by storage at −80°C until further use.
RNA-seq
Total RNA was extracted using the RNeasy Plant Mini Kits (Qiagen, USA), according to the manufacturer’s instructions. RNA quality was determined by BioAnalyzer (Agilent). Contaminating DNA was removed from the total RNA samples by treatment with Baseline-Zero DNase (Epicentre, Madison, WI, USA), and ribosomal RNA was removed using the Ribo-Zero rRNA Depletion Kit (Epicentre, Madison, WI, USA). Strand-specific RNA-seq libraries were synthesized using the Plant Leaf ScriptSeq Complete Kit (Epicentre, Madison, WI, USA). Three biological replicates were generated. Libraries were sequenced using Illumina protocols for 2×100-bp reads on an Illumina HiSeq 2500 at the New York University GenCore facility. The resulting total RNA-seq data was used to identify expressed regions of the rice genome rather than to quantify expression (for expression quantification, see GEO mRNA-seq below). Consequently, reads were 3’ trimmed for quality (q<20) and adapter sequences (Cutadapt 1.1163), and read pairs for which either end was shorter than 25 bps after trimming were rejected. Trimmed reads were aligned to the nuclear chromosomes of the soft-masked IRGSP1.0/MSU7 build of the rice genome (downloaded from Ensembl) using Bowtie264 (bowtie2–2.2.9) with option --sensitive-local. Alignments were converted to bam format (samtools 1.3), sorted, and converted to a bedGraph alignment format with bedtools v2.2565. Reads were subsequently converted to bigWig format (UCSC Kent tools66) for visualization in a custom UCSC Rice genome browser. A single sample was selected as an input in the fitCons approach. The choice of replicate was based on signal strength and sequencing coverage.
Small RNA-seq
For the extraction of small RNA, total RNA was first isolated using the Ambion Plant RNA Isolation Aid (Thermo Fisher Scientific, USA) and sRNA was subsequently extracted using the mirVana miRNA Isolation Kit (Thermo Fisher Scientific, USA). Thirty-five to 70 ng of small RNA served as input to generate libraries using the TruSeq Small RNA Library Prep Kit (Illumina, USA). Three biological replicates were generated. Libraries were sequenced using Illumina protocols for 2×50-bp reads on an Illumina HiSeq 2500 at the New York University GenCore facility. Reads were 3’ trimmed for quality (q<20) and adapter sequences (Cutadapt 1.11), and only reads longer than 15 bps were retained (-m 16). Trimmed reads were aligned to the nuclear chromosomes of the soft-masked IRGSP1.0/MSU7 build of the rice genome downloaded from Ensembl using Bowtie2 (bowtie2–2.2.9) with options --end-to-end, --sensitive and -k 100. Alignments were converted to bam format (samtools 1.3), sorted, and converted to a bedGraph alignment with bedtools v2.25. Reads were subsequently converted to bigWig format (UCSC Kent tools) for visualization in a custom UCSC Rice genome browser. In addition, for the purpose of inferring initial low-resolution chromatin states, signal was averaged over 40nt blocks for input into ChromHMM45. A single sample was selected as an input in the fitCons approach. The choice of replicate was based on signal strength and sequencing coverage.
DNA methylation
For methylation assessment using whole-genome bisulfite conversion, DNA was extracted using DNeasy Plant Mini Kits (Qiagen, USA) following the manufacturer’s protocol. Extracted DNA was sheared into 350-bp fragments using an S220 focused-ultrasonicator (Covaris). The Illumina Truseq DNA Kit (Cat #FC-121–3001) was used to construct the library and the Zymo Lightning Kit (Cat# D5030) to perform the bisulfite treatment. The KAPA Uracil Polymerase (Cat# KK2623) was used to amplify the library with 12 cycles. Two biological replicates were generated. Libraries were sequenced using Illumina protocols for 2×100-bp reads on an Illumina HiSeq 2500 at the New York University GenCore facility. For the purpose of assaying DNA methylation, reads were treated as two independent sets of forward and reverse reads, and were aligned using a basic pipeline generally suitable for all types of plant CG/CHG/CHH methylation (i.e. avoiding filters sometimes found in code designed for human-specific scenarios). Reads were pre-processed in silico by trimming bisulfite sequencing adapters, after which unconverted (methylated) cytosine bases were converted to thymine bases, with a record kept for each read of the location of these transformed bases. Reads were then aligned against both an A-G-T transformed IRGSP1.0/MSU7 genome (in which all cytosine bases had been replaced by thymine bases) and an A-C-T transformed genome (in which all guanine bases had been replaced by adenine bases) using Bowtie2 (bowtie2–2.2.9) with options --end-to-end, --sensitive -k 1. Finally, alignments were processed to combine their location with the offsets within the alignment of the unconverted (methylated) cytosines that had previously been recorded. These refined locations were accumulated in a bedGraph file and subsequently converted to bigWig format (UCSC Kent tools) for visualization in a custom UCSC Rice genome browser. For the purpose of inferring initial low-resolution chromatin states, methylation signal was averaged over 40-bp blocks for input into ChromHMM. A single sample was selected as an input in the fitCons approach. The choice of replicate was based sequencing coverage.
Chromatin accessibility
For ATAC-seq libraries, a dataset previously generated from leaf tissue of the same developmental stage in the Azucena background using the ATAC protocol38 was used (SRR2981235, SRR2981233, SRR2981221, SRR2981227, SRR2981231, SRR2981234). Reads were 3’ trimmed for quality (q<20) and adapters, and were aligned with Novoalign (V3.04.04, novocraft.com) with options -t 80 -a -i PE 500,400 -o SAM. Alignments were converted to bam format (samtools 1.3), sorted, and converted to a bedGraph alignment with bedtools v2.25 followed by conversion to bigWig format (UCSC Kent tools) for visualization in a custom UCSC Rice genome browser. ATAC peaks were called with MACS267 v. 2.1.1 with the genome size, q threshold and ATAC recommended parameters68 “-g 4.0e8 -q 0.025 --nomodel --shift −100 --extsize 200 -B”. For the purpose of inferring initial low-resolution chromatin states, signal was averaged over 40-bp blocks for input into ChromHMM. A single sample was selected as an input in the fitCons approach. The choice of replicate was based on signal strength and sequencing coverage.
ChIP-seq
Two grams of leaf tissue was fixed [Formaldehyde, 1% (v/v] for 15 min, after which glycine was added to final concentration of 125 mM (5 min incubation). Tissues were rinsed three times with cold, de-ionized water before being flash frozen in liquid nitrogen. Chromatin extraction and chromatin shearing were performed using the Universal Plant ChIP-seq Kit (Diagenode) following the manufacturer’s instructions. Protease inhibitor cocktail (Millipore Sigma) was added to the extraction buffer. The samples were sonicated for 4 min on a 30 s ON/30 s OFF cycle using Bioruptor Pico (Diagenode). Subsequent steps were also performed as in the Universal Plant Chip-seq Kit protocol. Immunoprecipitation was done using anti-acetyl-histone H3 (Lys 27) (H3K27ac, Cell Signaling Technology, cat. #4353S, lot #1), anti-trimethyl-histone H3 (Lys27) (H3K27me3, Millipore Sigma, cat. #07–449, lot #2919706), anti-trimethyl-histone H3 (Lys4) (H3K4me3, EMD Millipore, cat. #07–473, lot #2746331), and anti-acetyl-histone H3 (Lys18) (H3K18ac, Cell Signaling Technology, cat. #9675S, lot #1). Quality and fragment size of immunoprecipitated DNA and input samples were measured using agarose gel electrophoresis and TapeStation 2200 (Agilent). Three biological replicates were generated. Libraries were synthesized using the MicroPlex Library Preparation Kit v2 (Diagenode). Libraries were sequenced as 2×50-bp reads on an Illumina HiSeq 2500 instrument at the New York University GenCore facility. Reads were 3’ trimmed for quality (q<20) and adapter sequences (Cutadapt 1.11), and read pairs for which either end was shorter than 16 bps after trimming were rejected. Trimmed reads were aligned to the nuclear chromosomes of the soft-masked IRGSP1.0/MSU7 build of the rice genome (downloaded from Ensembl) using Bowtie2 (bowtie2–2.2.9) with option --end-to-end --sensitive. Alignments were converted to bam format (samtools 1.3), sorted, and converted to a bedGraph alignment format with bedtools v2.25. These were then converted to bigWig format (UCSC Kent tools) for visualization in a custom UCSC Rice genome browser. ChIP-seq peak calling was performed using MACS2 with input DNA used as a control and additional parameters “-g 4.0e8 --bw 200 -B -m 3 50”. For the purpose of inferring initial low-resolution chromatin states, signal was averaged over 40-bp blocks for input into ChromHMM. A single sample was selected as an input in the fitCons approach. The choice of replicate was based on signal strength and sequencing coverage.
PRO-seq
Nuclei isolation.
Nuclei isolation was performed as described in Hetzel et al.69, with modifications. Briefly, around 20 g of leaf tissue from 17-day old plants was collected in a cold room (4°C), placed in ice-cold grinding buffer immediately, and homogenized using a Qiagen TissueRuptor. Samples were filtered through a series of meshes, and pellets were washed twice. This was followed by homogenization, resuspension in storage buffer [10 mM Tris (pH 8.0), 5 mM MgCl2, 0.1 mM EDTA, 25% (vol/vol) glycerol, 5mM DTT], and snap freezing in liquid N2.
Nuclei sorting.
Nuclei were stained with DAPI, and loaded into a flow cytometer (Becton Dickinson FACSAria II). A total of around 15 million nuclei were sorted based on their size and the strength of the DAPI signal, and were subsequently collected in a tube with storage buffer. The nuclei were pelleted by centrifugation at 5000 g at 4°C for 10 min, and resuspended in 100 μl of storage buffer.
PRO-seq library preparation.
PRO-seq was performed as described by Mahat et al.30. This protocol generated strand-specific libraries with every read starting from the 3’ end of the RNA. Two biological replicates were generated. Amplified libraries were assessed for quality on a TapeStation prior to sequencing with 1×50-bp reads on a HiSeq2500 at the New York University GenCore facility. Reads were trimmed and carefully aligned against the soft-masked IRGSP1.0/MSU7 build of the rice genome downloaded from Ensembl and supplemented with chloroplast and mitochondrial plastid sequences (alignments to plastids were not used) using Novoalign (V3.04.04, novocraft.com) with options -o SAM -t 40 -r None -a TGGAATTCTCGGGTGCCAAGG -s 30 -l 25 (similar to bowtie2’s --local setting but with a more quality-informed alignment location for short reads). Alignments were converted to bam format (samtools 1.3), sorted, and finally converted to a bedGraph alignment format with bedtools v2.25. Reads were subsequently converted to strand-specific bigWig format (UCSC Kent tools) for visualization in a custom UCSC Rice genome browser and analysis by the dREG algorithm54. A single sample was selected as an input in the fitCons approach. The choice of replicate was based on signal strength and sequencing coverage.
Read preparation for genome assembly of wild rice Oryza australiensis
Plant material and methods for genome sequencing and assembly.
A voucher specimen of Oryza australiensis (IRGC #86534) was obtained from the International Rice Research Institute (IRRI). Leaf tissue for library construction was collected from 17-day-old, young plants growing in pot conditions in sterilized, premixed soil (50% perlite:vermiculite). Seeds had been previously incubated for 12 days at 50°C in the dark to break dormancy, and subsequently germinated. Plants were grown in climate-controlled growth chambers (11h days; 29.6°C/24.0°C day/night, 300–500 μmol quanta m−2 s−1; relative humidity 60%).
Illumina fragment library and sequencing.
Young leaf tissue was collected, and DNA was extracted using DNeasy Plant Mini Kits (Qiagen, USA) following the manufacturer’s protocol. About 1 μg of DNA was sheared, and the fragmented DNA was used to construct Illumina sequencing libraries. Fragment libraries, with a target insert size of 450 bp, were constructed using the Illumina TruSeq DNA PCR-Free Library Prep Kit and bead-purified (Agencourt AMPure XP beads, Beckman, USA). The fragment libraries were 2×250 bp-sequenced using a HiSeq2500 at the NGS sequencing core at Cold Spring Harbor Laboratory.
Assembly of wild rice genomes
Oryza australiensis reads were 3’ quality- (q<20) and adapter-trimmed using cutAdapt (v 1.11), and were assembled using DiscovarDeNovo (https://software.broadinstitute.org/software/discovar/blog/) with default options. Assembled scaffolds <3 kilobase (kb) in size were discarded along with those scaffolds with GC% > 0.55 (usually corresponding to microbial contaminants). The remaining scaffolds were trimmed for doubly assembled regions (a feature of some versions of the assembler that would cause it to occasionally follow the assembly graph in both directions, resulting in a concatenated forward and reverse sequence). This generated an assembly of size 785 megabase (Mb; 99.98% called bases, 0.02% gaps), in 39,193 scaffolds with a scaffold and contig N50 of 37 kb and 34 kb, respectively. Scaffold sizes ranged from 1.5 kb to 948 kb, similar to the size distributions for the other plant genome assemblies that were used for comparative genomics14.
Oryza officinalis was assembled through a hybrid assembly strategy. Paired-end reads were downloaded from the SRA (DRR000711, DRR003647, DRR003646), and were 3’ trimmed for adapters and low quality sequences (q<20) using cutAdapt (1.9.1). Reads were initially assembled with Ray70 (v 2.3.1) to generate a set of relatively short contigs that were fragmented in silico into a set of overlapping reads (2×100 bp in a 180-bp insert). The overlapping reads and original reads were combined with longer-insert 6-kb and 8-kb mate-pair reads (DRR003207, DRR003206) that were also processed for 3’ adapter trimming. Reads were converted to unaligned BAMs (Picard tools, https://broadinstitute.github.io/picard/) and assembled using AllPathsLG71 (version 52488), resulting in an assembly of size 399 Mb (85% called bases, 15% gaps), in 13,189 scaffolds, with a scaffold N50 of 64 kb, and scaffold lengths ranging from 0.8 kb to 450 kb.
greenINSIGHT
A detailed description of the method is given in Gronau et al.3 We adapted the pipeline for plant genomes (A. thaliana and O. sativa) by modifying those elements that were human-specific, including criteria for the selection of neutral sites, and by introducing several minor adjustments to account for the typically lower depth of sequencing across plant genomes, which causes an increase in noise in the population genetics component of the dataset. The greenINSIGHT pipeline is outlined in Supplementary Fig. 12, its input being a set of genomic alignments used to infer an ancestral probability, a set of base calls across a population of the focal species, and a set of sites that are inferred to be neutral from annotation and functional evidence.
There are three differences in greenINSIGHT relative to the human INSIGHT pipeline: (1) The INSIGHT approach benefits in several ways (e.g. improved alignments, more powerful inference of recent and ancient selection) from a balance between evidence for constraint derived from recent population divergence and that derived from mutations that have accumulated since the ancestral state Z. Consequently, Z was targeted to be the most recent common ancestor (MRCA) of the rice AA genomes, even though this necessarily introduces a risk for introgression. We considered an adjusted INSIGHT model in which the topology of flanking neutral models was dynamically adjusted, but this required the concatenation of neutral blocks in order for sufficient evidence for tree building to be generated. Therefore, we decided instead to interleave the flanking neutral regions as closely as possible with their matched test sites, since the most significant problems with INSIGHT in a species that experienced a high level of inter-specific genomic introgression arise when the flanking sequences have a different ancestry relative to their matched test sequences.
(2) Due to the low fidelity of alignments to TEs, flanking neutral sites had a different distribution in TEs relative to target regions. This created an asymmetry in the alignment fidelity between neutral and non-neutral sites in which the neutral sites likely saw different proportions of read noise. This was quite evident in the plot of Tajima’s D (Supplementary Fig. 13), where site classes that also had an enrichment for TE content showed a significant excess of rare variants. We consequently adjusted θ for neutral sites by blending the per-block θ with the mean θ across all neutral sites in the analysis using a sigmoid function that had a low impact for θ with a central tendency, but increasingly limited large departures in θ.
(3) In order for a base to be introduced into the human INSIGHT model, it required a valid ancestral state probability and a distribution of {A,C,T,G} base calls over virtually the entire population. In the greenINSIGHT scenario with relatively lower sequencing depth and potentially poorer alignments in some repetitive regions, we allowed slightly more missing population data (0–10%), and used the call-adjusted proportion of base counts to determine whether polymorphic bases were at a high or low population frequency.
A notable difference in the use of this pipeline in rice is the combination of extensive LD and short introns (mean intron size: rice ~400 bp, humans ~15 kb) in rice. This combination may introduce more indirect background selection into ρ in the rice model.
Chromatin state modeling
Chromatin states were inferred based on a set of chromatin marks (ATAC-seq, DNA methylation, H3K27ac, small-RNA associated, H3K4me3, H3K18ac, and H3K27me3) that had been found through earlier work in Arabidopsis (unpublished) to be informative of chromatin states both in coding and non-coding regions of plant genomes. Because the ChromHMM pipeline uses a single sample per covariate, the replicate with the highest signal-to-background was selected. This sample is available for viewing and downloading in the UCSC Browser. The hidden-Markov modeler ChromHMM was used essentially as described in Gulko et al.8 to binarize chromatin signals from 40-bp genomic windows - the mean size of regulatory elements previously suggested in plants14 - and produce a low-resolution map of chromatin states in the rice genome. We initially sought to use a parameter-count penalized log-likelihood ratio (LLR) for each model to determine the number of distinct chromatin states in the rice genome, cognizant of earlier principal component analyses (PCAs) that had suggested that plant genomes have very few states47 – a number of states similar to Drosophila48 - and more recent ChromHMM analyses that described a relatively complex patterning of states32. While there was no distinct inflection in the penalized LLR curve that could inform a high-confidence cutoff, it appeared that after c. 15 states, the addition of more states became relatively less informative. We therefore sought to support the selection of the number of states with the ChromHMM tool CompareModels46, which compares the correlation between the emissions of models with different numbers of states. Selecting a 50-state model as an over-parameterized reference model, we found that by the time 20 states were incorporated in the model, the mean state correlation with the closest state in the 50-state model exceeded 0.9. We therefore selected a 20-state ChromHMM model, but recognize that the optimum value here may not be independent of the number and type of chromatin marks used as input.
Determination of fitCons classes from ChromHMM states
The 20 chromatin states determined by ChromHMM were intersected with additional annotation and functional genomic datasets by setting bits in a 16-bit-wide bitmap of the genome depending on the class value and the binary combination of additional binary annotation. Functional genomic tracks and annotations were binarized for the bitmap at signal (alignment depth) thresholds chosen to differentiate presence/absence of signal against the track-specific noise background for alignment classes, and split annotation classes marked as 0/1: mRNA = 40 reads, PRO-seq = 7 reads, phastCons = 0.7 score, Exon = 0.5, and CDS = 0.5. This had the potential to generate 640 classes (note that class labels do not run continuously due to padding left in the bitmap for additional chromatin states). However, many of the states were either not found or found only very rarely, such that they could not be informatively used to infer a state-specific value of ρ. Consequently, states with coverage <20 kb were excluded and 246 final states were characterized by INSIGHT.
Genomic annotations
Neutral sites.
The collection of sites predicted to be neutral was obtained by eliminating from all genomic sites those likely to be under direct or indirect selection, including (i) annotated protein-coding genes and the 1,000-bp flanking regions on either side of IRGSP1.0.37 annotated genes; (ii) conserved noncoding elements (see below); (iii) locations associated with an open chromatin (ATAC) signal; (iv) sites with a coding potential predicted by a variety of other approaches including fgenesh, Phytozome XI, RAP-DB and blastp of all rice-lineage proteins against the genome; and (v) regions with a total expression depth >50 reads. Due to the limited intergenic annotation available in the rice genome compared to the human genome, this leaves open the possibility that sites in neutral regions could more often intersect functional domains in rice than in humans. As neutral regions form a baseline relative to which ρ is determined, this makes it possible that mostly-neutral classes in rice are more likely to be scored with a near-zero ρ in rice than in humans.
Genome annotations.
IRGSP 1.0.37 gene and ncRNA (pre-miRNA, lncRNA, snoRNA, rRNA, tRNA) annotations were used (Ensembl:ftp://ftp.ensemblgenomes.org/pub/plants/release-37/gff3/oryza_sativa) for annotation of the MSU7 assembly. Mature miRNA annotations were generated from the intersection of miRBase 22 (http://www.mirbase.org/) mature miRNA coordinates with IRGSP 1.0.37 pre-miRNA coordinates. Where gene IDs required conversion between the MSU7 and RAP-DB naming schemes (for example in the expression correlation analysis), the 2018 translation table from RAP-DB (https://rapdb.dna.affrc.go.jp/download/irgsp1.html) was used. Consensus annotations for genomic features were obtained from the featureBits program (http://www.soe.ucsc.edu/~kent/src/unzipped/hg/featureBits) from the intersection of IRGSP, MSU7, RAP-DB, and IOMAP annotations.
PhastCons and Conserved Noncoding Sequences (CNSs)
PhastCons1 (http://compgen.cshl.edu/phast/) scores of tribe-level inter-species conservation were generated as one input into the genome classification process. The process of multiple species alignment was carried out using the Kent alignment pipeline with minor adjustments, essentially as described in Haudry et al.14. Following the Multiz72 multiple alignment step, a global neutral evolutionary model was generated from the neutral sequence locations (described above) using phyloFit73 and input into phastCons (--target-coverage 0.11 --expected-length 50) along with the multiple sequence alignment to infer a per-base score for non-neutral evolution across the genome. Because both direct and indirect introgression into the reference genome from the set of non-reference genomes being compared can be readily mistaken as a signal of constraint, we avoided using non-reference rice genomes with evidence in the literature for extensive introgression with O. sativa. The inference of conservation at a medium resolution (>10 bps) can most clearly be made when neutral divergence has introduced substitutions at a higher rate in some parts of the genome than in others. However, for this inference these neutrally diverged regions need to be still similar enough to remain alignable to the reference genome. This creates an optimal neutral divergence level in the phylogeny where the total neutral branch length between the reference and non-reference species is 0.1 – 0.3. Since diploid, assembled rice genomes were not common in this range, we chose to assemble two additional rice genomes, O. australiensis and O. officinalis, to a locus-level (N50 = 10–50 kb). This brings the total number of genomes used for the comparative analysis to eight (see Supplementary Fig. 1). Once phastCons scores had been generated, a set of conserved noncoding sequences were inferred by selecting regions with a phastCons score >0.82 and a length longer than 11 bps (a combination intended to reduce the chance of coincidental undiverged alignments generating a conserved region) that did not overlap CDSs or CDS boundaries, and that did not overlap regions that had a possibility of having been protein-coding in the immediate evolutionary past (inferred through blastp of all rice tribe proteins against the O. sativa genome).
Enrichment of classes by annotation features
Genome-wide enrichment between genomic classes and annotated features used featureBits (Kent tools, UCSC) with bed file inputs of class and feature locations, and the optional parameter -enrichment for the estimation of enrichment relative to an independent and identically distributed assumption enabled.
ρ density by annotation
A genome-wide bedGraph of ρ values was created from the union of all 246 class-specific bedGraphs of per-class ρ. This was in turn intersected with the locations of seven annotations [neutral regions, distal (≥ 1 kb) CNSs, 500 bp upstream promoters, 5’ UTR, introns, CDSs, and 3’ UTRs] using bedTools2 in intersect mode with ‘awk’ used to generate a single line ρ score and annotation type for each base of the bedGraph ranges. The seven per-base ρ files were combined and displayed using the violin plot (geom_violin) function of the ggplot2 package with bandwidth set to 0.03, and the class median plotted as a single white point for each annotation.
Motif enrichment
Motif enrichment was determined using Homer (v. 4.10, http://homer.ucsd.edu/homer/) findMotifs with options -mset plants -len 6,7,8 enabled, and permuted sets of the input sequences used as controls.
Expression correlation analysis
Correlation between genomic class distributions and the expression of proximate genes used aggregated transcriptome profiles across all leaf tissues (excluding flag leaf) from NCBI GEO (GSE21494). Expression was used as per-array median-normalized values. For the immediate upstream, downstream and slightly more distal enhancer locations around each gene, featureBits (Kent tools, UCSC) was used to generate a count of total base coverage for each genomic class that was in turn correlated class-wise with expression.
Multiple regressions of gene expression and genomic class
A multiple linear regression model for all 246 classes was generated in SPSS (IBM statistics, version 20) by combining individual models of class density around expressed genes (see expression correlation analysis above) for rice chromosomes 2 – 12. To reduce the likelihood of over-fitting, the model was then tested for the correlation between predicted levels of log gene expression from class density with the actual expression of genes on chromosome 1 only (Fig. 2). To somewhat explore the possibility that different class distributions around silent and expressed genes are a consequence of gene expression (arising from the spread of activation epigenomic marks into upstream regions) rather than an indication of regulatory changes causative of expression, for the upstream model we masked the immediate 50-bps upstream promoter region where ATAC- and PRO-seq signals from highly expressed genes can leak into the promoter region. The contour aerial density plot was generated with Raw Graphs (http://app.rawgraphs.io/).
dREG
PRO-seq signal was analyzed on the Cornell community dREG GPU server for sites with an enhancer potential (https://dreg.dnasequence.org/). Signal was entered as alignment depth bigWig files (see PRO-seq section) separately for nascent RNA with a Crick-and-Watson origin, and with other options set to their defaults. This generated 58,920 candidate sites with a signal range from 0.32 to 1.46. Since many of these sites had only a very weak PRO-seq signal, we selected the top c. 5% of sites (3,600 sites) with signal ≥ 1.0 for further characterization. These were in turn refined for exactly 1,000 locations at least 1 kb distal of genes (featureBits) - while enhancers can be located within gene and promoter regions, without high resolution looping predictions we were unable to distinguish these sites from regular promoters and splice elements.
Chromatin mark correlations
For each annotation class (TE, upstream500, upstream25, CDS, intronic), chromatin signal for each chromatin mark used in the ChromHMM model was summed for each location and standardized for location length. To avoid spurious correlations in introns related to TE content, TEs were masked from intronic regions. Similarly, to avoid correlations characteristic of introns in TEs, only TEs 1 kb distal of genes were considered. Correlations between marks in each annotation class were then visualized using the R corrplot package (https://github.com/taiyun/corrplot).
Profiles of feature density
Profiles of bedGraph signal 1 kb either side of features of interest were generated by dividing the upstream and downstream regions into 50 × 20-bp blocks, and the interior of the feature into 5 equally sized blocks. Signal within and around the features was then totaled from the bedGraph files at each region to create (i) a composite profile of total signal integrated over all the features, and (ii) a representation of the distribution of signal at each location as a set of single lines where more intense color represents more signal and total signal is used to order the lines.
Minor allele frequency shift
For each of the 246 fitCons classes, the frequency distribution of the minor allele was calculated from a summary of alleles derived from the 3k RGP11 VCFs. To contrast these values with ρ generated for each class by INSIGHT a metric was generated that contrasted the standardized minor allele distribution of each class relative to the standardized minor allele frequency distribution of class 11, the largest neutral class covering 22% of the rice genome with a nominal ρ close to zero. The metric was generated by adding the excess of rare alleles in the class of interest in frequency classes 1 and 2 (i.e. sites where there were only 1 or 2 members of the populations with segregating variants) relative to class 11, to the excess of more common alleles in frequency classes 4, 5 and 6 in class 11 relative to the class of interest.
Gene Ontology
ArgriGO version 1.2 (http://bioinfo.cau.edu.cn/agriGO/) was used for GO analysis.
URLs
dREG, https://dreg.dnasequence.org;
3k RG project, https://www.ncbi.nlm.nih.gov/bioproject/PRJEB6180/
NCBI (accession PRJEB6180);
greenINSIGHT page, http://purugganan-genomebrowser.bio.nyu.edu/greenInsight/
Accession codes
All epigenomic data tracks, genome annotations, multiple alignments, conservation scores, fitCons scores and site classes are available for visualization and download on a local installation on the USCSC Genome Browser at http://purugganan-genomebrowser.bio.nyu.edu, as well as available for download from the NCBI SRA (<SRA identifier here>). The greenINSIGHT-specific code and data used to generate the greenINSIGHT online tool, as well as the code described in the Methods are available in the Additional Materials section at http://purugganan-genomebrowser.bio.nyu.edu/insightJuly2018/greenInsight.html.
Supplementary Material
Supplementary Table 1 | Information about the population data used in this study.
Supplementary Table 2 | Summary of sequencing data.
Supplementary Table 3 | Correlation matrices by types of annotation.
Supplementary Table 4 | Overlap between CpG and non-CpG context across the genome for methylation profiles.
Supplementary Table 5 | Summary of 246 FitCons classes.
Supplementary Table 6 | Overview of the 246 classes distribution across the 12 chromosomes.
Supplementary Table 7 | Resampling analysis to validate class coherence.
Supplementary Table 8 | Correlation of class occurrence with gene expression in leaf tissue.
Supplementary Table 9 | Folded SFS for 1,476 O. sativa individuals.
Supplementary Table 10 | Gene ontology overrepresentation of genes associated with the noncoding "conserved classes".
Supplementary Table 11 | 1,000 dREG site coordinates and class enrichment.
Supplementary Figure 1 | Phylogeny of the 8-way alignment of rice genomes.
Supplementary Figure 2 | PRO-seq read distribution within and around protein-coding genes.
Supplementary Figure 3 | Emission parameter correlation comparison heat map.
Supplementary Figure 4 | ρ scores validation between randomized fitCons classes and actual fitCons classes.
Supplementary Figure 5 | Log expression predicted from fitCons class distribution.
Supplementary Figure 6 | Intron ρ scores and effect of nearby coding sequences.
Supplementary Figure 7 | Three categories of intergenic fitCons classes highlighted in this study.
Supplementary Figure 8 | Motif analysis for the Conserved classes.
Supplementary Figure 9 | Motif analysis for the Open Chromatin classes.
Supplementary Figure 10 | Motif analysis for the Enhancer Candidates classes.
Supplementary Figure 11 | Outline of the greenINSIGHT pipeline.
Supplementary Figure 12 | Tajima's D for all fitCons classes.
ACKNOWLEDGEMENTS
We thank the New York University Center for Genomics and Systems Biology GenCore Facility and the NGS sequencing core at Cold Spring Harbor Laboratory for sequencing support. We thank Dr. Olivia Wilkins and Dr. Charles Danko for their valuable suggestions with the ATAC and PRO-seq protocols, respectively. This work was supported primarily by a grant from the Zegar Family Foundation, as well as some support from the National Science Foundation Plant Genome Research Program and the NYU Abu Dhabi Research Institute to M.D.P., NSF Career (MCB-1552455), NIH-MIRA (R35GM124806), and USDA-Hatch (1012915) to X.Z., (R35GM127070) to A.S., and fellowships from the Gordon and Betty Moore Foundation/Life Sciences Research Foundation (GBMF2550.06) to S.C.G., and the Natural Sciences and Engineering Research Council of Canada (PDF-502464-2017) to Z.J-L.
Footnotes
The authors declare no competing interests.
REFERENCES
- 1.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schrider DR & Kern AD Inferring selective constraint from population genomic data suggests recent regulatory turnover in the human brain. Genome Biol. Evol 7, 3511–3528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gronau I, Arbiza L, Mohammed J & Siepel A Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol. Biol. Evol 30, 1159–1171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald JH & Kreitman M Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Sawyer SA & Hartl DL Population genetics of polymorphism and divergence. Genetics 132, 1161 LP–1176 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bustamante CD et al. Natural selection on protein-coding genes in the human genome. Nature 437, 1153–1157 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Smith NGC & Eyre-Walker A Adaptive protein evolution in Drosophila. Nature 415, 1022–1024 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Gulko B, Hubisz MJ, Gronau I & Siepel A A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet 47, 276–283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gulko B & Siepel A An evolutionary framework for measuring epigenomic information and estimating cell-type-specific fitness consequences. Nat. Genet 51, 335–342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wing RA, Purugganan MD & Zhang Q The rice genome revolution: from an ancient grain to Green Super Rice. Nat. Rev. Genet 19, 505–517 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Wang W et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557, 43–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein JC et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet 50, 285–296 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Cao J et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet 43, 956–963 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Haudry A et al. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet 45, 891–898 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Huang X et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutaker RM et al. Genomic history and ecology of the geographic spread of rice. bioRxiv 748178 (2019). doi: 10.1101/748178 [DOI] [PubMed] [Google Scholar]
- 17.Josephs EB, Lee YW, Stinchcombe JR & Wright SI Association mapping reveals the role of purifying selection in the maintenance of genomic variation in gene expression. Proc. Natl. Acad. Sci 112, 15390–15395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flowers JM et al. Natural selection in gene-dense regions shapes the genomic pattern of polymorphism in wild and domesticated rice. Mol. Biol. Evol 29, 675–687 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Caicedo AL et al. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet. 3, 1745–1756 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradnam KR & Korf I Longer first introns are a general property of eukaryotic gene structure. PLoS One 3, e3093 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigau M, Juan D, Valencia A & Rico D Intronic CNVs and gene expression variation in human populations. PLOS Genet. 15, e1007902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess DG, Xu J & Freeling M Advances in understanding cis regulation of the plant gene with an emphasis on comparative genomics. Curr. Opin. Plant Biol 27, 141–147 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Freeling M, Rapaka L, Lyons E, Pedersen B & Thomas BC G-boxes, bigfoot genes, and environmental response: characterization of intragenomic conserved noncoding sequences in Arabidopsis. Plant Cell 19, 1441–1457 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi HI, Hong JH, Ha JO, Kang JY & Kim SY ABFs, a family of ABA-responsive element binding factors. J. Biol. Chem 275, 1723–1730 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Lu T et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 20, 1238–1249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng T et al. Differentially expressed microRNA cohorts in seed development may contribute to poor grain filling of inferior spikelets in rice. BMC Plant Biol. 14, 196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 2015, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng S et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci 107, 8689 LP–8694 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahat DB et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwak H, Fuda NJ, Core LJ & Lis JT Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science (80-. ) 339, 950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y et al. PCSD: A plant chromatin state database. Nucleic Acids Res. 46, D1157–D1167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan W et al. Dynamic control of enhancer activity drives stage-specific gene expression during flower morphogenesis. Nat. Commun 10, 1–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen M et al. Expression variations of miRNAs and mRNAs in rice (Oryza sativa). Genome Biol. Evol 8, 3529–3544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zong W, Zhong X, You J & Xiong L Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol. Biol 81, 175–188 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Lozano R et al. RNA polymerase mapping in plants identifies enhancers enriched in causal variants. bioRxiv (2018). doi: 10.1101/376640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia J et al. Detecting and characterizing microRNAs of diverse genomic origins via miRvial. Nucleic Acids Res. 45, e176–e176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkins O et al. EGRINs (Environmental Gene Regulatory Influence Networks) in rice that function in the response to water deficit, high temperature, and agricultural environments. Plant Cell 28, 2365–2384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan F et al. Analysis of chromatin regulators reveals specific features of rice DNA methylation pathways. Plant Physiol. 171, 2041–2054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C, Lu F, Cui X & Cao X Histone methylation in higher plants. Annu. Rev. Plant Biol 61, 395–420 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Liu N, Fromm M & Avramova Z H3K27me3 and H3K4me3 chromatin environment at super-induced dehydration stress memory genes of arabidopsis thaliana. Mol. Plant 7, 502–513 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Fang H, Liu X, Thorn G, Duan J & Tian L Expression analysis of histone acetyltransferases in rice under drought stress. Biochem. Biophys. Res. Commun 443, 400–405 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Du Z et al. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol. Plant 6, 1463–1472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee T, Zhai J & Meyers BC Conservation and divergence in eukaryotic DNA methylation. Proc. Natl. Acad. Sci 107, 9027 LP–9028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ernst J & Kellis M Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc 12, 2478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roudier F et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 30, 1928–1938 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sequeira-Mendes J et al. The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell 26, 2351–2366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu C et al. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 26, 1057–1068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo H & Moose SP Conserved noncoding sequences among cultivated cereal genomes identify candidate regulatory sequence elements and patterns of promoter evolution. Plant Cell 15, 1143–1158 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berendzen KW et al. Bioinformatic cis-element analyses performed in Arabidopsis and rice disclose bZIP- and MYB-related binding sites as potential AuxRE-coupling elements in auxin-mediated transcription. BMC Plant Biol. 12, 125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu L, Xu W, Hu X, Liu H & Lin Y W-box and G-box elements play important roles in early senescence of rice flag leaf. Sci. Rep 6, 20881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding M et al. Enhancer RNAs (eRNAs): New insights into gene transcription and disease treatment. J. Cancer 9, 2334–2340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, Chu T, Choate LA & Danko CG Identification of regulatory elements from nascent transcription using dREG. Genome Res. 29, 293–303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Danko CG et al. Dynamic evolution of regulatory element ensembles in primate CD4+ T cells. Nat. Ecol. Evol 2, 537–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savisaar R & Hurst LD Exonic splice regulation imposes strong selection at synonymous sites. Genome Res. 28, 1442–1454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cannavò E et al. Shadow Enhancers Are Pervasive Features of Developmental Regulatory Networks. Curr. Biol 26, 38–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prescott SL et al. Enhancer Divergence and cis-Regulatory Evolution in the Human and Chimp Neural Crest. Cell 163, 68–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mezmouk S & Ross-Ibarra J The pattern and distribution of deleterious mutations in maize. G3 Genes, Genomes, Genet. 4, 163–171 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wallace JG, Rodgers-Melnick E & Buckler ES On the Road to Breeding 4.0: Unraveling the Good, the Bad, and the Boring of Crop Quantitative Genomics. Annu. Rev. Genet 52, 421–444 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Moyers BT, Morrell PL & McKay JK Genetic costs of domestication and improvement. J. Hered 109, 103–116 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Morrell PL, Buckler ES & Ross-Ibarra J Crop genomics: Advances and applications. Nat. Rev. Genet 13, 85–96 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12 (2011). [Google Scholar]
- 64.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kent WJ BLAT--the BLAST-like alignment tool. Genome Res. 12, 656–64 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raurell-Vila H, Ramos-Rodríguez M & Pasquali L Assay for Transposase Accessible Chromatin (ATAC-Seq) to Chart the Open Chromatin Landscape of Human Pancreatic Islets BT - CpG Islands: Methods and Protocols. in (eds. Vavouri T & Peinado MA) 197–208 (Springer; New York, 2018). doi: 10.1007/978-1-4939-7768-0_11 [DOI] [PubMed] [Google Scholar]
- 69.Hetzel J, Duttke SH, Benner C & Chory J Nascent RNA sequencing reveals distinct features in plant transcription. Proc. Natl. Acad. Sci (2016). doi: 10.1073/pnas.1603217113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boisvert S, Raymond F, Godzaridis É, Laviolette F & Corbeil J Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 13, R122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Butler J et al. ALLPATHS : De novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green ED et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14, 708–715 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siepel A & Haussler D Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol 21, 468–488 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 | Information about the population data used in this study.
Supplementary Table 2 | Summary of sequencing data.
Supplementary Table 3 | Correlation matrices by types of annotation.
Supplementary Table 4 | Overlap between CpG and non-CpG context across the genome for methylation profiles.
Supplementary Table 5 | Summary of 246 FitCons classes.
Supplementary Table 6 | Overview of the 246 classes distribution across the 12 chromosomes.
Supplementary Table 7 | Resampling analysis to validate class coherence.
Supplementary Table 8 | Correlation of class occurrence with gene expression in leaf tissue.
Supplementary Table 9 | Folded SFS for 1,476 O. sativa individuals.
Supplementary Table 10 | Gene ontology overrepresentation of genes associated with the noncoding "conserved classes".
Supplementary Table 11 | 1,000 dREG site coordinates and class enrichment.
Supplementary Figure 1 | Phylogeny of the 8-way alignment of rice genomes.
Supplementary Figure 2 | PRO-seq read distribution within and around protein-coding genes.
Supplementary Figure 3 | Emission parameter correlation comparison heat map.
Supplementary Figure 4 | ρ scores validation between randomized fitCons classes and actual fitCons classes.
Supplementary Figure 5 | Log expression predicted from fitCons class distribution.
Supplementary Figure 6 | Intron ρ scores and effect of nearby coding sequences.
Supplementary Figure 7 | Three categories of intergenic fitCons classes highlighted in this study.
Supplementary Figure 8 | Motif analysis for the Conserved classes.
Supplementary Figure 9 | Motif analysis for the Open Chromatin classes.
Supplementary Figure 10 | Motif analysis for the Enhancer Candidates classes.
Supplementary Figure 11 | Outline of the greenINSIGHT pipeline.
Supplementary Figure 12 | Tajima's D for all fitCons classes.