

Abstract
We present a non-redundant benchmark, coined PepPro, for testing peptide-protein docking algorithms. Currently, PepPro contains 89 non-redundant experimentally determined peptide-protein complex structures, with peptide sequence lengths ranging from 5 to 30 amino acids. The benchmark covers peptides with distinct secondary structures, including helix, partial helix, a mixture of helix and β-sheet, β-sheet formed through binding, β-sheet formed through self-folding, and coil. In addition, unbound proteins’ structures are provided for 58 complexes, and can be used for testing the ability of a docking algorithm handling the conformational changes of proteins during the binding process. PepPro should benefit the docking community for the development and improvement of peptide docking algorithms. The benchmark is available at (http://zoulab.dalton.missouri.edu/PepPro_benchmark).
Keywords: peptide docking, protein-peptide docking, benchmark, protein-peptide complexes, protein-peptide interactions
INTRODUCTION
Peptide-mediated interactions play crucial roles in cellular processes by regulating up to 40% of all protein-protein interactions (PPIs) [1]. Targeting PPIs for therapeutic purposes has gained increased attention in recent years [2-3]. As natural compounds, peptides are excellent candidates to target protein-protein interfaces [4-5]. Interaction details of a peptide with a targeting protein are important for the peptide-based drug design. However, determining peptide-protein complex structures using experimental methods (e.g. X-ray and NMR) is costly and time-consuming. Recently, a number of in silico approaches have been developed as a complementary strategy for studying peptide-protein interactions.
The exiting methods for the prediction of peptide-protein complex structures can be categorized into three classes: template-based modeling, molecular docking, and molecular dynamics (MD) simulation. The template-based methods are computationally efficient, but suffer from limited available peptide-protein templates [7-8]. On the other hand, MD simulations require intensive computations and are impractical for peptide sequence-based structure prediction and large-scale applications [8-9]. Molecular docking offers a balance between accuracy and computational efficiency [10-20]. Current existing protein-peptide complex structure prediction methods were carefully reviewed by Ciemny et al. [21].
A peptide-protein docking method mainly consists of two components, a sampling algorithm and a scoring function. The sampling algorithm generates putative peptide binding modes for a given protein, starting from the peptide sequence and the 3D atomic structure of the protein. The scoring function attempts to select near-native binding modes from these putative binding modes. One major challenge in peptide-protein docking is the flexibility problem - for both the peptide and the protein - resulting in large degrees of freedom for sampling. The second challenge is the scoring function, which involves the inter- and intra-energies of the peptide-protein complex, the solvent effect, and the entropic effect.
To facilitate the study of peptide-protein interactions, several peptide-protein structural datasets have been developed [22-25]. Two of them are suitable for the development of peptide-protein sampling/scoring algorithms. The first dataset is peptiDB, which focuses on short peptides with sequence lengths ranging from 5 to 15. The commonly-used peptiDB dataset consists of 103 peptide-protein complex structures; 69 of them have unbound protein structures [23]. It is noteworthy that peptiDB contains only 61 truly non-redundant peptide-protein complexes (around 40 of them have experimentally determined unbound protein structures), but most published studies employed the whole (redundant) dataset. The other example is a recently developed dataset, LEADS-PEP [25], which also focuses on short peptides with peptide lengths ranging from 3 to 12 residues. LEADS-PEP contains 53 peptide-protein complex structures, but unbound protein structures are not provided in its current version.
These useful peptide docking benchmarks restrict peptide lengths to within 15 residues. However, longer peptides (more than 15 a.a.) commonly exist in nature. According to the data (Jan. 26th, 2017) in the Protein Data Bank (PDB) [26], there were 9286 entries having at least one chain with a peptide length ranging from 5 to 30 residues, and about 40% of them fell into the range between 16 to 30. The restriction of the peptide length (less than or equal to 15 a.a.) in the existing peptide docking benchmarks mainly avoids the difficulty of conformational sampling for long, highly flexible peptides, because the sampling space grows rapidly as the peptide length increases. Fortunately, this barrier has gradually declined with the newly developed novel sampling algorithms [10-21] and the rapidly increasing computational power of modern computers. As it can be seen in our recent study based on peptiDB, the novel peptide docking method MDockPeP successfully generated near-native binding modes in 95.0% of the bound docking cases and in 92.2% of the unbound docking cases, respectively [15]. There has never been a better time to step forward towards developing novel docking algorithms for long peptides (more than 15 a.a.). Consequently, a new peptide docking benchmark with a larger range of peptide lengths is urgently needed.
Here, we present a nonredundant peptide-protein complex structural dataset, referred to as PepPro, with peptide lengths ranging from 5 to 30. PepPro consists of 89 peptide-protein complex structures and 58 unbound protein structures. The dataset can be divided into several subsets based on secondary structures of bound peptides: α-helix (H), partial α-helix (pH), α-helix/β-sheet (HE), β-sheet formed through binding (bE), β-sheet formed through self-folding (sE), and coil (C). The interface root-mean-square deviation (I-RMSD) of bound and unbound protein structures are provided to reflect the conformational changes on peptide binding. The dataset would benefit the development and improvement of peptide docking algorithms for the docking community.
MATERIALS AND METHODS
Data collection
The PepPro benchmark was constructed using a semi-automatic pipeline. First and foremost, peptide-protein complex structures were collected from the PDB using the following criteria: (1) The structure was experimentally determined by X-ray diffraction techniques. (2) The resolution was better than 2.5 Å. (3) The Rfree value was below 0.3. (4) The pH value was between 6.0 and 8.0. (5) The complex structure contained two or three protein chains. (6) There was at least one chain with a sequence length between 5 and 30. (7) The structure did not contain any modified residues. Finally, a total of 1198 PDB entries were downloaded as of 26/01/2017.
Next, the chain with a sequence length between 5 and 30 was defined as a peptide for each PDB entry. The chain directly interacting with the peptide (i.e., at least one atom pair across the peptide-protein interface within 5.0 Å distance) were defined as the protein partner for the peptide. The peptide-protein complex was discarded in the following cases: (1) The percentage of solved peptide sequence length (i.e., the ratio of the number of amino acids having coordinates in the PDB file to the full sequence length of the peptide) was less than 70%. (2) The peptide was broken, having backbone atoms with missing coordinates in the middle of the peptide rather than at the terminals. (3) The peptide contained covalent bonds between non-adjacent residues. (4) Small molecules (with molecular weight ≥ 140 g/mol) or ions appeared on the peptide-protein interfaces (i.e., within 5.0 Å distance to any atom in the peptide and to any atom in the protein). (5) The peptide was covalently bound to the protein.
Furthermore, in order to remove the entries in which only a few contacts appeared between a peptide and its protein partner, the percentage of buried surface area of the peptide upon binding was used as a threshold criterion. More specifically, the solvent-accessible surface area (SASA) of the peptide alone (the protein partner was deleted from the complex structure) was calculated using the program Naccess V2.1.1 [27], referred to as SASApep. The SASA of the peptide in the peptide-protien complex was also calculated and referred to as SASApep’. Thus, the buried percentage of the peptide was calculated by ΔSASA/SASApep, where ΔSASA = (SASApep - SASApep’). The entry was discarded if the value of the buried percentage was < 25%. For the case in which the protein partner contains two chains, the entry was also discarded if the contribution of ΔSASA from any protein chain was less than one third of the total ΔSASA. Notably, the maximum number of protein partner chains in the PepPro benchmark was set to 2. Redundancies were removed for the remaining PDB entries (712), as described in the next subsection.
Treatment of redundancy
Peptides interact with proteins usually through select conserved residues, which correspond to the hot-spot residues in the peptide-protein complex structures [23,28]. These peptide sequences are also known as linear motifs, in which a few conserved residues contribute the majority of the free energy of binding [29]. Furthermore, proteins within a superfamily often bind peptides with the same linear motif. A typical example is the superfamily of nuclear receptors (NR), which bind peptides with the LXXLL binding motif (L is leucine and X stands for any residues), resulting in 10 redundant entries in the peptiDB benchmark when only the sequence identity was used as the threshold. Proteins with low sequence similarity (< 30%) could share a similar fold and also a similar binding site for the binding of a peptide partner, as observed for proteins from the NR superfamily. Therefore, to efficiently remove redundancies for peptide-protein complexes, both the sequence identity and the structural similarity need to be considered for protein partners.
In the preparation of the PepPro benchmark, protein sequence identities were calculated using the global sequence alignment program EMBOSS Needle [30]. Structural similarities were calculated using the program TM-align (TM-Score) [31]. A greedy clustering strategy was employed to extract representative peptide-protein complex structures for the benchmark. Specifically, peptide-protein complexes were sorted in decreasing order of the peptide sequence length. Entries with the same peptide sequence length were further sorted in decreasing order of the protein sequence length. If entries happened to contain the same sequence length for both peptides and proteins, they were sorted in increasing order of the Rfree value (i.e., the measure of the quality of the atomic model obtained from crystallographic data). Then, a new list (referred to as the representative list) was created to store the selected representative non-redundant peptide-protein complex structures. For each query entry in the sorted list, both sequence identities and TM-Scores for the protein partner (against protein partners in the representative list) were calculated. If there was not a similar protein partner (the sequence similarity was ≥ 30% or TM-Score ≥ 0.5) found in the representative list, the query entry was set as a new representative and added to the representative list. Noticeably, this clustering strategy generated a representative list in which protein partners were distinct from each other (based on the sequence and the structure), and the peptide in a representative complex was longer than the peptides in the entries with a similar protein partner. In total there were 94 non-redundant peptide-protein complex structures that were generated in this step.
Next, we manually reviewed each peptide-protein complex structure generated in the previous step. Five entities were discarded. One was removed because the conformation of the peptide was stabilized by an ion (PDB entry: 3wxa). Others were discarded due to artificial errors in PDB files or the crystal packing problem (i.e., interactions observed in crystal structural data but not biologically relevant). As in the PDB entry 4ep3, the protein partner was artificially set as one chain, which should be two chains instead (as in the PDB entry 4qja which was kept in the database). Another example is the PDB entry 4p6x, in which the protein partner of a peptide (chain D) contains two chains (chain IDs: CI). However, only one chain (C) is the true receptor and the other chain (I) exists due to crystal packing. Upon completion the database contains 89 nonredundant peptide-protein complex structures.
Unbound protein structures
In addition to bound structures, a docking benchmark usually provides unbound structures that are used for testing the ability of a docking algorithm to handle the conformational changes that occur during the binding process [32-33]. In docking studies, the 3D structures of two partners in the experimentally determined complex are defined as bound structures, and unbound structures refer to apo structures or structures in a different complex. For the peptide-protein docking process, the starting point of a peptide is the linear sequence of amino acids, and the peptide structure is totally flexible in the docking process. Therefore, there is no need to provide unbound structures for peptides in our dataset. For the protein partners, we selected unbound structures from free forms (in which no ligands occupied the peptide binding site) or protein-small molecule complexes collected from the PDB, and excluded complexes in which the binding site was occupied by another peptide or protein. The selection process placed top priority on the free structure with the smallest Rfree value. If there was no free structure available, the protein structure in the protein-small molecule complex with the smallest Rfree value was selected as the unbound structure. In total, unbound protein structures were found for 58 out of 89 peptide-protein complexes.
RESULTS AND DISCUSSION
The final PepPro dataset contains 89 nonredundant peptide-protein complex structures. Among them, 77 entries contain one-chain protein partners, and the protein partners of the remaining 12 entries are comprised of two chains. Unbound protein structures are available for 58 complexes. Corresponding PDB entries and chain ids are reported in Table 1. In addition, the name of the protein partner is also reported for each entry in the dataset. The sequence lengths (number of amino acids with resolved coordinates in PDB files) of protein partners range from 59 to 860, and their distributions are shown in Fig. 1A. The protein sequence lengths of about 70% (62 out of 89) entries fall into the range of 100 and 400 amino acids. 18 entries contain small protein partners with the sequence length ≤ 100, and the remaining 9 entries contain large protein partners (> 400 amino acids). For the peptides in the dataset, the sequence length ranges from 5 to 30 amino acids. Importantly, for more than half (47 out of 89) of the entries, the peptide sequence length is longer than 15 (shown in Fig. 1B). The corresponding chain ID and the sequence for each peptide are reported in Table 1. It is worth mentioning that coordinates for residues (< 30% of the peptide sequence length, see the section “Data collection”) at the terminal ends of peptides could be missing in the PDB file for some entries, which are marked in the column of “sequence” in Table 1. The peptide sequences (both full sequences and sequences with coordinates) in FASTA files are available at the web site “http://zoulab.dalton.missouri.edu/PepPro_benchmark”, in which both bound and unbound structures (in PDB format) of the protein partners are also provided.
Table 1.
Benchmarking structures for peptide-protein docking.
| PDB IDa |
Peptide |
Bound protein partner |
Unbound protein structure |
|||||
|---|---|---|---|---|---|---|---|---|
| Sequenceb | Lc | SSd | Protein name | L | PDB IDa | Irmsd_b | Irmsd_h | |
| 1avf_Q:J | AVVKVPLKKFKSIRETMKEKGLLGEF | 26 | HE | Gastricsin | 323 | - | ||
| 1d4t_B:A | KSLTIYAQVQK | 11 | bE | T cell signal transduction molecule SAP | 104 | - | ||
| 1dkd_E:A | SWMTTPWGFLHP | 12 | sE | GroEL | 146 | 1kid_A | 0.8 | 1.6 |
| 1fv1_C:AB | NPVVHFFKNIVTPRTPPPSQ | 20 | C | Major histocompatibility complex | 369 | - | ||
| 1gux_E:B | DLYCYEQLN | 9 | C | Retinoblastoma protein | 141 | 2r7g_C | 0.4 | 1.3 |
| 1hc9_C:A | WRYYESSLLPYPD | 13 | bE | α-bungarotoxin | 74 | - | ||
| 1j2x_B:A | SEADEMAKALEAELNDLM | 18 | H | Transcription initiation factor IIF | 67 | 1i27_A | 0.6 | 1.1 |
| 1lb6_B:A | KQEPQEIDF | 9 | bE | TNF receptor-associated factor 6 | 155 | 1lb4_A | 0.4 | 1.0 |
| 1oai_B:A | DSGFSFGSK | 9 | C | Nuclear RNA export factor | 59 | 1go5_A | 1.0 | 1.7 |
| 1oj5_B:A | LPPTEQDLTKLLLE | 14 | H | Steroid receptor coactivator 1A | 105 | - | ||
| 1skg_B:A | VAFRS | 5 | C | Phospholipase A2 | 121 | - | ||
| 1sqk_B:A | DLPKVAENLKSQLEGFNQDKLKNAS | 25 | pH | Actin, α skeletal muscle | 360 | 2q0u_A | 0.5 | 0.8 |
| 1t0j_C:B | GAQQLEEDLKGYLDWITQAE | 20 | H | Voltage-gated calcium channel subunit β2a | 187 | 1t0h_B | 0.3 | 1.2 |
| 1u00_P:A | ELPPVKIHC | 9 | bE | Chaperone protein hscA | 227 | - | ||
| 1uhb_P:AB | GKDSCQGDS | 9 | C | Trypsin | 223 | 2a31_A | 0.3 | 0.7 |
| 1uti_D:A | GQPPLVPPRKEKMRGK | 16 | pH | GRB2-related adaptor protein 2 | 57 | - | ||
| 1wkw_B:A | PGGTRIIYDRKFLMECRNSP | 20 | pH | Eukaryotic translation initiation factor 4E | 191 | 5gw6_A | 0.4 | 0.8 |
| 2b1j_C:A | MGDSILSQAEIDALLN | 16 | H | Chemotaxis protein cheY | 128 | 3rvq_A | 2.4 | 3.3 |
| 2bba_P:A | TNYLFSPNGPIARAW | 15 | pH | Ephrin type-B receptor 4 | 185 | - | ||
| 2bz8_C:AB | PARPPKPRPRR | 11 | C | SH3-domain kinase binding protein 1 | 114 | - | ||
| 2c5k_P:T | KSLRVSSLNKDRRLLLREFYNLEN | 24 | H | T-snare affecting a late golgi compartment protein 1 | 89 | 2c5j_A | 0.8 | 1.5 |
| 2g30_P:A | DDGLDEAFSRLAQSRT | 16 | H | AP-2 complex subunit β-1 | 233 | 2iv9_A | 0.5 | 1.9 |
| 2hpl_B:A | DDLYG | 5 | C | PNGase | 100 | 2hpj_A | 0.2 | 0.3 |
| 2ihs_D:B | DINNNNNIVEDVERKREFYI | 20 | pH | Gustavus, CG2944-PF | 195 | - | ||
| 2nm1_B:A | EDMFAKLKDKFFNEINK | 17 | H | Botulinum neurotoxin type B | 430 | 1s0e_A | 0.4 | 1.2 |
| 2okr_C:A | IKIKKIEDASNPLLLKRRKKARAL | 24 | pH | Mitogen-activated protein kinase 14 | 339 | 3gc7_A | 1.8 | 2.1 |
| 2otw_E:AB | GQQQQQQQQQQG | 12 | C | Antibody Fv | 233 | - | ||
| 2peh_C:A | KRKSRWDETP | 10 | C | Splicing factor 45 | 104 | 2pe8_A | 1.4 | 2.5 |
| 2puy_E:B | ARTKQTARKS | 10 | bE | PHD finger protein 21A | 60 | 2yql_A | 1.2 | 1.9 |
| 2pv1_B:A | WEYIPNV | 7 | C | Chaperone surA | 103 | 1m5y_A | 0.5 | 1.2 |
| 2pv2_E:AB | NFTLKFWDIFRK | 12 | H | Chaperone surA | 206 | - | ||
| 2qn6_C:A | SSEKEYVEMLDRLYSKLP | 18 | H | Translation initiation factor 2 γ subunit | 393 | 4rjl_A | 0.4 | 1.6 |
| 2qos_A:C | LRYDSTAERLY | 11 | sE | Complement protein C8 γ | 173 | 2ova_A | 0.8 | 2.3 |
| 2qxv_B:A | KTMFSSNRQKILERTETLNQEWKQRRIQPV | 30 | H | Embryonic ectoderm development | 352 | - | ||
| 2r9q_Y:B | VEVPLAGAV | 9 | C | 2'-deoxycytidine 5'-triphosphate deaminase | 342 | - | ||
| 2rl0_E:D | GQVTTESNLVEFDEESTK | 18 | bE | Fibronectin | 89 | - | ||
| 2whx_C:A | ADLSLEKAANVQWD | 14 | bE | Serine protease / NTPase / Helicase NS3 | 582 | - | ||
| 2x0x_D:A | YLVGQIDSEVDTDDLSNFQL | 20 | HE | Ribonucleotide-diphosphate Reductase 1 subunit α | 728 | - | ||
| 2xs1_B:A | SREKPYKEVTEDLLHLNSLF | 20 | H | Programmed cell death 6-interacting protein | 697 | 2oev_A | 0.9 | 1.7 |
| 2xum_S:A | HLEVVKLLLEHGADVDAQDK | 20 | pH | Hypoxia-inducible factor 1-α inhibitor | 349 | 3od4_A | 0.4 | 0.8 |
| 2ybf_B:A | SKYRKKHKSEFQLLVDQARKGYKKIAG | 27 | H | Ubiquitin-conjugating enzyme E2 B | 150 | 2yb6_A | 0.7 | 1.3 |
| 3at0_B:A | GSWNSGSSGTGSTGNQ | 16 | bE | Clumping factor B | 317 | 3au0_A | 0.3 | 0.6 |
| 3awr_C:A | GPRLSRLLSSAGC | 13 | H | Mitochondrial import receptor subunit TOM20 homolog | 73 | 5az9_A | 1.9 | 2.7 |
| 3bef_C:AB | NDKYEPFWE | 9 | C | Prothrombin | 283 | 5afy_LH | 1.2 | 1.7 |
| 3d9u_B:A | AVPIAQ | 6 | bE | Baculoviral IAP repeat-containing protein 2 | 92 | 1qbh_A | 1.3 | 3.3 |
| 3dab_B:A | SQETFSDLWKLLPEN | 15 | H | Mdm4 protein | 88 | 3lbj_E | 0.5 | 0.8 |
| 3dy0_B:A | RSQRLVFNRPFLMFIVDNNILFLGKVNRP | 29 | sE | N-terminus Plasma serine protease inhibitor | 328 | - | ||
| 3h8a_E:AB | QSPMPLTVAAASPELASGKVWIRYPIVR | 28 | pH | Enolase | 860 | 1e9i_AB | 0.4 | 0.6 |
| 3hbv_Z:P | AKASQAA | 7 | bE | Secreted protease C | 380 | 3hb2_P | 0.5 | 1.2 |
| 3ik5_B:A | AYQQGQNQLYNELNLGRR | 18 | H | Protein Nef | 119 | - | ||
| 3kj0_B:A | GSGGRPEIWYAQELRRIGDEFNAYYAR | 27 | H | Induced myeloid leukemia cell differentiation protein Mcl-1 | 157 | 2mhs_A | 1.5 | 1.9 |
| 3kut_C:A | SNLNPNAAEFVPGVKYG | 17 | C | Polyadenylate-binding protein 1 | 84 | 1g9l_A | 2.1 | 2.6 |
| 3l81_B:A | TYKFFEQ | 7 | bE | AP-4 complex subunit mu-1 | 250 | - | ||
| 3lu9_C:B | ATNATLDPRSFLLRNPNDKYEPFWE | 25 | C | Prothrombin | 251 | 3u69_H | 0.6 | 1.3 |
| 3mhp_C:AB | KTEQPLSPYTAYDDLKPPSSPSPTKP | 26 | C | Ferredoxin, NADP reductase | 585 | 1qfz_BA | 0.4 | 1.1 |
| 3n3x_B:A | SDDDMG | 6 | C | Ribosome inactivating protein | 246 | - | ||
| 3njf_B:A | PQIINRPQN | 9 | bE | Peptidase | 112 | - | ||
| 3o37_E:A | ARTKQTARKS | 10 | bE | Transcription intermediary factor 1-α | 175 | 4yat_A | 0.6 | 1.4 |
| 3plv_C:A | SLSIEETNELRASLGLKLIPP | 21 | pH | Ubiquitin-like modifier HUB1 | 80 | 1m94_A | 0.7 | 2.2 |
| 3r7g_B:A | KSLYKIKPRHDSGIKAKISMKT | 22 | HE | Protein spire homolog 1 | 154 | 2ylf_A | 1.3 | 1.8 |
| 3ro2_B:A | RNSFYMGTCQDEPEQLDDWNRIAELQQR | 28 | pH | G-protein-signaling modulator 2 | 328 | - | ||
| 3ryb_B:A | SLSQSLSQS | 9 | bE | Oligopeptide-binding protein oppA | 563 | 3fto_A | 4.0 | 4.2 |
| 3so6_Q:A | NSINFDNPVYQKTT | 14 | bE | LDL receptor adaptor protein | 137 | - | ||
| 3ukx_C:B | GSRRRRRRKRKREWDDDDDPPKKRRRLD | 28 | C | Importin subunit α-2 | 426 | 4u5u_A | 0.5 | 0.8 |
| 3wg5_C:AB | NVIVLMLPME | 10 | bE | Stomatin-specific protease, 1510-N | 438 | 3bpp_A | 3.2 | 3.3 |
| 4aom_T:A | KNIPSLLRVQAHIRKKMV | 18 | H | Myosin A tail domain interacting protein | 143 | - | ||
| 4dj9_B:A | ETQVVLINAVKDVAKALGDLISATKAAAG | 29 | H | Vinculin | 242 | 1rke_A | 3.4 | 3.5 |
| 4eto_P:AB | DAMNREVSSLKNKLRR | 16 | H | Protein S100-A4 | 179 | 2q91_AB | 0.3 | 0.6 |
| 4ext_B:C | RTANILKPLMSPPSREEIMATLL | 23 | HE | Mitotic spindle assembly checkpoint protein MAD2B | 198 | - | ||
| 4hh6_Z:A | KKWDSVYASLFEKINLKK | 18 | H | Putative type VI secretion protein | 157 | 4hh5_A | 0.7 | 2.7 |
| 4htp_C:A | DVPQWEVFFKR | 11 | pH | DNA ligase 4 | 221 | 3w5o_A | 0.3 | 0.8 |
| 4j1v_G:A | ALPAWARPDYNPPLVESWRR | 20 | pH | MOB kinase activator 1A | 166 | 4jiz_A | 0.5 | 1.2 |
| 4k0u_B:A | RTFRQVQSSISDFYD | 15 | H | Lipoprotein OutS | 95 | 3utk_A | 0.6 | 0.9 |
| 4m5s_B:A | GERTIPITRE | 10 | bE | α-crystallin B | 87 | 2y1y_A | 0.6 | 1.1 |
| 4oni_C:A | QGAASRPAILYALLSSSLK | 19 | H | Human nuclear receptor LRH1 | 241 | 3tx7_B | 1.1 | 2.0 |
| 4q5u_C:A | ARKEVIRNKIRAIGKMARVFSVLR | 24 | H | Calmodulin | 145 | 4bw7_A | 15.0 | 14.6 |
| 4qja_P:AB | RPGNFLQSRL | 10 | bE | HIV-1 protease | 198 | 3hvp_A | 3.0 | 3 |
| 4qqi_X:A | KAFVHMPTLPNLDFHKT | 17 | C | Ankyrin repeat family A protein 2 | 176 | 3so8_A | 1.1 | 1.7 |
| 4uwx_D:B | TPRSARLERMAQALALQAGSP | 21 | H | Protein diaphanous homolog 1 | 230 | 2bnx_A | 0.4 | 1.0 |
| 4x3h_B:A | RIPSYRYRY | 9 | bE | Activity-regulated cytoskeleton-associated protein | 79 | - | ||
| 4xoe_B:A | ADVTITVNGKVVAK | 14 | bE | FimH protein | 279 | - | ||
| 4yl6_B:A | MDEQEALNSIMNDLVALQMNRR | 22 | H | Malcavernin | 88 | 4ykd_A | 1.4 | 2.0 |
| 4yz6_B:A | ELPIARRASLHRFLEKRKDRVT | 22 | H | Transcription factor MYC3 | 175 | - | ||
| 5cqx_C:AB | HENIDWGEPKDKEVW | 15 | C | Endoribonuclease MazF | 224 | 5ck9_AB | 1.4 | 1.8 |
| 5crw_B:A | GKTKEGVLYVG | 11 | C | Protein disulfide-isomerase | 242 | 2djk_A | 1.4 | 2.0 |
| 5epp_B:A | SPEEMRRQRLHRFDS | 15 | H | Transitional endoplasmic reticulum ATPase | 170 | 3hu3_A | 1.3 | 2.2 |
| 5f67_C:A | GPGSRGKSTVTGRMISGWL | 19 | sE | Inactivation-no-after-potential D protein | 98 | - | ||
| 5fzt_B:A | PELDDILYHVKGMQRIVNQWSEK | 23 | H | TALIN-1 | 306 | 5ic0_A | 1.4 | 2.3 |
| 5gtu_B:A | GQQDLMINNPLSQDEGSLWNKFFQDKE | 27 | pH | Vacuolar protein sorting-associated protein 29 | 186 | 1w24_A | 0.3 | 0.8 |
PDB entries are followed by chain IDs of peptides and their protein partners. In the PDB files that we provided (see the supplementary files), peptide and protein chain IDs are renamed as P and A (or A and B for two protein chains), respectively.
Peptide sequences. Residues with missing coordinates in PDB files are marked with underlines.
Sequence lengths.
Secondary Structures of peptides in complex structures.
Figure 1.
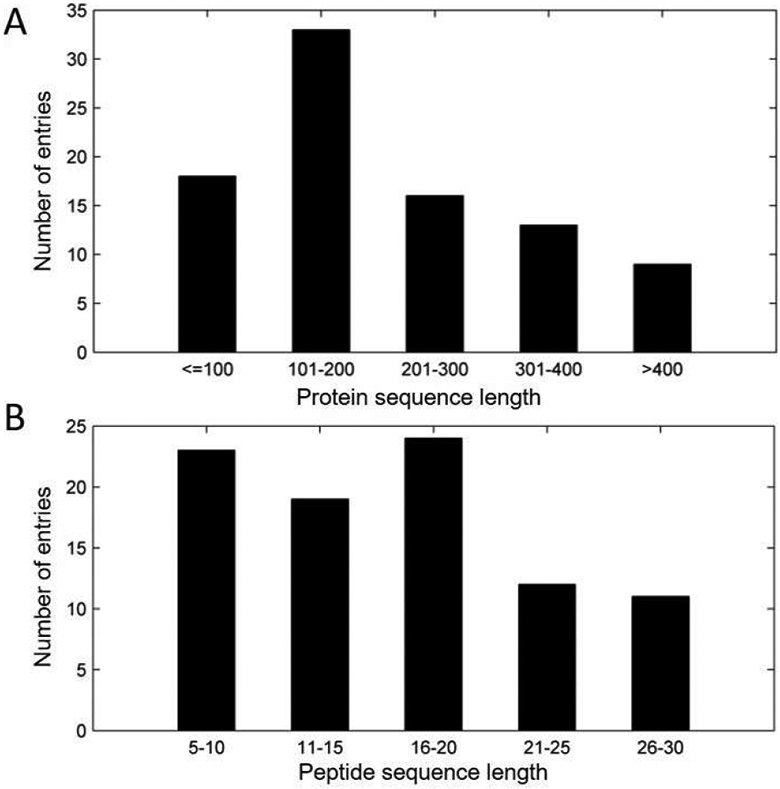
Distributions of protein (A) and peptide (B) sequence lengths for entries in the PepPro dataset.
As aforementioned, peptides are highly flexible. Both tertiary structures and secondary structures of peptides could dramatically change during the binding process with protein partners. For peptide-protein docking methods, the information residing within a peptide’s secondary structure can also be used as constraints in the sampling processes [14]. Here, we classified entries in the PepPro dataset according to the secondary structures of peptides in the peptide-protein complexes. The program DSSP [34] was employed to calculate secondary structures of peptides based on 3D structures of peptide-protein complexes, a step that can be replaced by using the secondary structure classification in the HEADER sections of the downloaded PDB files. For clarity, we simplified the default DSSP secondary structure classification for residues. Specifically, residues predicted as α-helix, 3-helix (310 helix), and 5 helix (π-helix) were all defined as helix residues. Residues predicted as a coil, isolated β-bridge, hydrogen-bonded turn, and bend were all defined as coil residues. Residues predicted as an extended strand that participates in a β ladder were defined as β-strand residues. Then a peptide was classified using the following criteria:
If half or more than half of residues (≥ 50%) in a peptide were predicted as helix residues and the remaining residues were predicted as coil residues, the peptide was predicted as a helix (H).
If less than half of residues (< 50%) in a peptide were predicted as helix residues and the remaining residues were predicted as coil residues, the peptide was predicted as a partial helix (pH).
If both helix residues and β-strand residues were found, the peptide was predicted as a mixture of helix and β-strand (HE).
If only β-strand residues and coil residues were found in a peptide, and the β-strand residues existed only in the peptide-protein complex structure, the peptide was predicted as a β-sheet formed through binding (bE).
If only β-strand residues and coil residues were found in a peptide, and the β-strand residues existed in the peptide-alone structure (remove the protein partner from the complex structure), the peptide was predicted as a β-sheet formed through self-folding (sE).
A peptide was predicted as a coil (C) if only coil residues were found.
The classification results are reported in Table 1, and the corresponding distributions are shown in Fig. 2A. Peptides in 31.5% of entries formed helices (H) and in 14.5% of entries formed partial helices (pH). Fig. 2B and C present examples for helical peptides and partial helical peptides, respectively. In very few entries (4.5%), peptides contained both helix residues and β-strand residues, and an example is shown in Fig. 2D. Peptides containing β-strand residues and coil residues (no helix residues) were grouped into two classes, bE (22.5%) and sE (4.5%). For entries in the bE class, the backbone hydrogen bonds of β-sheets were formed between the peptide and the protein partner, as shown in Fig. 2E. For entries in the sE class, the backbone hydrogen bonds of β-sheets were formed within the peptides (β-hairpins, shown in Fig. 2F). The remaining entries (22.5%) were grouped into the coil class (C), in which peptides contained only coil residues. An example is also shown in Fig. 2G.
Figure 2.

(A) Distributions of entries in the dataset according to classifications of peptide secondary structures in peptide-protein complexes. An example is presented for each class in (B-G). Proteins are colored gray. Peptides in (B) and (C) are helix (H, colored red; PDB entry: 3kj0, chain B) and partial helix (pH, colored magenta; PDB entry: 1sqk, chain B), respectively. (D) shows an example of the mixture of helix and β-strand (HE, colored orange; PDB entry: 3r7g, chain B). (E) and (F) show two different types of β-sheets, bE (colored blue; PDB entry: 1d4t, chain B) and sE (colored cyan; PDB entry: 2qos, chain A), respectively. Backbone hydrogen bonds of the β-sheets in (E) are formed between the peptide and the protein partner, and those in (F) are formed within the peptide. (G) shows an example of coil (C, colored green; PDB entry: 1fv1, chain C).
In addition to investigating secondary structures of peptides in complexes, we also calculated conformational changes of protein partners upon binding. Specifically, for entries with available unbound protein structures, I-RMSDs (i.e., the root-mean-square deviation of interface residues) between bound and unbound protein structures were calculated as follows. Interface residues were defined using a distance cutoff, namely at least one atom pair across the peptide-protein interface within 4.0 Å distance. Here, I-RMSDs were calculated based on both backbone atoms (I-RMSD_b) and heavy atoms (I-RMSD_h). The Kabsch algorithm [35] was employed to calculate the optimal rotation matrix for minimizing the RMSDs. Values of I-RMSDs are reported in Table 1 for each unbound protein structure. For clarity, I-RMSD data are also shown in Fig. 3A, in which proteins are sorted by corresponding I-RMSD_b values. For most entries (60%), protein unbound structures are close to bound structures with I-RMSD_b values smaller than 1.0 Å. Meanwhile, significant conformational changes (1.0 Å ≤ I-RMSD_b < 2.0 Å) were found for 28% of entries, and dramatic conformational changes (I-RMSD_b ≥ 2.0 Å) were found for 7% of entries. Fig. 3A also plots values of I-RMSD_h, which are normally larger than corresponding I-RMSD_b values. The large value of I-RMSD_h is mainly contributed to the highly flexible side chains of the interface residues, making unbound docking challenging even when using low I-RMSD_b unbound protein structures. Fig.3 B shows an example of the conformational changes between bound (PDB entry: 2g30, chain A) and unbound (PDB entry: 2iv9, chain A) protein structures. Although the value of I-RMSD_b is as low as 0.5 Å, side chain conformations are distinct, having an I-RMSD_h value of 1.9 Å. Another example with a large I-RMSD_b value (2.4 Å) between bound (PDB entry: 2b1j, chain A) and unbound (PDB entry: 3rvq, chain A) protein structures is shown in Fig. 3C. The corresponding value of I-RMSD_h is 3.3 Å.
Figure 3.

(A) I-RMSDs of bound and unbound protein structures. Both the backbone I-RMSD and the heavy atom I-RMSD are plotted. Proteins are sorted by values of the backbone I-RMSD. (B) and (C) show two examples of interface residues in bound (green) and unbound (magenta) protein structures. The proteins are matched by the UCSF Chimera version 1.11 [36]. (B) Values of I-RMSD_b and I-RMSD_h between bound (PDB entry: 2g30, chain A) and unbound (PDB entry: 2iv9, chain A) protein structures in (B) are 0.5 Å and 1.9 Å, respectively. (C) Values of I-RMSD_b and I-RMSD_h between bound (PDB entry: 2b1j, chain A) and unbound (PDB entry: 3rvq, chain A) protein structures in (B) are 2.4 Å and 3.3 Å, respectively.
CONCLUSION
In this study, we constructed a peptide-protein structural dataset, referred to as PepPro. The dataset was comprised of 89 non-redundant peptide-protein complex structures, with peptide sequence lengths ranging from 5 to 30 amino acids. Peptide secondary structures in a bound state were investigated and used as criteria to classify entries in the PepPro dataset. Furthermore, unbound protein structures were provided for 58 complexes, and their conformational changes against bound protein structures were also analyzed. The dataset was designed as a benchmark for testing and improving peptide-protein docking algorithms.
Supplementary Material
Acknowledgements
The authors thank Benjamin Merideth and Marshal Huang for editing the manuscript. This work was supported by NIH R01GM109980 (PI: XZ), NIH R01HL126774 and NIH R01HL142301 (PI: Jianmin Cui) to XZ. The computations were performed on the high performance computing infrastructure supported by NSF CNS-1429294 (PI: Chi-Ren Shyu) and the HPC resources supported by the University of Missouri Bioinformatics Consortium (UMBC).
REFERENCES
- 1.Petsalaki E, Russell RB, Curr. Opin. Biotechnol 2008, 19, 344–350. [DOI] [PubMed] [Google Scholar]
- 2.Wells JA, McClendon CL, Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- 3.London N, Raveh B, Schueler-Furman O, Curr. Opin. Chem. Biol 2013, 17, 952–959. [DOI] [PubMed] [Google Scholar]
- 4.Craik DJ, Fairlie DP, Liras S, Price D, Chem. Biol. Drug Des 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- 5.Fosgerau K, Hoffmann T, Drug Discov. Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- 6.Verschueren E, Vanhee P, Rousseau F, Schymkowitz J, Serrano L, Structure 2013, 21, 789–797. [DOI] [PubMed] [Google Scholar]
- 7.Lee H, Heo L, Lee MS, Seok C, Nucleic Acids Res. 2015, 43, W431–W435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niv MY, Weinstein H, J. Am. Chem. Soc 2005, 127, 14072–14079. [DOI] [PubMed] [Google Scholar]
- 9.Antes I, Proteins 2010, 78, 1084–1104. [DOI] [PubMed] [Google Scholar]
- 10.Raveh B, London N, Zimmerman L, Schueler-Furman O, PloS one 2011, 6, e18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trellet M, Melquiond AS, Bonvin AM, PloS one 2013, 8, e58769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schindler CE, de Vries SJ, Zacharias M, Structure 2015, 23, 1507–1515. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Shimon A, Niv MY, Structure 2015, 23, 929–940. [DOI] [PubMed] [Google Scholar]
- 14.Kurcinski M, Jamroz M, Blaszczyk M, Kolinski A, Kmiecik S, Nucleic Acids Res. 2015, 43, W419–W424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan C, Xu X, Zou X, Structure 2016, 24, 1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X, Yan C, Zou X, J Comput Chem 2018, 39: 2409–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alam N, Goldstein O, Xia B, Porter KA, Kozakov D, Schueler-Furman O, Plos Comput Biol 2017, 13: e1005905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Porter KA, Xia B, Beglov D, Bohnuud T, Alam N, Schueler-Furman O, Kozakov D, Bioinformatics 2017, 33: 3299–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou P; Jin BW,; Li H,; Huang SY, Nucleic Acids Res. 2018, 46: W443–W450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou P, Li BT, Yan YM, Jin BW, Wang LB, Huang SY, J Chem Inf Model 2018, 58: 1292–1302. [DOI] [PubMed] [Google Scholar]
- 21.Ciemny M, Kurcinski M, Kamel K, Kolinski A, Alam N, Schueler-Furman O, Kmiecik S, Drug Discov Today 2018, 23, 1530–1537. [DOI] [PubMed] [Google Scholar]
- 22.Vanhee P, Reumers J, Stricher F, Baeten L, Serrano L, Schymkowitz J, Rousseau F, Nucleic Acids Res. 2009, 38, D545–D551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.London N, Movshovitz-Attias D, Schueler-Furman O, Structure 2010, 18, 188–199. [DOI] [PubMed] [Google Scholar]
- 24.Das AA, Sharma OP, Kumar MS, Krishna R, Mathur PP, Genomics Proteomics Bioinformatics 2013, 11, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hauser AS, Windshügel B, J. Chem. Inf. Model 2016, 56, 188–200. [DOI] [PubMed] [Google Scholar]
- 26.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubbard SJ, Thornton JM, NACCESS, Version 2.1.1, computer program, Department of Biochemistry and Molecular Biology, University College London, 1996. [Google Scholar]
- 28.London N, Raveh B, Schueler-Furman O, Curr. Opin. Struct. Biol 2013, 23, 894–902. [DOI] [PubMed] [Google Scholar]
- 29.Van Roey K, Uyar B, Weatheritt RJ, Dinkel H, Seiler M, Budd A, Gibson TJ, Davey NE, Chem. Rev 2014, 114, 6733–6778. [DOI] [PubMed] [Google Scholar]
- 30.Rice P, Longden I, Bleasby A, Trends Genet. 2000, 16, 276–277. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Skolnick J, Nucleic Acids Res. 2005, 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vreven T, Moal IH, Vangone A, Pierce BG, Kastritis PL, Torchala M, Chaleil R, Jiménez-García B, Bates PA, Fernandez-Recio J, Bonvin AM, J. Mol. Biol 2015, 427, 3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang SY, Zou X, J. Comput. Chem 2013, 34, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabsch W, Sander C, Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W, Acta Crystal. 1976, 32A: 922–923. [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J Comput Chem 2004, 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.