

A healthy endothelium prevents the development of atherosclerosis through protective effects on vasomotion, platelet adhesion, leukocyte trafficking, anti-inflammatory and anti-oxidant properties [1]. Nitric oxide and endothelin-1, autocrine and paracrine factors produced by endothelial cells, have opposing effects on smooth muscle cells contraction. The net balance between these pleiotropic molecules contributes to the regulation of local vascular tone. Nitric oxide (NO), the most potent vasodilatory molecule produced in the arterial wall, mediates endothelium-dependent relaxation (EDR). NO arises from the conversion of l-arginine to l-citrulline by the enzymatic action of an NADPH-dependent NO synthase (NOS) [2]. The endothelium produces NO by constitutive expression of the endothelial isoform of NOS (NOS3), which is activated by shear stress [3]. NO has a variety of functions, but its action as the predominant endothelium-derived relaxing factor (EDRF) is the most important for the maintenance of vascular homeostasis. Endothelin-1 (ET-1), which is encoded by the preproendothelin-1 gene (EDN1), functions as an opposing force on vascular tone, mediating vasoconstriction of vascular smooth muscle cells through binding to endothelin ETA receptors [4]. ET-1 links causally to coronary artery disease. ETA receptor (EDNRA) blockade inhibits whereas endothelium-restricted overexpression of EDN1 increases experimental atherosclerosis in mice [5,6], and non-coding variants that regulate EDN1 and EDNRA expression associate with human disease in genome-wide associate studies [7,8].
Given the opposite but complementary roles of NO and ET-1 it is not surprising that they are co-regulated by the same factors. In endothelial cell dysfunction and later stage atherosclerosis eNOS expression increases and the enzyme becomes uncoupled, generating the highly oxidant species superoxide instead of NO [9,10]. Risk factors for atherosclerosis such as dyslipidemia, diabetes, hypertension and smoking all reduce NO expression in cultured endothelial cells and impair EDR [11,12]. The opposite holds true for ET-1 function and expression, which increases in endothelial cell dysfunction and atherosclerosis [4,13]. There is some evidence that NO and ET-1 directly regulate each other to achieve vascular tone homeostasis. Stimulating the production of NO in endothelial cells can reduce ET-1 expression and production [14]. Similarly, ET-1 can directly induce the uncoupling of eNOS [10] whereas blocking ETA receptors restores NO-dependent vascular function in mice with atherosclerosis [6]. These multiple mechanisms of counter-regulation between NO and ET-1 demonstrate the close control of vascular tone in health and disease. Identifying more molecular pathways that affect this tight balance is important for identifying therapies that affect the arterial wall.
In the current issue of Atherosclerosis, Rafnsson et al. [15] identify the direct effect of ET-1 on arginase expression and activity as a new mechanism that links ET-1 to NO function and EDR. With samples from the large human Biobank of Karolinska Endarterectomies (BiKE) the authors show that ET-1 and arginase pathway genes demonstrate similar patterns of expression. In RNA extracted from 177 carotid plaques there was higher arginase 2 (ARG2) and EDNRA gene expression compared with non-atherosclerotic iliac artery controls. Comparing symptomatic patients (defined as those who have experienced transient ischemic attack, minor stroke, or amaurosis fugax) to asymptomatic patients in their registry demonstrated higher EDN1 and EDNRB (ETB receptor) expression in the plaques as well as augmented mRNA expression of ARG1, ARG2, EDNRA and EDNRB in the PBMCs. Immunohistochemical studies showed co-localization of arginase-1 and arginase-2 and ET-1 in the necrotic core of the plaque. The proteins seemed to also co-localize with macrophage marker CD68, suggesting the participation of plaque macrophages in the regulation of arterial tone in regions with atheromatous lesions. To validate the functional importance of these findings, the authors show that ET-1 stimulates ARG2 expression and activity (Fig. 1) in cultured human carotid artery endothelial cells and the THP-1 human macrophage cell line. The change in expression of arginase-2, but not arginase-1 in response to ET-1 is greater in ECs than in macrophages. Only in macrophages, however, did ET-1 stimulate the production of superoxide, as measured by ESR using 1-hydroxy-3-methoxycarbonyl*−2,2,5,5-tetramethylpyrrolidine as a spin trap, an effect that was abrogated by an arginase inhibitor.
Fig. 1.
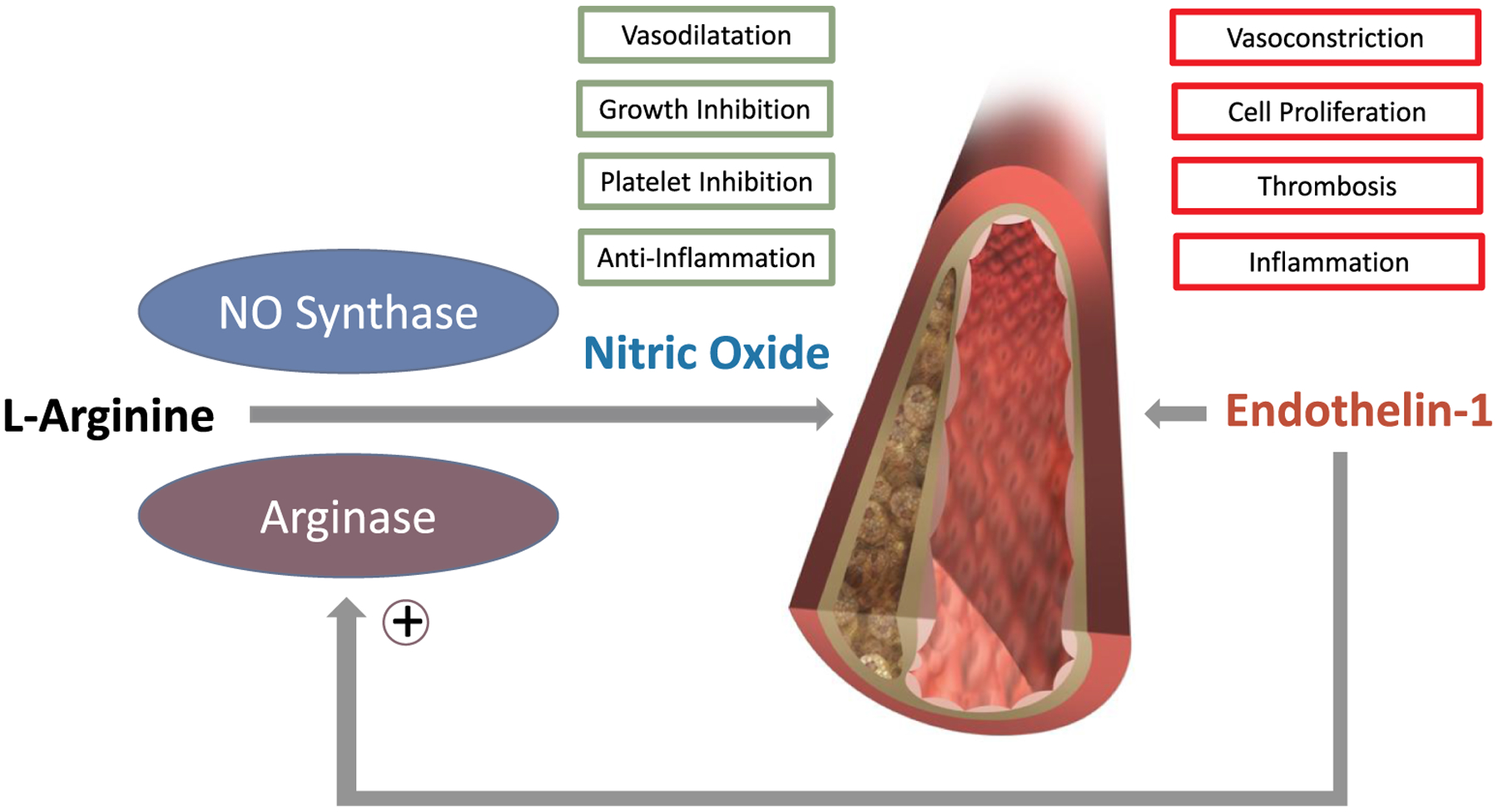
The opposite effects of NO and ET-1 on vascular function are counterbalanced in healthy tissue and dysregulated in atherosclerosis.
A new mechanism of regulation is the ability of ET-1 in vascular endothelial cells and macrophages to induce arginase expression, which then competes for l-arginine substrate with NO synthase, thereby reducing NO bioavailability.
The findings by Rafnsson et al. [15] support the opposite but complementary actions of ET-1 and NO in the development and progression of atherosclerosis. The abundant expression of ET-1 and its two receptors, ETA and ETB in late stage carotid plaques confirms the role of this potent vasoconstrictive pathway, as previously demonstrated in cultured ECs and atherosclerotic arteries [16]. One novel function of ET-1 appears to be upregulation of arginase-2 expression and function in cells found in the atherosclerotic plaque. Arginase is known to be a critical reciprocal regulator of NO production by competing with eNOS for the substrate l-arginine in endothelial cells (Fig. 1). By co-localizing ET-1 and arginase-2 expression to the necrotic core of the plaque and functional experiments in ECs and THP-1 macrophage the authors link ET-1 to decoupling of eNOS from production of NO to superoxide production via the depletion of arginase in both cell types [15]. The finding that ET-1 regulates arginase-2 expression and activity as well as arginase-2-derived superoxide production thereby inhibiting NO bioavailabilty is unexpected and novel. This observation adds to the understanding of the role of ET-1 in atherosclerosis and, possibly, other vascular diseases. Importantly, the effects seen in the cell studies were distinct, seen in either endothelial cells or macrophages. If relevant for human atherosclerosis, arginase-2-dependent effects of ET-1 could contribute to the anatomic and functional heterogeneity of atherosclerotic lesions [4,13]. Local arterial spasm favored by imbalance in the net actions of NO and ET-1 could explain why lesions of the same size and degree of luminal encroachment vary widely in their clinical expression and the temporal dispersion of events.
This study also has some limitations. Vascular smooth muscle cells determine atherosclerotic plaque progression but were not studied for their regulation of the ET-1/arginase pathway. Prior studies on arginase in VSMCs suggest they play an important role in mediating similar effects attributed to ECs and macrophages by Rafnsson et al. [15]. The aorta of rabbits with atherosclerosis exhibit increased expression of arginase-1 and arginase-2 [17]. Arginase promotes proliferation of vascular smooth muscle cells and is stimulated by oxidized LDL in both endothelial and intimal smooth muscle cells [18]. Moreover, angiotensin II, which accelerates atherosclerosis progression, stimulates vascular smooth muscle cell proliferation and fibrosis through arginase-1, but not arginase-2 [19]. These findings suggest that the mechanism for the beneficial effects of ETA receptor-selective endothelin receptor antagonists (ERAs) in mice [6] and patients [20] with atherosclerosis may results in part from reduction in arginase expression in multiple vascular cells.
Future work to understand the exact cells which exhibit dysregulated ET-1, arginase-1, arginase-2 and NO function will be important for understanding how homeostasis goes awry in atherosclerotic arteries. Rafnsson et al. find that ET-1 expression is relevant in at least endothelial cells and macrophages, two cell types involved in the formation of fatty streaks as early as in utero [21]. Determining if certain subtypes of these cells, such as foam cells or VSMC-derived macrophages, display different patterns of gene regulation will be important. Indeed, expression of the arginase isoforms differs in macrophages with different functional polarization (“M1 vs. M2”) [22]. Advancements in droplet-based single cell RNA-sequencing now permit even finer analyses of cellular heterogeneity in human vascular disease [23]. Two recent papers have identified signatures of immune cells in human plaques [24] and VSMCs in mice with hypercholesterolemia due to loss of apolipoprotein E [25]. In both cases distinct subsets of cells displayed characteristics implicated in plaque progression. These single cell transcriptional profiling methods allow for unbiased identification of all pathways upregulated in atherosclerosis. Rafnsson et al. [15] focus on the ET-1/NO axis, but unbiased analyses for mechanisms upregulated at different stages of disease will prove informative for uncovering new biology. Large repositories of human vascular tissue like the BiKE registry are an excellent resource for studies to profile the cells and biological pathways responsible for disease using new sequencing and bioinformatic technology.
Finally, the question as to whether and how endothelin receptor antagonists (ERAs), which limit atherosclerosis progression in mice and patients [6,20] might affect arginases deserves further study. It would not surprise if ERAs mediate some of their protective effects on atherosclerosis via inhibition of arginase. Moreover, it is possible if not likely that the improvement of NO-dependent vasodilatation seen after selective ETA receptor blockade in atherosclerosis [6] involves opposing effects on both NO synthase and arginases which both share the same substrate: l-arginine (Fig. 1). The links between the ET-1/arginase/NO axis and expression of disease-causing genes can be unexpectedly complicated, but remain integral for the development of new therapies for vascular disease.
Acknowledgments
Supported by the National Heart, Lung, and Blood Institute (HL128810, HL148483 and HL152423 to R.M.G; R01HL080472 and 1R01HL134892 to P.L.), the American Heart Association (18CSA34080399 to P.L.), the RRM Charitable Fund (to P.L.) and the Swiss National Science Foundation (Nr. 108 258 and 122 504 to M.B.)
Footnotes
Declaration of competing interest
Dr. Libby has a financial interest in Xbiotech, a company developing therapeutic human antibodies. Dr. Libby’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies.
References
- [1].Gimbrone MA, Garcia-Cardeña G, Endothelial cell dysfunction and the pathobiology of atherosclerosis, Circ. Res 118 (2016) 620–636, 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vanhoutte PM, Zhao Y, Xu A, Leung SWS, Thirty years of saying NO: sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator, Circ. Res 119 (2016) 375–396, 10.1161/CIRCRESAHA.116.306531. [DOI] [PubMed] [Google Scholar]
- [3].Harrison DG, Cellular and molecular mechanisms of endothelial cell dysfunction, J. Clin. Investig 100 (1997) 2153–2157, 10.1172/JCI119751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Barton M, Yanagisawa M, Endothelin: 30 years from discovery to therapy, Hypertension 74 (2019) 1232–1265, 10.1161/HYPERTENSIONAHA.119.12105. [DOI] [PubMed] [Google Scholar]
- [5].Li MW, Mian MOR, Barhoumi T, Rehman A, Mann K, et al. , Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice, Arterioscler. Thromb. Vasc. Biol 33 (2013) 2306–2315, 10.1161/ATVBAHA.113.302028. [DOI] [PubMed] [Google Scholar]
- [6].Barton M, Haudenschild CC, d’Uscio LV, Shaw S, Münter K, Lüscher TF, Endothelin ETA receptor blockade restores NO-mediated endothelial function and inhibits atherosclerosis in apolipoprotein E-deficient mice, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 14367–14372, 10.1073/pnas.95.24.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, et al. , A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression, Cell 170 (2017) 522–533, 10.1016/j.cell.2017.06.049e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nikpay M, Goel A, Won H-H, Hall LM, Willenborg C, et al. , A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease, Nat. Genet 47 (2015) 1121–1130, 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kawashima S, Yokoyama M, Dysfunction of endothelial nitric oxide synthase and atherosclerosis, Arterioscler. Thromb. Vasc. Biol 24 (2004) 998–1005, 10.1161/01.ATV.0000125114.88079.96. [DOI] [PubMed] [Google Scholar]
- [10].Oelze M, Knorr M, Kröller-Schön S, Kossmann S, Gottschlich A, et al. , Chronic therapy with isosorbide-5-mononitrate causes endothelial dysfunction, oxidative stress, and a marked increase in vascular endothelin-1 expression, Eur. Heart J 34 (2013) 3206–3216, 10.1093/eurheartj/ehs100. [DOI] [PubMed] [Google Scholar]
- [11].Vanhoutte PM, Boulanger CM, Endothelium-dependent responses in hypertension, Hypertens. Res 18 (1995) 87–98, 10.1291/hypres.18.87. [DOI] [PubMed] [Google Scholar]
- [12].Creager MA, Cooke JP, Mendelsohn ME, Gallagher SJ, Coleman SM, et al. , Impaired vasodilation of forearm resistance vessels in hypercholesterolemic humans, J. Clin. Investig 86 (1990) 228–234, 10.1172/JCI114688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barton M, Traupe T, Haudenschild CC, Endothelin, hypercholesterolemia and atherosclerosis, Coron. Artery Dis 14 (2003) 477–490, 10.1097/01.mca.0000091441.11300.c1. [DOI] [PubMed] [Google Scholar]
- [14].Mitsutomi N, Akashi C, Odagiri J, Matsumura Y, Effects of endogenous and exogenous nitric oxide on endothelin-1 production in cultured vascular endothelial cells, Eur. J. Pharmacol 364 (1999) 65–73, 10.1016/s0014-2999(98)00806-1. [DOI] [PubMed] [Google Scholar]
- [15].Rafnsson A, Matic LP, Lengquist M, Mahdi A, Shemyakin A, et al. , Endothelin-1 increases expression and activity of arginase 2 via ETB receptors and is co-expressed with arginase 2 in human atherosclerotic plaques, Atherosclerosis 292 (2020) 215–223, 10.1016/j.atherosclerosis.2019.09.020. [DOI] [PubMed] [Google Scholar]
- [16].Winkles JA, Alberts GF, Brogi E, Libby P, Endothelin-1 and endothelin receptor mRNA expression in normal and atherosclerotic human arteries, Biochem. Biophys. Res. Commun 191 (1993) 1081–1088, 10.1006/bbrc.1993.1327. [DOI] [PubMed] [Google Scholar]
- [17].Hayashi T, Esaki T, Sumi D, Mukherjee T, Iguchi A, Chaudhuri G, Modulating role of estradiol on arginase II expression in hyperlipidemic rabbits as an atheroprotective mechanism, Proc. Natl. Acad. Sci. U.S.a 103, (2006), pp. 10485–10490, 10.1073/pnas.0603918103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH, OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling, Atherosclerosis 214 (2011) 279–287, 10.1016/j.atherosclerosis.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bhatta A, Yao L, Toque HA, Shatanawi A, Xu Z, et al. , Angiotensin II-induced arterial thickening, fibrosis and stiffening involves elevated arginase function, PLoS One 10 (2015), 10.1371/journal.pone.0121727e0121727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoon MH, Reriani M, Mario G, Rihal C, Gulati R, Lennon R, et al. , Long-term endothelin receptor antagonism attenuates coronary plaque progression in patients with early atherosclerosis, Int. J. Cardiol 168 (2013) 1316–1321, 10.1016/j.ijcard.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Napoli C, D’Armiento FP, Mancini FP, Postiglione A, Witztum JL, et al. , Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions, J. Clin. Investig 100 (1997) 2680–2690, 10.1172/JCI119813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, et al. , Macrophage activation and polarization: nomenclature and experimental guidelines, Immunity 14 (2014) 14–20, 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, et al. , Single-cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell Populations, Circulation 140 (2019) 147–163, 10.1161/CIRCULATIONAHA.118.038362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, et al. , Single-cell immune landscape of human atherosclerotic plaques, Nat. Med 25 (2019) 1576–1588, 10.1038/s41591-019-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, et al. , Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis, Nat. Med 25 (2019) 1280–1289, 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]