

Abstract
All kingdoms of life use the transient 5′-deoxyadenosyl radical (5′-dAdo•) to initiate a wide range of difficult chemical reactions. Because of its high reactivity, the 5′-dAdo• must be generated in a controlled manner to abstract a specific H atom and avoid unproductive reactions. In radical S-adenosylmethionine (SAM) enzymes, the 5′-dAdo• is formed upon reduction of SAM by an [Fe4S4] cluster. An organometallic precursor featuring an Fe–C bond between the [Fe4S4] cluster and the 5′-dAdo group was recently characterized and shown to be competent for substrate radical generation, presumably via Fe–C bond homolysis. Such reactivity is without precedent for Fe-S clusters. Here, we show that synthetic [Fe4S4]–alkyl clusters undergo Fe–C bond homolysis when the alkylated Fe site has a suitable coordination number, thereby providing support for the intermediacy of organometallic species in radical SAM enzymes. Addition of pyridine donors to [(IMes)3Fe4S4–R]+ clusters (R = alkyl or benzyl; IMes = 1,3-dimesitylimidazol-2-ylidene) generates R•, ultimately forming R–R coupled hydrocarbons. This process is facile at room temperature and allows for the generation of highly reactive radicals including primary carbon radicals. Mechanistic studies, including use of the 5-hexenyl radical clock, demonstrate that Fe–C bond homolysis occurs reversibly. Using these experimental insights and kinetic simulations, we evaluate the circumstances in which an organometallic intermediate can direct the 5′-dAdo• toward productive H-atom abstraction. Our findings demonstrate that reversible homolysis of even weak M–C bonds is a feasible protective mechanism for the 5′-dAdo• that can allow selective X–H bond activation in both radical SAM and adenosylcobalamin enzymes.
Graphical Abstract
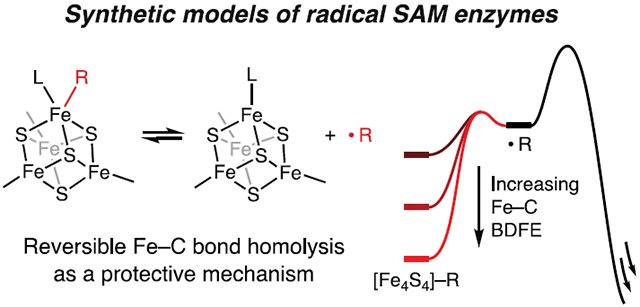
INTRODUCTION
Nature utilizes radical chemistry for a wide range of challenging reactions including cofactor maturation, antibiotic synthesis, and secondary metabolism.1–4 The most common radical initiator is the 5′-deoxyadenosyl radical (5′-dAdo•), which is generated in adenosylcobalamin and radical S-adenosylmethionine (SAM) enzymes.1,3 Because the 5′-dAdo• is a primary carbon radical, it can abstract H atoms from strong X–H bonds and initiate reactions that proceed through high-energy intermediates.5 Although this enables thermodynamically and kinetically challenging reactions to take place, it introduces an additional difficulty: how to generate and utilize the 5′-dAdo• in a controlled fashion so as to avoid unproductive reactivity.
The 5′-dAdo• is derived from relatively unreactive precursors, either by homolysis of the Co-C bond in adenosylcobalamin1 or by reductive cleavage of an S-C bond in SAM (Figure 1).3,6,7 In both adenosylcobalamin and radical SAM enzymes, a dramatic rate enhancement for production of the 5′-dAdo• is observed only when all reaction components are present (i.e., substrate and adenosylcobalamin for adenosylcobalamin enzymes8–11 or substrate, SAM, and a reduced [Fe4S4] cluster for radical SAM enzymes12). But even with this safety mechanism operative, it is possible for the 5′-dAdô to react unproductively, for example, by abstracting the wrong H atom from the substrate or the protein. How selective H-atom abstraction occurs upon generation of the 5′-dAdo• is therefore a critical subject of investigation. Structural modeling and spectroscopic studies of adenosylcobalamin enzymes suggest that the 5′ carbon must traverse several angstroms (ca. 6 Å) between the Co center and the target substrate H atom.13–16 In contrast, structural and spectroscopic studies of radical SAM enzymes with SAM and substrate bound reveal that the substrate X–H bond is positioned in close proximity to the 5′-dAdo carbon (ca. 4 Å, corresponding to only ca. 3 Å to the target H atom; Figure 1, inset).17,18 Based on these findings, it was proposed17 that the closer proximity of the substrate's target H atom and the 5′-carbon in radical SAM enzymes compared to adenosylcobalamin enzymes may explain why radical SAM enzymes outnumber19,20 adenosylcobalamin enzymes by a factor of ~104: because radical SAM enzymes position the substrate X–H bond adjacent to the nascent 5′-dAdo•, their active sites are predisposed to perform selective H-atom abstraction upon generation of the 5′-dAdo•.
Figure 1.
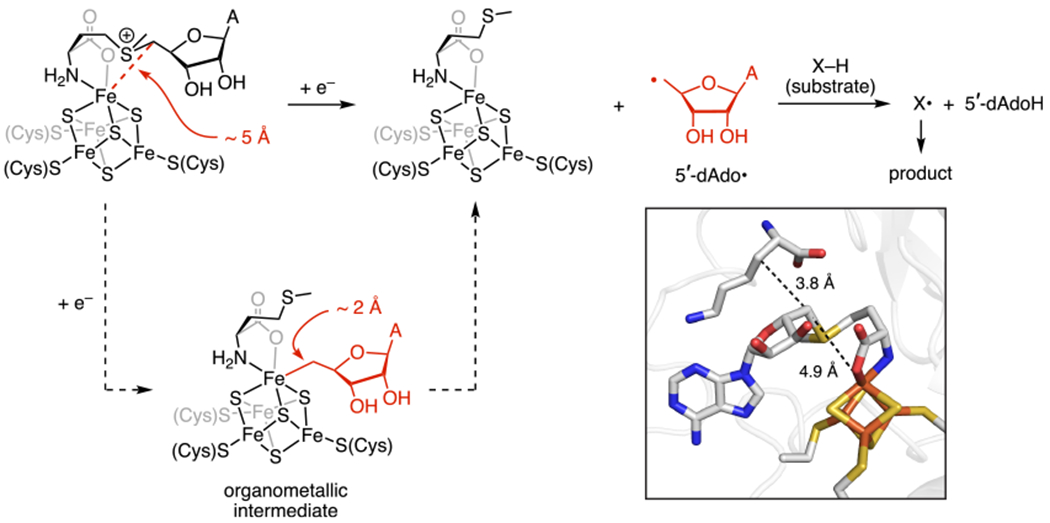
Proposed mechanism for 5′-dAdo• formation in radical SAM enzymes showing the bond-distance changes incurred upon Fe–C bond formation and homolysis. (Inset) Structure of the lysine 2,3-aminomutase active site (PDB ID: 2A5H). From the available structural and spectroscopic data, the 5′ carbon appears to be primed for selective H-atom abstraction, but formation and homolysis of an Fe–C bond would require significant rearrangement in the active site with the 5′ carbon moving away from the substrate X–H bond to form an Fe–C bond and back toward the substrate to undergo H-atom abstraction.
This narrative has been somewhat complicated by the discovery of an unexpected intermediate in 5′-dAdo• formation: an organometallic species—observed in a wide range of radical SAM enzymes—that features a bond between an Fe of the [Fe4S4] cluster and the 5′-dAdo carbon, akin to the Co—C bond in adenosylcobalamin (Figure 1).21,22 Efforts to understand this species’ precise geometric and electronic structure are ongoing, and its role as an intermediate in 5′-dAdo• generation raises several questions. First, because Fe–C bond homolysis is an unprecedented reaction for Fe–S clusters, it is unclear if and how the Fe–C bond in an alkylated [Fe4S4] cluster can be sufficiently weakened to generate primary carbon radicals. Second, the careful positioning of the 5′ carbon near the target H atom—certainly an important factor for selectivity—must also be considered in light of the geometric changes required to form and break an Fe–C bond (Figure 1);23 if such dramatic rearrangements occur during catalysis, then rigid positioning of the substrate and SAM cannot fully account for the selectivity of radical SAM enzymes. Therefore, other mechanisms for achieving selectivity must be considered, particularly those that take into account the dynamic nature of the active site.
We herein address these questions with a combination of model chemistry and kinetic simulations. In contrast to synthetic models of adenosylcobalamin enzymes, which have provided insights into the structure and reactivity of adenosylcobalamin,24–33 there are no functional models of the organometallic intermediate in radical SAM enzymes (i.e., alkylated [Fe4S4] clusters that generate alkyl radicals). Using synthetic [Fe4S4]–alkyl clusters, we show that even highly reactive, primary carbon radicals can be generated from [Fe4S4]–alkyl clusters and in doing so delineate the coordination-chemistry requirements for Fe–C bond homolysis. Notably, we find that Fe–C bond homolysis is both facile and reversible. Using kinetic simulations, we then evaluate the circumstances in which reversible Fe–C bond homolysis can affect the selectivity of H-atom abstraction. Our findings suggest that, by slowing the rates of all reactions of the 5′-dAdo•, an organometallic intermediate may be key to achieving selective reactivity in radical SAM enzymes.
RESULTS AND DISCUSSION
Synthesis of Alkylated [Fe4S4] Clusters.
We recently reported the first [Fe4S4]–alkyl cluster, which is stable with respect to Fe–C bond homolysis at room temperature.34 Our current study centers on the chemistry of alkylated [Fe4S4] clusters supported by the N-heterocyclic carbene IMes (IMes = 1,3-dimesitylimidazol-2-ylidene); the complement of three strongly binding IMes ligands increases the stability of these clusters and makes them suitable for reactivity and mechanistic studies. Synthetic access to IMes-supported [Fe4S4]–alkyl clusters is afforded by treatment of (IMes)3Fe4S4Cl (1)35 with the appropriate Grignard reagent to provide (IMes)3Fe4S4(benzyl) (2) and (IMes)3Fe4S4(octyl) (3) in 64 and 66% yield, respectively (Figure 2). The 1H NMR resonances from the IMes ligands in 2 and 3 are similar to those observed for 1. The α protons of the benzyl and octyl groups are observed at 223 and 258 ppm for 2 and 3, respectively; the β protons of the octyl group in 3 are observed at 22 ppm. EPR spectroscopic analysis establishes that both clusters have an S = 1/2 ground spin state (Figures S53 and S54). The one-electron oxidized clusters [(IMes)3Fe4S4(benzyl)][BArF4] ([2]+, [BArF4] = [tetrakis-(3,5-bis(trifluoromethyl)phenyl)borate]) and [(IMes)3Fe4S4(octyl)][BArF4] ([3]+) were synthesized by reaction of 2 or 3 with [Cp2Co][BArF4]. Upon oxidation, the resonances in the 1H NMR spectrum corresponding to the α protons of the benzyl and octyl groups shift upfield to 68 ppm for [2]+ and 75 ppm for [3]+; the β protons in [3]+ also shift upfield to −3.5 ppm. These shifts are similar to the 1H NMR shifts observed in the previously reported [Fe4S4]2+–Etcluster34 and are indicative of a diamagnetic ground state with thermally populated, paramagnetic excited states. Clusters 2 and [2]+ have been crystallographically characterized (Figures 2 (inset) and S80); their metrical parameters are similar to those of 1 and of the previously reported alkyl cluster.34
Figure 2.
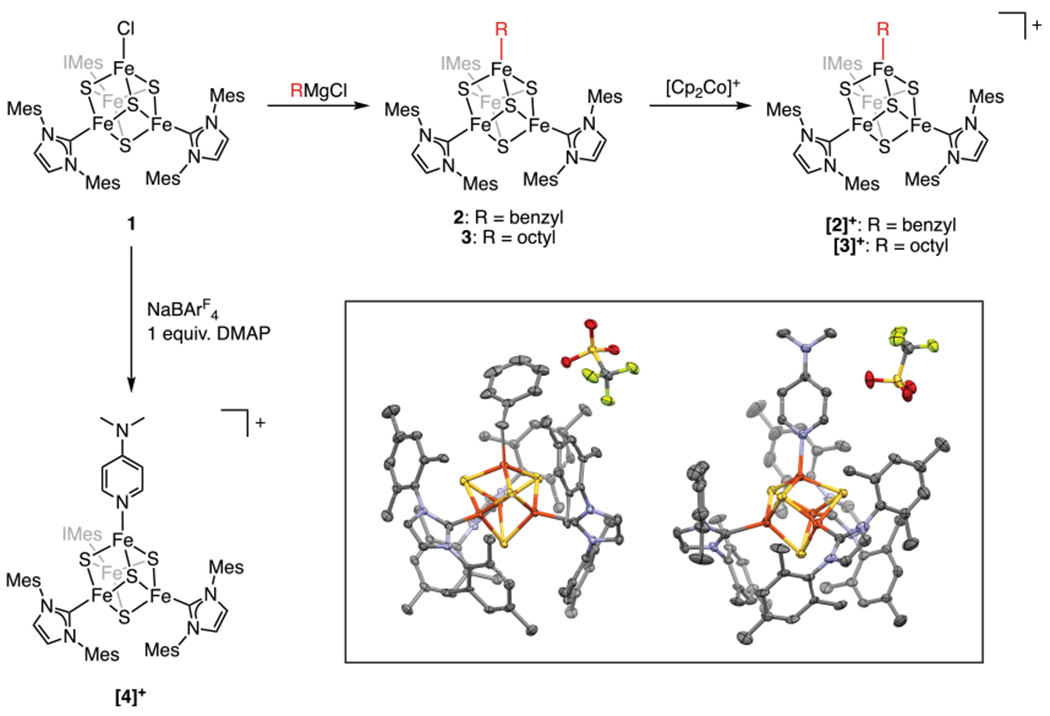
Synthesis of alkylated [Fe4S4]+/2+ clusters and an [Fe4S4]+–DMAP adduct. (Inset) Thermal ellipsoid (50%) plot of [2][OTf] (left) and [4][OTf] (right). Color scheme: carbon (gray), iron (orange), sulfur (yellow), nitrogen (blue), oxygen (red), fluorine (green).
Observation of Fe–C Bond Homolysis.
In comparison to the organometallic intermediate characterized in radical SAM enzymes—which is consumed in minutes at 170 K21— the alkylated clusters described above are strikingly stable. We attribute this to the coordination number of the alkylated Fe site: homolysis of the Fe–C bond would lead to a high-energy species with a three-coordinate Fe site. In contrast, homolysis of the Fe–C bond in the organometallic intermediate in radical SAM enzymes, which is thought to have a six-coordinate Fe site, would generate an energetically feasible, five-coordinate Fe site. We therefore hypothesized that addition of an exogenous ligand to the synthetic clusters could lead to Fe–C bond homolysis through an intermediate with a five-coordinate Fe site. Indeed, reaction of [2]+ with an excess of a pyridine, such as 4-N,N-dimethylaminopyridine (DMAP), results in a rapid reaction at room temperature to generate the reduced [Fe4S4]+ cluster [(IMes)3Fe4S4(DMAP)][BArF4] ([4]+, 95 ± 3%) and bibenzyl (92 ± 3% yield) (Scheme 1A). The identity of [4]+ was confirmed by its independent synthesis and characterization (Figure 2).
Scheme 1. Formation of Radicals at [Fe4S4] Clustersa.
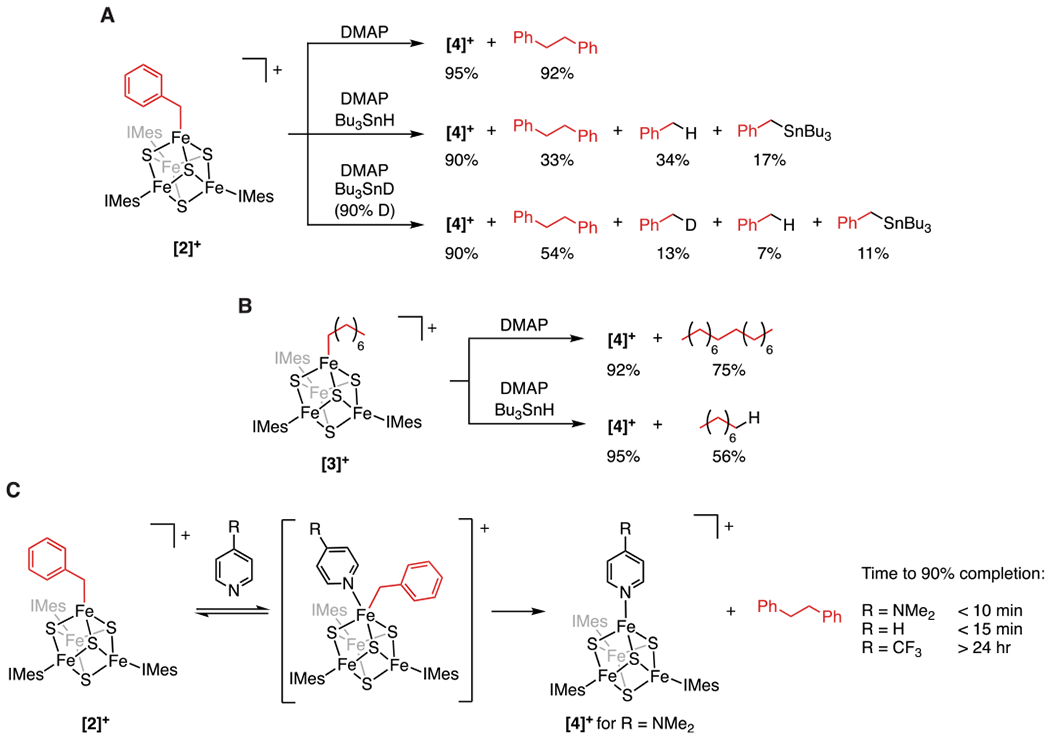
a(A) Generation and trapping of benzyl radicals upon treatment of [2]+ with DMAP. (B) Generation and trapping of octyl radicals upon treatment of [3]+ with DMAP. (C) Reaction of [2]+ with substituted pyridines at room temperature.
Fe–C bond homolysis also occurs with other alkyl groups including those whose corresponding radicals are much more reactive than the benzyl radical. For example, when DMAP is added to [3]+, [4]+ (92 ± 3%) and hexadecane (75 ± 8% yield) are rapidly formed (Scheme 1B). The rate of C–C bond formation scales with the substituent Hammett parameters;36 when DMAP (σp = −0.83) is added to [2]+, the reaction is complete in less than 10 min, whereas the reaction of [2]+ with pyridine (σp = 0.0) results in ca. 90% conversion in 15 min and that of [2]+ with 4-trifluoromethylpyridine (CF3-py, σp = 0.54) takes over 24 h to reach 90% completion (Scheme 1C). The rates increase with the donicity of the substituted pyridine, consistent with pyridine coordination preceding Fe–C bond homolysis. Fe–C bond homolysis is not limited to pyridinyl donors; addition of quinuclidine generates the quinuclidine adduct (98 ± 3%) and bibenzyl (90 ± 4%).
The reactions of [2]+ and [3]+ with exogenous ligands are consistent with donor-induced Fe–C bond homolysis followed by a combination of the radical with another benzyl or alkyl group to form bibenzyl or hexadecane (the C–C bond-forming step could occur by one or more pathways involving a radical; see Supporting Information (SI), p. S39). Such mechanisms involving M–C bond homolysis would align our findings with those observed for adenosylcobalamin model complexes24,29,33,37 and those proposed for organometallic intermediates in radical SAM enzymes.21,22,38 However, these observations do not rule out other conceivable mechanisms that proceed without formation of organic radicals (i.e., those in which bibenzyl or hexadecane is formed through concomitant Fe–C bond cleavage and C–C bond formation). No intermediates were observable by NMR or EPR spectroscopies, so to distinguish between these mechanistic possibilities, we performed a series of experiments to assay for the generation of organic radicals.
As an initial test, we carried out the reaction of [2]+ and [3]+ with DMAP in the presence of a radical trap. We chose Bu3SnH because it does not react with either cluster in the absence of DMAP, and the expected organic products (toluene for [2]+ and octane for [3]+) are readily detected by 1H NMR spectroscopy and GC–MS, respectively. When DMAP is added to [2]+ in the presence of 20 equiv of Bu3SnH, the reduced cluster [4]+ is formed in less than 10 min. Alkyl groups are detected as a mixture of toluene (34 ± 3%), bibenzyl (33 ± 3%), and Bu3SnBn (17 ± 3%) (Scheme 1A). That both bibenzyl and toluene were observed in this reaction indicates incomplete trapping of the benzyl radicals by Bu3SnH and suggests that the pathways that lead to bibenzyl and toluene occur with competitive rates. Addition of DMAP to [3]+ in the presence of Bu3SnH induces similar reactivity with the production of octane (56 ± 6%; Scheme 1B). The reaction of [2]+ with DMAP was also carried out in the presence of Bu3SnD (90% D), which resulted in the formation of bibenzyl (54 ± 3%), toluene (7 ± 3%), d1-toluene (13 ± 3%), and Bu3SnBn (11 ± 3%) (Scheme 1A). Formation of d1-toluene indicates that Bu3SnD is the deuterium-atom source, and the lower deuterium incorporation into toluene (1:1.9 H/D) compared to Bu3SnH/D (1:9 H/D) is indicative of a kinetic isotope effect for H/D atom abstraction from Bu3SnH/D.
Because radical traps can alter the major mechanistic pathway of the reaction—and therefore not provide definitive evidence of free-radical intermediates—we pursued the use of radical clocks as a complementary test for the generation of free radicals. For these purposes, we wanted a radical clock that rearranges on a time scale similar to or faster than the rate of other reactions involving organic radicals, which we estimated based on the following observations. In the reaction of [2]+ with DMAP, the benzyl radical is only partially trapped by Bu3SnH, suggesting that other reactions leading to bibenzyl formation are competitive with trapping. Because H–SnBu3 abstraction by an organic radical occurs at ~106 s−1,39 we predicted that a radical clock with a similar rate constant would exhibit at least partial rearrangement under the Fe–C bond homolysis conditions. We therefore selected the 5-hexenyl radical clock, which rearranges to the cyclopentylmethyl radical with a rate constant of 1.8 × 105 s−1 (20 °C),40–42 and assayed for organic products derived from the 5-hexenyl and/or cyclopentylmethyl radicals.
To test these predictions, we prepared (IMes)3Fe4S4(5-hexenyl) (5) and [(IMes)3Fe4S4(5-hexenyl)][BArF4] ([5]+) using the methodology described above. The 1H NMR spectra of 5 and [5]+ are similar to those of 3 and [3]+; the α and β protons of the 5-hexenyl group shift from 256 and 24.5 ppm to 74 and −4.0 ppm, respectively, upon oxidation of 5 to [5]+. Addition of excess DMAP to [5]+ leads to immediate formation of [4]+ and coupled organic fragments. Integration of the 1H NMR spectrum suggests that approximately 72 ± 2% of the alkyl groups are cyclized, while 28 ± 2% of the alkyl groups still contain alkene resonances (Figures S50 and S51). The observation of both cyclized and uncyclized organic products strongly supports that the Fe–C bond undergoes homolysis upon addition of DMAP to [5]+ and suggests that the rates of subsequent steps involving the organic radical are indeed competitive with that of cyclization.
Evidence for Reversible Fe–C Bond Homolysis.
We also carried out the reaction of [5]+ with an excess of CF3-py. As expected, the addition of CF3-py induces consumption of [5]+ and formation of the CF3-py adduct, [6]+. This reaction takes more than 24 h to reach completion (much slower than when DMAP is added) because of the diminished equilibrium for CF3-py binding. Intriguingly, a second species, characterized by peaks in the 1H NMR spectrum at 75 and −3 ppm, increases in concentration over the course of this reaction (Figure 3). Given the similarity of the 1H NMR resonances to those of the other [Fe4S4]2+−alkyl clusters described herein, we hypothesized that this new species was [(IMes)3Fe4S4(cyclopentylmethyl)][BArF4] ([7]+), containing a cyclopentylmethyl group derived from the 5-hexenyl radical; its identity was confirmed by independent synthesis of [7]+. We note that [5]+ does not convert to [7]+ in the absence of added pyridine. The conversion of [5]+ to [7]+ via organic radicals demonstrates that Fe–C bond homolysis is reversible, whereby pyridine binding to [5]+ induces formation of the transient 5-hexenyl radical, which rearranges to the cyclopentylmethyl radical and recombines with [6]+ to give [7]+ (Scheme 2A).
Figure 3.
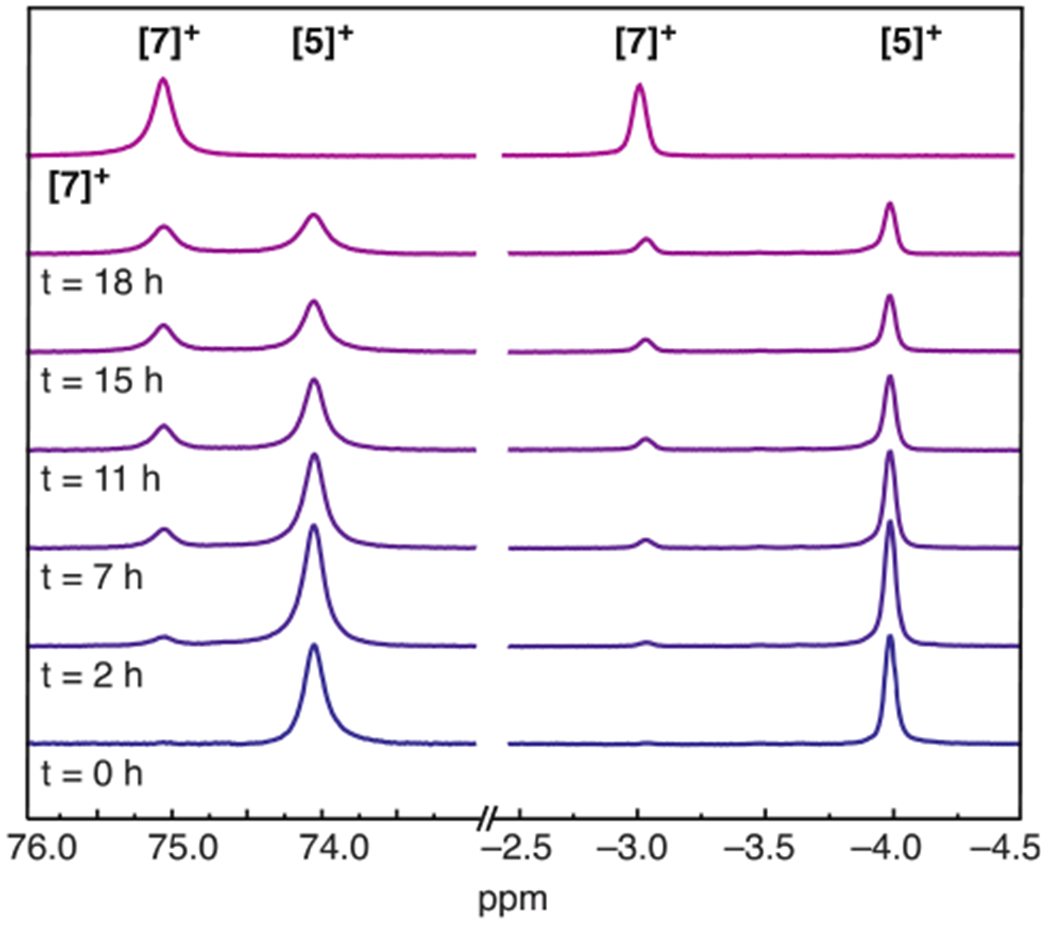
Partial 1H NMR spectrum of the reaction between [5]+ and CF3-py showing the decay of [5]+ and the growth of [7]+ during the course of the reaction. The top trace is an authentic sample of [7]+.
Scheme 2. Evidence for Reversible Fe–C Bond Homolysisa.
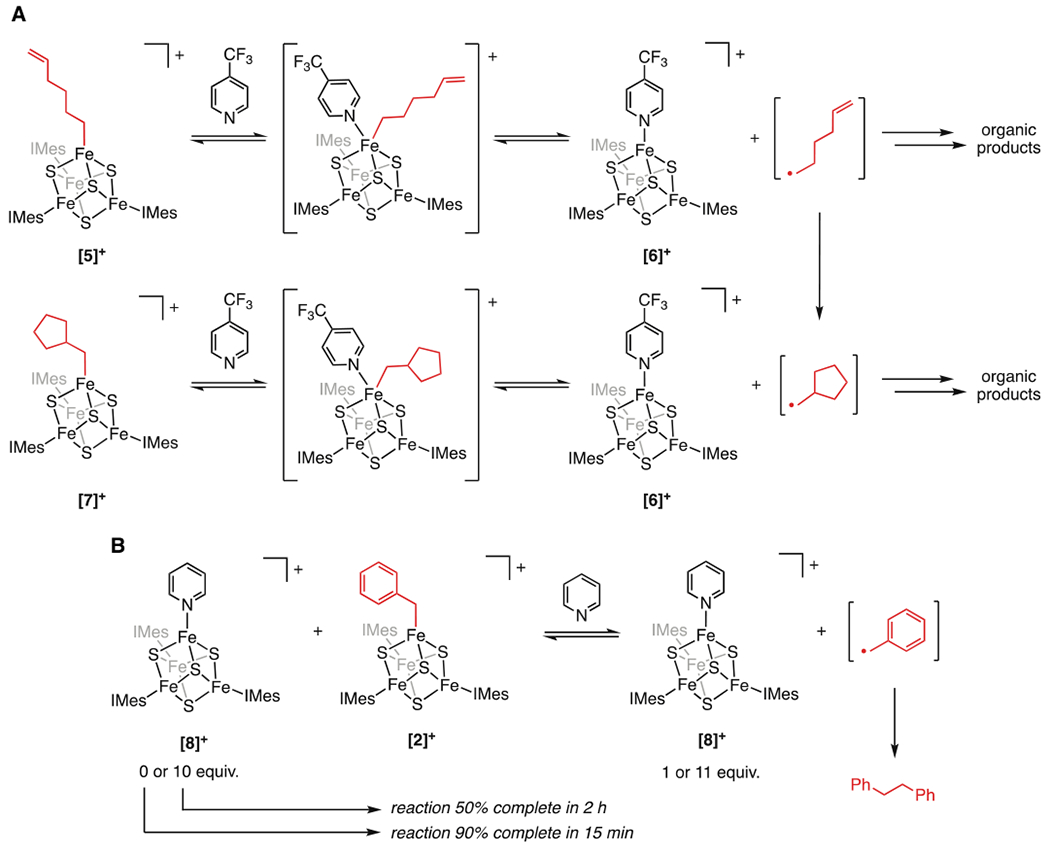
a(A) Proposed mechanism for the conversion of [5]+ to [7]+. (B) Reaction of [2]+ with pyridine in the presence of an excess of [8]+ decreases the rate of bibenzyl formation.
Based on these findings, we predicted that Fe–C bond homolysis in the reaction of [2]+ with pyridines should also be reversible, and that if this was the case, [(IMes)3Fe4S4(pyridine)][BArF4] ([8]+) would inhibit the radical termination steps that lead to bibenzyl. To test this, we added pyridine to [2]+ in the presence and absence of additional [8]+ (Scheme 2B); we chose to perform this test with pyridine and [8]+ rather than DMAP and [4]+ to slow the reaction so that the effect of the additional pyridine adduct would be easier to observe by 1H NMR spectroscopy. In the absence of added [8]+, the reaction of [2]+ and pyridine to yield [8]+ and bibenzyl is ~90% complete in 15 min. Strikingly, in the presence of 10 equiv of added [8]+, the reaction is only ~50% complete after 2 h. Taken together, these observations demonstrate rapid and reversible alkyl radical formation from [Fe4S4] clusters. That [Fe4S4] clusters exhibit this reactivity, which has also been observed in other open-shell metal complexes,37,43–46 most notably adenosylcobalamin models, further deepens the connections between the chemistry of adenosylcobalamin and radical SAM enzymes.
Kinetic Simulations of Reversible Fe–C Bond Homolysis in Radical SAM Enzymes.
Our results demonstrate that even primary alkyl radicals can be produced from [Fe4S4]–alkyl clusters, providing support for the intermediacy of organometallic species in radical SAM enzymes.21,22,38 We now consider how an organometallic intermediate that generates an alkyl radical can impact catalysis in radical SAM enzymes. First, an organometallic precursor to the 5′-dAdo• would clearly decrease the population of the 5′-dAdo•, with a stronger Fe–C bond shifting the homolysis equilibrium toward the organometallic species. An Fe–C bond that is too strong could shut down 5′-dAdo• production entirely, but our results suggest that radical SAM enzymes may avoid this scenario by virtue of the coordination geometry at the unique Fe site: coordination of the amine (and likely the carboxylate) of methionine renders the apical Fe site five- or six-coordinate. As we have shown here, a coordination number greater than four for the alkylated Fe results in a dramatically weakened Fe–C bond.
However, even a weak Fe–C bond would significantly deplete the population of the 5′-dAdo•. This would decrease the rates of all subsequent steps, but how would it impact selectivity for productive versus unproductive reactions? Of course, if the 5′-dAdo• and substrate are perfectly positioned for abstraction of only the desired H atom, the selectivity for the productive pathway would be perfect, and an organometallic intermediate would have no impact on selectivity. Even if the 5′-dAdo• can undergo unproductive reactions, such as abstracting the wrong H atom from the substrate or protein, the selectivity would depend only on the relative rates of the productive and unproductive pathways (Figure 4A,B).
Figure 4.
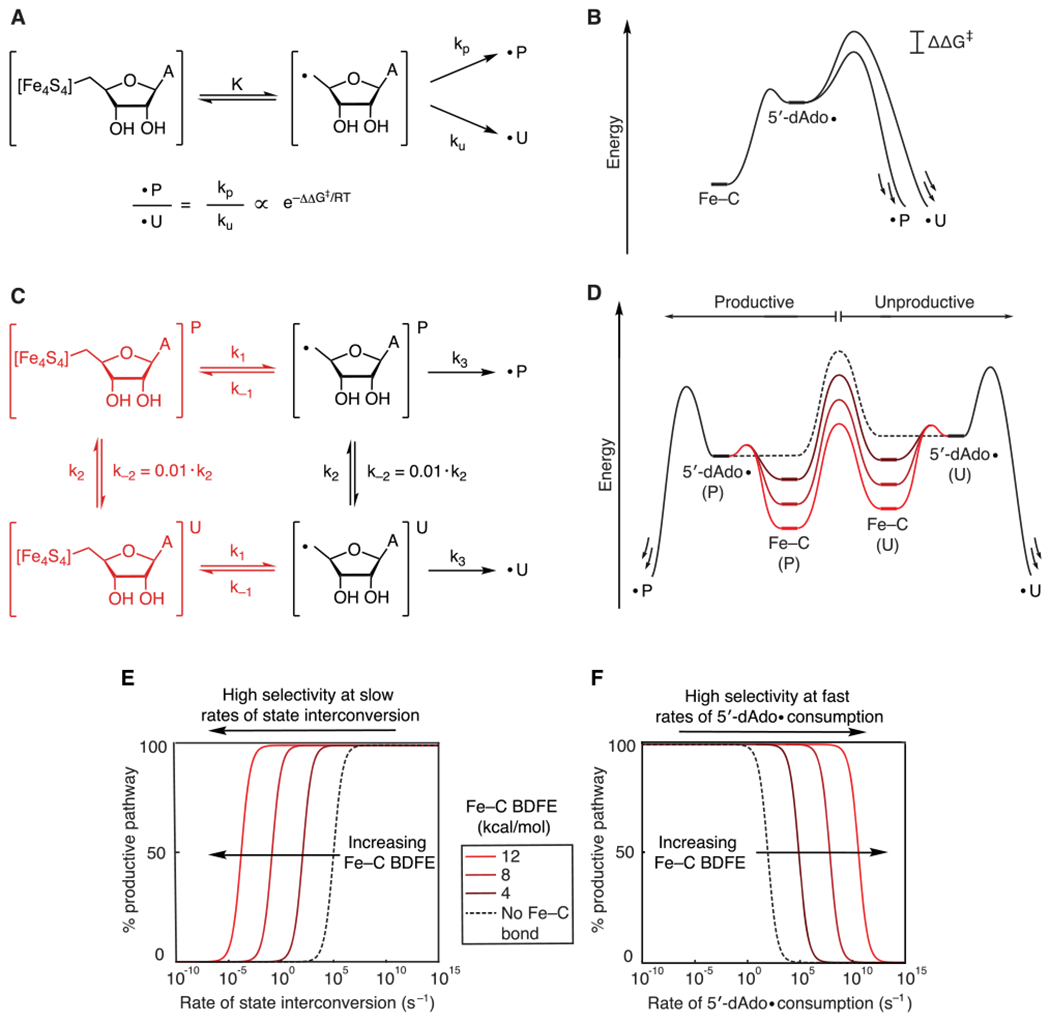
(A) Kinetic model and (B) energy diagram for a system in which the 5′-dAdo• reacts through a branching path with fixed rates of X–H abstraction. The presence of the organometallic intermediate has no effect on the selectivity. (C) Kinetic model that invokes interconversion between productive (P) and unproductive (U) states in the absence (black) and presence (black and red) of an organometallic intermediate. (D) Quantitative energy diagram for the system depicted in C showing the effect of the organometallic intermediate using barriers calculated from the rate constants listed below. As the Fe–C bond strength increases, the barrier to state interconversion becomes lower than the barrier to homolysis and X–H bond activation. (E) Simulations showing the selectivity of the reaction as a function of the rate of state interconversion and Fe–C bond strength. (F) Simulations showing the selectivity of the reaction as a function of the rate of X–H bond activation and Fe–C bond strength. k−1 (1011 s−1), k2 (102 s−2), and k3 (105 s−2) were held constant, and k2 was varied to give the indicated Fe–C bond strengths.
An organometallic intermediate can only affect the selectivity of the H-atom abstraction step if an additional, kinetically coupled process is operative. Multiple selectivity-influencing processes can be envisioned including bond rotations, rearrangements in the active site, or larger conformational changes. Regardless of their precise nature, such processes will affect the selectivity of the reaction only if they can outcompete X–H bond activation. Under these conditions, an organometallic intermediate can affect selectivity by slowing the rates of X–H bond activation to allow such processes to occur.
We evaluated this hypothesis using a simplified kinetic model consisting of a radical SAM enzyme for which the selectivity of X–H bond activation is dictated by whether the protein is in a productive state (denoted P and representing states leading to the substrate radical) or an unproductive state (denoted U and representing states leading to any other reaction outcome) (Figure 4C). In choosing rate constants for these simulations, we assumed that recombination of the 5′-dAdo• with the cluster is rapid (k−1 = 1011 s−1), that interconversion between protein states is slow (k2 = 102 s−1 and k−2 = 1 s−1, favoring the productive state by ~2 kcal/mol), and that Fe–C bond homolysis occurs slower than X–H bond activation (k1 = 103 s−1, k3 = 105 s−1). Further discussion of these rate constants is in the SI. Under these conditions, the slow rate of state interconversion favors X–H bond activation from the initial state; if the radical is formed in the unproductive state, state interconversion must outcompete unproductive X–H bond activation for productive reactivity to occur. We therefore initialized our simulations with the 5′-dAdo• in the unproductive state as a worst-case scenario to determine if an organometallic intermediate can recover selectivity for the productive reaction.
With these rate constants and in the absence of the organometallic intermediate, 99% of the radical is funneled to undesired pathways because, as noted above, H-atom abstraction is much faster than state interconversion. However, if an organometallic intermediate is formed, the H-atom abstraction step is sufficiently slowed such that the reaction occurs with 99% selectivity for the productive pathway (note that given the difference in energy of the productive and unproductive states, the maximum selectivity under thermodynamic control is 99%). Thus, if SAM is cleaved when the protein is in an unproductive state (e.g., if the initial substrate positioning is somehow perturbed), formation of the Fe–C bond could buy time for the protein to convert to a state that yields productive reactivity. Figure 4D illustrates the energetics of this process: introducing an Fe–C bond raises the barrier to X–H bond activation relative to state interconversion. This shifts the reaction from being under kinetic control, in which X–H bond activation outcompetes state interconversion, to being under Curtin–Hammett control, in which the relative X–H bond activation barriers control the selectivity.
We also probed how selectivity is affected by the individual kinetic parameters. Figure 4E shows the reaction outcome as a function of the rate of state interconversion with a range of Fe–C bond strengths; as the Fe–C bond strength is increased, higher selectivity for the productive reaction can be obtained at slower rates of state interconversion. This demonstrates how even a weak Fe–C bond in an organometallic intermediate can allow for relatively slow processes to impact the selectivity of the reaction. An analogous effect is observed when the rate of (both productive and unproductive) X–H bond activation is varied (Figure 4F); increasing the Fe–C bond strength allows for high selectivity even with very fast rates of X–H bond activation.
Overall, these results support a mechanism by which reversible formation of an organometallic intermediate can affect the selectivity in radical SAM reactions: it increases the range of kinetic parameters (X–H abstraction rates and state interconversion rates) for which interconversion between productive and unproductive states can occur, allowing these processes to outcompete what may be intrinsically very fast unproductive reactions.
Connection between Reversible M–C Bond Homolysis in Radical SAM and Adenosylcobalamin Enzymes.
The protective mechanism described above is agnostic with respect to the identity of the metal fragment and therefore may apply equally well to adenosylcobalamin enzymes for which reversible Co–C bond cleavage has been observed.45–47 Evidence for reversible Co–C bond homolysis was provided by rate measurements using protiated and deuterated substrates, which showed an H/D kinetic isotope effect on the apparent rate of Co–C bond homolysis that results from kinetic coupling of Co–C bond homolysis and H-atom abstraction.47–50 Similar kinetic experiments carried out with radical SAM enzymes could determine if reversible Fe–C bond cleavage occurs during turnover of radical SAM enzymes. The observation of a kinetic isotope effect for consumption of the organometallic intermediate when the substrate is deuterated would simultaneously provide evidence that the organometallic intermediate is on-path for productive chemistry and that Fe–C bond homolysis is reversible in enzymatic systems.
CONCLUSION
The observation of facile Fe–C bond homolysis from [Fe4S4]–alkyl clusters demonstrates the feasibility of 5′-dAdo• formation from organometallic intermediates in radical SAM enzymes and links the chemistry of radical SAM and adenosylcobalamin model systems. Similarly to Co–C bond homolysis in adenosylcobalamin model systems, Fe–C bond homolysis from [Fe4S4]–alkyl clusters occurs reversibly to generate primary carbon radicals, but unlike in adenosylcobalamin models, homolysis of Fe–C bonds in [Fe4S4]–alkyl clusters is facile at room temperature. Overall, these findings suggest that in both adenosylcobalamin enzymes and radical SAM enzymes, reversible homolysis of M–C bonds can allow for selective X–H bond activation by kinetically coupling reactions of the 5′-dAdo• to dynamic processes within or beyond the protein active site. Studies of the dynamics of these proteins will help illuminate how radical SAM and adenosylcobalamin enzymes utilize reversible M–C bond homolysis as a strategy to control the 5′-dAdo• and will contribute to our understanding of the differences between these two enzyme families.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Peter Muller for assistance with XRD experiments, Dr. Fang Wang for assistance with GC–MS measurements, and Profs. Alexander Radosevich and Alison Wendlandt for critical feedback on the manuscript. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM136882 and by fellowships for A.C.B. from the National Science Foundation (Graduate Research Fellowship #1122374) and the Fannie and John Hertz Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c05590.
Experimental procedures, spectra, additional discussion, and script for kinetic simulations (PDF)
Crystallographic data for 2 (CIF)
Crystallographic data for [2][OTf] (CIF)
Crystallographic data for [4][OTf] (CIF)
The authors declare no competing financial interest.
Contributor Information
Alexandra C. Brown, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
Daniel L. M. Suess, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
REFERENCES
- (1).Banerjee R; Ragsdale SW The Many Faces of Vitamin B12: Catalysis by Cobalamin-Dependent Enzymes. Annu. Rev. Biochem 2003, 72 (1), 209–247. [DOI] [PubMed] [Google Scholar]
- (2).Booker SJ; Grove TL Mechanistic and Functional Versatility of Radical SAM Enzymes. F1000 Biology Reports. 2010, 2, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-Adenosylmethionine Enzymes. Chem. Rev 2014, 114 (8), 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Landgraf BJ; McCarthy EL; Booker SJ Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem 2016, 85 (1), 485–514. [DOI] [PubMed] [Google Scholar]
- (5).Owing to its high reactivity, the 5′-dAdo• radical has only recently been observed. For details, see Yang H; McDaniel EC; Impano S; Byer AS; Jodts RJ; Yokoyama K; Broderick WE; Broderick JB; Hoffman BM. The Elusive 5′-Deoxyadenosyl Radical: Captured and Characterized by Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Spectroscopies. J. Am. Chem. Soc 2019, 141 (30), 12139–12146. and Sayler RI; Stich TA; Joshi S; Cooper N; Shaw JT.; Begley TP.; Tantillo DJ; Britt RD. Trapping and Electron Paramagnetic Resonance Characterization of the 5′dAdo• Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate. ACS Cent. Sci 2019, 5 (11), 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Frey PA Lysine 2,3-Aminomutase: Is Adenosylmethionine a Poor Man’s Adenosylcobalamin? FASEB J. 1993, 7 (8), 662–670. [DOI] [PubMed] [Google Scholar]
- (7).Frey PA; Magnusson OT S-Adenosylmethionine: A Wolf in Sheep's Clothing, or a Rich Man’s Adenosylcobalamin? Chem. Rev 2003, 103 (6), 2129–2148. [DOI] [PubMed] [Google Scholar]
- (8).Tamao Y; Blakley RL Direct Spectrophotometric Observation of an Intermediate Formed from Deoxyadenosylcobalamin in Ribonucleotide Reduction. Biochemistry 1973, 12 (1), 24–34. [DOI] [PubMed] [Google Scholar]
- (9).Finlay TH; Valinsky J; Mildvan AS; Abeles RH Electron Spin Resonance Studies with Dioldehydrase. Evidence for Radical Intermediates in Reactions Catalyzed by Coenzyme B12. J. Biol. Chem 1973, 248 (4), 1285–1290. [PubMed] [Google Scholar]
- (10).Berkovitch F; Behshad E; Tang KH; Enns EA; Frey PA; Drennan CL A Locking Mechanism Preventing Radical Damage in the Absence of Substrate, as Revealed by the X-Ray Structure of Lysine 5,6-Aminomutase. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (45), 15870–15875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zelder O; Beatrix B; Leutbecher U; Buckel W Characterization of the Coenzyme-B12–Dependent Glutamate Mutase from Clostridium Cochlearium Produced in Escherichia Coli. Eur. J. Biochem 1994, 226 (2), 577–585. [DOI] [PubMed] [Google Scholar]
- (12).Lieder KW; Booker S; Ruzicka FJ; Beinert H; Reed GH; Frey PA S-Adenosylmethionine-Dependent Reduction of Lysine 2,3-Aminomutase and Observation of the Catalytically Functional Iron-Sulfur Centers by Electron Paramagnetic Resonance. Biochemistry 1998, 37 (8), 2578–2585. [DOI] [PubMed] [Google Scholar]
- (13).Gruber K; Reitzer R; Kratky C Radical Shuttling in a Protein: Ribose Pseudorotation Controls Alkyl-Radical Transfer in the Coenzyme B12 Dependent Enzyme Glutamate Mutase. Angew. Chem., Int. Ed 2001, 40 (18), 3377–3380. [DOI] [PubMed] [Google Scholar]
- (14).Shibata N; Tamagaki H; Hieda N; Akita K; Komori H; Shomura Y; Terawaki SI; Mori K; Yasuoka N; Higuchi Y; Toraya T Crystal Structures of Ethanolamine Ammonia-Lyase Complexed with Coenzyme B12 Analogs and Substrates. J. Biol. Chem 2010, 285 (34), 26484–26493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).LoBrutto R; Bandarian V; Magnusson OT; Chen X; Schramm VL; Reed GH 5′-Deoxyadenosine Contacts the Substrate Radical Intermediate in the Active Site of Ethanolamine Ammonia-Lyase: 2H and 13C Electron Nuclear Double Resonance Studies. Biochemistry 2001, 40 (1), 9–14. [DOI] [PubMed] [Google Scholar]
- (16).Canfield JM; Warncke K Geometry of Reactant Centers in the CoII-Substrate Radical Pair State of Coenzyme B12-Dependent Ethanolamine Deaminase Determined by Using Orientation-Selection-ESEEM Spectroscopy. J. Phys. Chem. B 2002, 106 (34), 8831–8841. [DOI] [PubMed] [Google Scholar]
- (17).Horitani M; Byer AS; Shisler KA; Chandra T; Broderick JB; Hoffman BM Why Nature Uses Radical SAM Enzymes so Widely: Electron Nuclear Double Resonance Studies of Lysine 2,3-Aminomutase Show the 5′-DAdo• “Free Radical” Is Never Free. J. Am. Chem. Soc 2015, 137 (22), 7111–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vey JL; Drennan CL Structural Insights into Radical Generation by the Radical SAM Superfamily. Chem. Rev 2011, 111 (4), 2487–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sofia HJ; Chen G; Hetzler BG; Reyes-Spindola JF; Miller NE Radical SAM, a Novel Protein Superfamily Linking Unresolved Steps in Familiar Biosynthetic Pathways with Radical Mechanisms: Functional Characterization Using New Analysis and Information Visualization Methods. Nucleic Acids Res. 2001, 29 (5), 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Holliday GL; Akiva E; Meng EC; Brown SD; Calhoun S; Pieper U; Sali A; Booker SJ; Babbitt PC Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug and Play” Domain. Methods Enzymol 2018, 606, 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Horitani M; Shisler K; Broderick WE; Hutcheson RU; Duschene KS; Marts AR; Hoffman BM; Broderick JB Radical SAM Catalysis via an Organometallic Intermediate with an Fe-[5′-C]-Deoxyadenosyl Bond. Science 2016, 352 (6287), 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Byer AS; Yang H; McDaniel EC; Kathiresan V; Impano S; Pagnier A; Watts H; Denler C; Vagstad AL; Piel J; Duschene KS; Shepard EM; Shields TP; Scott LG; Lilla EA; Yokoyama K; Broderick WE; Hoffman BM; Broderick JB Paradigm Shift for Radical S-Adenosyl-L-Methionine Reactions: The Organometallic Intermediate Ω Is Central to Catalysis. J. Am. Chem. Soc 2018, 140 (28), 8634–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nicolet Y Structure-Function Relationships of Radical SAM Enzymes. Nat. Catal 2020, 3 (4), 337–350. [Google Scholar]
- (24).Daikh BE; Finke RG Unprecedented and Reversible Cobalt-to-Carbon Alkyl Bond Rearrangement in the Coenzyme B12 Model Complex C6H5CH2CoIII[C2(DO)(DOH)Pn]I: Synthesis, Structural Characterization, and Mechanistic Studies. J. Am. Chem. Soc 1991, 113 (11), 4160–4172. [Google Scholar]
- (25).Garr CD; Finke RG Adocobalamin (AdoCbl or Coenzyme B12) Co-C Bond Homolysis Radical-Cage Effects: Product, Kinetic, Mechanistic, and Cage Efficiency Factor (Fc) Studies, plus the Possibility That Coenzyme B12-Dependent Enzymes Function as “Ultimate Radical Cages” and “Ultimate Radical Traps”. Inorg. Chem. 1993, 32 (20), 4414–4421. [Google Scholar]
- (26).Hay BP; Finke RG Thermolysis of the Co-C Bond of Adenosylcobalamin. 2. Products, Kinetics, and Co-C Bond Dissociation Energy in Aqueous Solution. J. Am. Chem. Soc. 1986, 108 (16), 4820–4829. [Google Scholar]
- (27).Koenig T; Finke RG The Cage Effect and Apparent Activation Parameters for Bond Homolysis. J. Am. Chem. Soc 1988, 110 (8), 2657–2658. [Google Scholar]
- (28).Finke RG; Hay BP Thermolysis of Adenosylcobalamin: A Product, Kinetic, and Cobalt-Carbon (C5′) Bond Dissociation Energy Study. Inorg. Chem 1984, 23 (20), 3041–3043. [Google Scholar]
- (29).Halpern J Some Aspects of Organocobalt Chemistry Related to Vitamin B12. Ann. N. Y. Acad. Sci 1974, 239 (1), 2–21. [DOI] [PubMed] [Google Scholar]
- (30).Tsou TT; Loots M; Halpern J Kinetic Determination of Transition Metal-Alkyl Bond Dissociation Energies: Application to Organocobalt Compounds Related to B12 Coenzymes. J. Am. Chem. Soc 1982, 104 (2), 623–624. [Google Scholar]
- (31).Ng FTT; Rempel GL; Halpern J Ligand Effects on Transition-Metal-Alkyl Bond Dissociation Energies. J. Am. Chem. Soc 1982, 104 (2), 621–623. [Google Scholar]
- (32).Halpern J Determination of Transition Metal-Alkyl Bond Dissociation Energies from Kinetic Measurements. Polyhedron 1988, 7 (16–17), 1483–1490. [Google Scholar]
- (33).Halpern J Determination and Significance of Transition Metal-Alkyl Bond Dissociation Energies. Acc. Chem. Res 1982, 15 (8), 238–244. [Google Scholar]
- (34).Ye M; Thompson NB; Brown AC; Suess DLM A Synthetic Model of Enzymatic [Fe4S4]-Alkyl Intermediates. J. Am. Chem. Soc 2019, 141 (34), 13330–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brown AC; Suess DLM Controlling Substrate Binding to Fe4S4 Clusters through Remote Steric Effects. Inorg. Chem 2019, 58 (8), 5273–5280. [DOI] [PubMed] [Google Scholar]
- (36).Hansch C; Leo A; Taft RW A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91 (2), 165–195. [Google Scholar]
- (37).Daikh BE; Finke RG The Persistent Radical Effect: A Prototype Example of Extreme, 105 to 1, Product Selectivity in a Free-Radical Reaction Involving Persistent ·CoII[Macrocycle] and Alkyl Free Radicals. J. Am. Chem. Soc 1992, 114 (8), 2938–2943. [Google Scholar]
- (38).Dong M; Kathiresan V; Fenwick MK; Torelli AT; Zhang Y; Caranto JD; Dzikovski B; Sharma A; Lancaster KM; Freed JH; Ealick SE; Hoffman BM; Lin H Organometallic and Radical Intermediates Reveal Mechanism of Diphthamide Biosynthesis. Science 2018, 359 (6381), 1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Newcomb M Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49 (6), 1151–1176. [Google Scholar]
- (40).Newcomb M Radical Kinetics and Clocks. Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd: Chichester, UK, 2012. [Google Scholar]
- (41).Walling C; Cioffari A Cyclization of 5-Hexenyl Radicals. J. Am. Chem. Soc 1972, 94 (17), 6059–6064. [Google Scholar]
- (42).Ash CE; Hurd PW; Darensbourg MY; Newcomb M Competing Nucleophilic Displacement and Radical Chain Reduction in Reactions of Transition-Metal Hydride Anions with Alkyl Bromides. J. Am. Chem. Soc 1987, 109 (11), 3313–3317. [Google Scholar]
- (43).Fischer H The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev 2001, 101 (12), 3581–3610. [DOI] [PubMed] [Google Scholar]
- (44).Fischer H Unusual Selectivities of Radical Reactions by Internal Suppression of Fast Modes. J. Am. Chem. Soc 1986, 108 (14), 3925–3927. [Google Scholar]
- (45).Branchaud BP; Yu GX An Example of the Persistent Radical Effect in Cobaloxime-Mediated Radical Alkyl-Alkenyl Cross Coupling. Organometallics 1993, 12 (11), 4262–4264. [Google Scholar]
- (46).Byungho S; Goff HM Free Radical-Mediated Electron Transfer in Organometallic Complexes: Homolysis and Alkyl Group Crossover Reactions for Alkyliron(II) Porphyrins. Inorg. Chim. Acta 1994, 226 (1–2), 231–235. [Google Scholar]
- (47).Chen D; Abend A; Stubbe JA; Frey PA Epimerization at Carbon-5′ of (5′R)-[5′-2H]Adenosylcobalamin by Ribonucleoside Triphosphate Reductase: Cysteine 408-Independent Cleavage of the Co-C5′ Bond. Biochemistry 2003, 42 (15), 4578–4584. [DOI] [PubMed] [Google Scholar]
- (48).Marsh ENG; Meléndez GDRR Adenosylcobalamin Enzymes: Theory and Experiment Begin to Converge. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824 (11), 1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Marsh ENG; Ballou DP Coupling of Cobalt-Carbon Bond Homolysis and Hydrogen Atom Abstraction in Adenosylcobalamin-Dependent Glutamate Mutase. Biochemistry 1998, 37 (34), 11864–11872. [DOI] [PubMed] [Google Scholar]
- (50).Cheng MC; Marsh ENG Isotope Effects for Deuterium Transfer between Substrate and Coenzyme in Adenosylcobalamin-Dependent Glutamate Mutase. Biochemistry 2005, 44 (7), 2686–2691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.