

Abstract
Type I and III interferons (IFNs) activate similar downstream signaling cascades but unlike type I IFNs, type III IFNs (IFNλ) do not elicit strong inflammatory responses in vivo. Here we examined the molecular mechanisms underlying this disparity. Type I and III IFNs displayed kinetic differences in expression of IFN-stimulated genes and proinflammatory responses, with type I IFNs preferentially stimulating expression of the transcription factor IRF1. Type III IFNs failed to induce IRF1 expression due to low IFNλ receptor abundance and insufficient STAT1 activation on epithelial cells, and thus did not activate the IRF1 proinflammatory gene program. Rather, IFNλ stimulation preferentially induced factors implicated in tissue repair. Our findings suggest that IFN receptor compartmentalization and abundance confer a spatiotemporal division of labor wherein type III IFNs control viral spread at the site of the infection while restricting tissue damage; the transient induction of inflammatory responses by type I IFNs recruits immune effectors to promote protective immunity.
Keywords: interferons, interferon regulatory factor 1, chemokines, inflammation, epithelial cells
eTOC Blurb
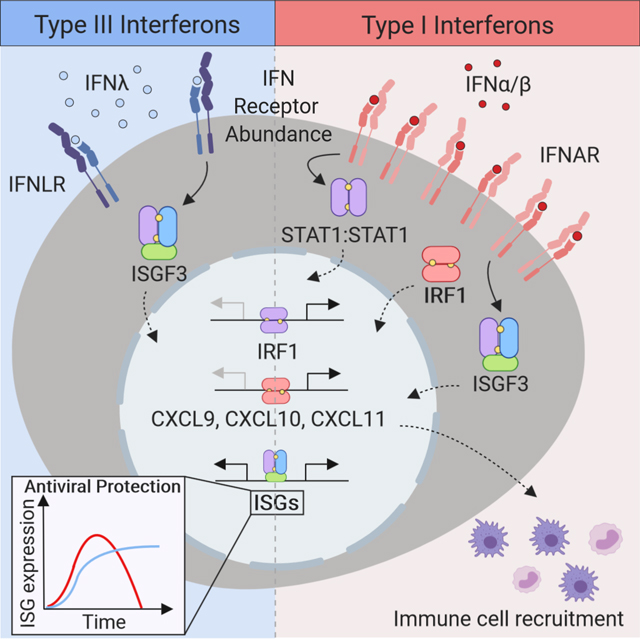
Type I IFNs but not type III IFNs (IFNλ) promote inflammation at the site of infection. Forero et al. find that the differential expression of proinflammatory genes results from selective induction of the transcription factor IRF1 by type I IFNs. Type III IFNs induce a tissue-repair program, suggesting a division of labor that spans proinflammatory and tissue repair functions to promote protective immunity.
Graphical abstract
INTRODUCTION
Control of viral replication and spread is largely executed by interferons (IFNs) through the induction of an antiviral state in infected and neighboring uninfected cells. IFNs are directly induced as a result of viral recognition by pattern recognition receptors and serve as the first line of defense by promoting the expression of antiviral effector IFN-stimulated genes (ISG). The IFNs that activate intrinsic antiviral programs are classified into type I and III IFN families. The human type I IFN family consists of 13 IFNα subtypes, IFNβ, IFNκ, IFNω, and IFNε. Type I IFNs exert their biological activity through the activation of a JAK-STAT signaling cascade downstream of the type I IFN receptor (IFNΑR) formed by the IFNΑR1 and IFNΑR2 subunits. The essential role of type I IFNs in eliciting robust innate and adaptive immune responses to control viral infection has been extensively demonstrated in Ifnal−/− mice (Hwang et al., 1995; Muller et al., 1994) across several viral infections. The type III IFN family includes IFNλ1, IFNλ2, IFNλΑ3, and IFNλ4, which bind to the IFNλ, receptor composed of IFNLR1 and IL-10R2 (Kotenko et al., 2003; Sheppard et al., 2003). Type III IFNs confer strong antiviral protection via the activation of JAK-STAT signaling cascades and the induction of an overlapping set of ISGs (Ank et al., 2008; Dumoutier et al., 2004; Hong et al., 2016; Zhou et al., 2007). Single nucleotide polymorphisms in the IFNL locus, affecting IFNL3 mRNA stability and disrupting the IFNL4 open reading frame, strongly associate with impaired clearance of hepatitis C virus (HCV) and response to antiviral therapy (Ge et al., 2009; McFarland et al., 2014; Prokunina-Olsson et al., 2013; Suppiah et al., 2009; Tanaka et al., 2009; Thomas et al., 2009). These findings highlight the evolutionary requirement for both type I and III IFNs for the control of viral infections.
We now understand the general pathways of induction, response, and activation of antiviral activities of type III IFNs, and a few key broadly distinguishing features of this family have emerged (Hemann et al., 2017; Lazear et al., 2019). While IFNΑR expression is ubiquitous, IFNLR1 expression is mostly restricted to mucosal surfaces (Ank et al., 2008) and mediates control of viral infections at these sites (Baldridge et al., 2017; Galani et al., 2017; Jewell et al., 2010; Nice et al., 2015). The induction of ISG expression in response to type III IFN is delayed relative to that elicited by type I IFN in cells expressing both IFNΑR and IFNLR1 (Jilg et al., 2014; Marcello et al., 2006). In contrast to the responses elicited by type I IFNs, type III IFNs fail to mount a robust inflammatory response in epithelial barriers (Davidson et al., 2016; Galani et al., 2017). These observations suggest that these two IFN families have evolved to carry out specialized functions and coordinate antiviral responses spatiotemporally, presumably to benefit the host.
Here we examined the molecular mechanisms underlying the distinct responses to type I and III IFNs at mucosal surfaces. We found that the transcription factor IFN regulatory factor 1 (IRF1) was induced primarily by type I, but not by type III IFNs in epithelial cells. IRF1 activated the transcription of a specific set of genes, including pro-inflammatory chemokines. The differential induction of IRF1 was dictated by the amount of IFNLR1 expressed by the cell. IFNλ, induced a distinct transcriptional program that lacked inflammatory gene activation and rather featured expression of genes associated with the maintenance barrier integrity. We propose that type III IFNs control viral spread at the site of the infection, restricting tissue damage by limiting inflammatory responses and initiating epithelial repair. In contrast, the transient induction of inflammatory responses by type I IFNs serve to recruit immune effectors to the site of infection to promote protective immunity.
RESULTS
The magnitude of the response to IFNs is regulated at the transcriptional level.
Type I and III IFNs induce a largely overlapping antiviral gene signature in human hepatocytes (Jilg et al., 2014) with quantitative and temporal differences in induction. However, the induction of proinflammatory genes seen in response to type I IFN is absent with type III IFN treatment (Davidson et al., 2016; Galani et al., 2017; Kim et al., 2017). To probe the mechanisms contributing to such differences, we studied the transcriptional response to either type I or III IFN treatment in immortalized human hepatocytes. Type I IFN (IFNλ) induced an early but transient induction of ISGs, while type III IFN (IFNλ3) induced a delayed, prolonged accumulation of ISG15, MX1, and OAS1 mRNA (Figure 1A) which was reflected in the expression of ISG15 protein at early (Figure 1B) and late (Figure1C) timepoints. These differences were evident using doses of IFN that induced comparable proximal activation of STAT1 downstream of both IFNΑR and IFNLR, as determined by its phosphorylation and nuclear localization at 30 mins after IFN-treatment (Figure S1A and B). Electromobility shift assays (EMSA) using an ISG15-ISRE DNA probe (Figure S1C) showed that an early DNA:protein complex is induced by IFNβ and is absent by 20h post-treatment. This complex is also induced IFNλ3 by 3h and sustained up to 20h post-treatment (Figure 1D). Inhibition of complex formation was achieved through co-incubation with antibodies against the subunits of the ISGF3 transcriptional complex: IRF9, STAT1, and STAT2 (Figure S1D). This indicated that ISGF3-mediated transcription drives gene expression downstream of both IFN families, but transactivation of gene expression by IFNλ3 is not subject to early inhibitory mechanisms described for type I IFN (Blumer et al., 2017).
Figure 1 – The antiviral response to IFNλ is delayed relative to IFNβ.

IFN-stimulated gene (ISG) expression following IFN treatment in PH5CH8 cells. (A) Induction of ISG15, MX1, and OAS1 mRNA following IFNβ or IFNλ3 for the indicated times. Mean changes ± SD in gene expression were determined relative to mock-treated cells (value of 1) and normalized to HPRT. (B-C) Immunoblot analysis of ISG15 post IFN treatment. Statistical significance of stimulation and time-dependent gene expression changes were analyzed using two-way ANOVA. (D) EMSA with nuclear extracts from IFNβ or IFNλ3 treated PH5CH8 co-incubated with radiolabeled ISG15 ISRE probe. Data are representative of 2 independent experiments. (E) Violin plots indicate the relative mRNA expression of total RNA (F) and polyribosome associated mRNA across 41 ISGs at the indicated times post IFN-treatment. Statistical significance was determined by Mann-Whitney test. Solid bars indicate the median and quartiles. (G) Heatmap representation of log2 transformed relative expression of individual ISG mRNA after IFN treatment (left) and relative ratio of polysome-bound mRNA (right). Red color indicates increases while blue indicates a decrease in gene expression and polysome association. (H) Assessment of antiviral protection in cells preincubated with IFNβ, IFNλ3, or both for the indicated time prior to infection with VSV at a multiplicity of infection (moi) of 0.1. Schematic of experimental design and representative image of crystal violet uptake assay (top) and quantification of dye-uptake (bottom). Uninfected, untreated cells were used as negative controls (100% protection). Infected, untreated cells served as positive control (no protection). Data represents mean protection across 3 independent experiments ± SEM. Across all experiments, cells were stimulated with 25 IU/ml of IFNβ or 100 ng/ml IFNλ3. Unless otherwise indicated, data is representative of 3 independent experiments. See also Figure S1.
IFNβ promoted a burst of mRNA induction across 41 core ISGs that peaked at 8h and resolved over time (Figure 1E). In IFNλ3 treated cells, the induction of ISGs was delayed but persisted across all timepoints. Translation control is critical to the induction of IFN-mediated protein expression (Kroczynska et al., 2014). Overall, the rate of polyribosome association across the core ISGs, was consistent with the kinetics of transcription induced by either IFN (Figure 1F). This suggested that there were no overt differences in the translational control of these ISGs (Figure 1G) or the overall efficiency of mRNA translation were observed in response to either IFN (Figure S1E). Differences in the induction or activation of transcriptional regulators, rather than translation control, might account for the temporal differences in expression of ISGs by type I and III IFN despite the activation of similar proximal signaling cascades.
IFN-induced antiviral activities are mediated by the products of ISGs (Schoggins et al., 2011). Thus, we investigated whether the temporal alteration in ISG induction between IFNβ and IFNλ3 was consistent with the antiviral activities of either IFN. Pre-incubation with IFNβ for 6h minimized the cytopathic effect (CPE) induced by vesicular stomatitis virus (VSV) infection, but the antiviral protection waned after 24h. In contrast, the protective effects of IFNλ were delayed, reaching their peak at 24h and sustained up to 72h. Dual pre-treatment with IFNβ and IFNλΑ up to 72h prior to infection, led to sustained, additive antiviral protection (Figure 1H). Together these data demonstrate that the intersecting antiviral activities of type I and III IFNs are consistent with the timing of ISG transcription and protein accumulation. Furthermore, through the distinct regulation of ISG expression, type I and III IFNs fulfilled a non-redundant role in conferring prolonged resistance against viral infection.
The transcription factor IRF1 is induced following type I IFN, but not type III IFN treatment.
Although our transcriptional and translational profiling of ISGs supports the overlapping induction of ISGs, we observed a significant disparity in the induction of the chemokine gene, CXCL10 (Figure 1G). IFNβ stimulated CXCL10 expression, whereas IFNλ treatment failed to do so (Figure S1F). We observed unique co-regulation of the family of proinflammatory chemokines genes that signal through CXCR3, CXCL9, CXCL10 and CXCL11 (Groom and Luster, 2011), by IFNβ treatment (Figure 2A). This coincided with synthesis and secretion of CXCL10 protein by IFNβ, but not IFNλ3 treated cells (Figure 2B). These observations are consistent with a significant induction of CXCL10 mRNA and polyribosome association upon type I IFN treatment (Figure 1G). The selective induction of CXCL10 suggests a type I IFN-biased transcriptional regulatory mechanism.
Figure 2 – IRF1 is differentially induced by type I and III IFNs.

(A) Relative gene expression of CXCL9, CXCL10, and CXCL11 mRNA measured by qPCR following IFNβ or IFNλ3 treatments of PH5CH8 cells at the indicated timepoints. Data is representative of means ± SD (B) Average CXCL10 protein production ± SEM after treatment of PH5CH8 cells with IFNβ or IFNλ3 across 2 independent experiments. Statistical significance determined by two-way ANOVA. (C) Immunoblot analysis of IRF1, (D) IRF7 and IRF9 and β-Actin expression after IFN treatment of PH5CH8 cells. (E) Immunoblot analysis of IRF1 and β-Actin expression following stimulation with IFNα2 (250 IU/ml), IFNβ (25 IU/ml) or IFNλ1–3 (100 ng/ml) for 4h in PH5CH8 cells. (F) Average CXCL10 and ISG15 mRNA expression following stimulation with IFNα2 (250 IU/ml), IFNβ (250 IU/ml), or IFNλ1–3 (100 ng/ml) for 4h in PH5CH8 cells across 4 independent experiments ± SEM. (G) Firefly luciferase activity of IRF1 promoter after treatment with IFNβ (25 IU/ml), IFNλ1–3 (100 ng/ml), or IFNγ (5 ng/ml) for 6hrs. Statistical significance was determined using one-way ANOVA. Data represents the mean ± SEM across 5 independent experiments. (H-I) Average relative gene expression changes of IRF1, CXCL10 and CXCL10 of total mRNA (top row) and 4sU labeled newly synthesized mRNA (bottom row) following IFN treatment across 3 independent experiments. Changes in mRNA expression are represented relative to mock-treated cells (value 1) and normalized to HPRT. Unless otherwise indicated, cells were stimulated with 25 IU/ml of IFNβ or 100 ng/ml IFNλ3. Unless otherwise indicated, data is representative of the mean ± SEM across 3 independent experiments. See also Figure S2.
IRFs are transcription factors activated in response to pattern recognition receptor activation and IFN sensing that regulate gene expression through binding to IFN-stimulated response elements (ISRE) (Ikushima et al., 2013). IRFs participate in the control of CXCL9, CXCL10, and CXCL11 expression with IFNγ treatment, as IFNγ, a type II IFN that is produced by immune cells (Billiau and Matthys, 2009; Kanda et al., 2007). Disruption of the ISRE motif in the CXCL10 promoter abrogated luciferase reporter activity in response to both IFNβ or IFNγ and TNFα, while IFNλ3 treatment had no effect on reporter activity (Figure S2A). IRF1, IRF7, and IRF9, exert regulatory functions downstream of IFN receptor signaling (Honda and Taniguchi, 2006). IRF1 or IRF7 overexpression is sufficient to promote the induction of CXCL10, CXCL11, ISG15 mRNA (Figure S2B). IRF1 protein was robustly induced by IFNβ, while IFNλ3 treatment induced significantly lower IRF1 expression (Figure 2C). IRF1 induction (2–8h) preceded IRF7 protein expression (4–24h) post-IFNβ treatment. In IFNλ3-treated cells, the expression of IRF7 was delayed while sustained at low levels (8–72h). No significant temporal or quantitative differences were apparent in the accumulation of IRF9 following treatment with either IFN (Figure 2D). As IFNβ, IFNα2 treatment led to IRF1 induction but neither IFNλ1 nor IFNλ2 stimulation induced robust IRF1 expression (Figure 2E). This was consistent with the induction of CXCL10 mRNA by type I IFNs at 4h post-stimulation, but not by type III IFNs despite their collective ability to induce ISG15 transcription (Figure 2F).
The increase in IRF1 protein expression was due to the transcriptional induction of IRF1, confirmed through IRF1 luciferase promoter reporter assays that demonstrated specific increase in luciferase activity only upon stimulation with IFNβ and IFNγ, but not IFNλ1–3 (Figure 2G). IFN-induced de novo RNA synthesis of IRF1 by metabolically labelling newly transcribed RNA with 4-thiouridine (4sU) following IFN stimulation (Figure 2H). The de novo mRNA (bottom) and total mRNA (top) transcription of IRF1 followed similar patterns. De novo IRF1 transcription was at its peak at 2h post-IFNβ treatment and decreased over time. Early synthesis of CXL10 and CXCL11 mRNA was apparent after IFNβ treatment and declined by 8h. On the other hand, the transcription of MX1, which was transient in IFNβ, continued up to 24h after IFNλ3 sensing (Figure 2I). The dose-dependent induction of CXCL10 in IFNΑR1 or IFNLR1 deficient cells by exogenous IRF1 expression, demonstrated that IFN-mediated signal transduction is dispensable for IRF1-mediated transactivation of CXCL10 (Figure S2C). Taken together, these data demonstrate a selective induction of IRF1 by type I IFN treatment, which in turn is capable of inducing IRF1-dependent genes such as the CXCR3 ligands. On the other hand, IRF1 is largely refractory to type III IFNs and this correlates with muted chemokine responsiveness.
IFNLR1 abundance determines the threshold of STAT1-driven IRF1 expression.
To determine if the ISGF3 complex is required for type I IFN-mediated IRF1 induction we tested the requirement of STAT1, STAT2 and IRF9 for IFNβ-induced IRF1 mRNA expression in hepatocytes. Using RNA interference, we observed that suppression of STAT1 and STAT2 reduced the basal levels of IRF1 mRNA (Figure 3A, S3A). Decrease in IRF9, STAT1 and STAT2 led to a significant decrease in IRF1 mRNA upon IFNβ treatment. We confirmed this in STAT1-deficient PH5CH8 cells, where we saw complete ablation of IFNβ-dependent induction of IRF1 (Figure 3B) with a concomitant loss of CXCL10 mRNA expression (Figure 3C). STAT2 expression was also required for IRF1 expression in response to IFNβ, but not IFNγ (Figure 3D), as STAT2-deficiency resulted in a severe impairment in the activation of STAT1 (pSTAT Y701) by IFNβ (Figure 3E and Figure S3C). Moreover, neither STAT1 nor STAT2 loss affected the induction of IRF1 in the response to IFNλ. This phenotype was replicated in 2fTGH cells, where loss of STAT1 (U3A) or STAT2 (U6A) results in abrogation of IRF1 responsiveness to IFNβ treatment (Figure 3F) and STAT2-deficiency affects STAT1 activation (Figure S3C). These data suggested that the primary transcription factor required for IRF1 induction is STAT1, whereas STAT2 is required for its maximal activation in response to IFNβ treatment.
Figure 3 – Robust STAT1 activation is required for the induction of IRF1.

(A) Requirement of ISGF3 complex subunits in the induction of IRF1. PH5CH8 cells transfected with 20nM of IRF9, STAT1, and STAT2-targeting siRNA, or scramble siRNA and mock-treated (black) or IFNβ-treated for 6h. Relative mean ± SD expression changes of IRF1 mRNA is plotted relative to control siRNA transfected cells (100%). (B) Immunoblot IRF1, STAT1, STAT2, and β-Actin following of Wild-type (WT) and STAT1-deficient PH5CH8 cells with IFNβ or IFNλ3 for 4h. (C) Relative mean ± SD gene expression changes of IRF1 and CXCL10 mRNA expression by qPCR following 4h of IFNβ or IFNλ3 treatment. (D) Immunoblot analysis of IRF1 β-Actin following treatment with IFNβ or IFNλ3 or IFNγ in WT and STAT2-deficient PH5CH8 cells. (E) Immunoblot analysis of phosphorylated STAT1 (Y701), total STAT1, and β-Actin in WT and STAT2-deficientPH5CH8 cells treated with IFNβ (25 IU/ml or 250IU/ml), IFNλ3, or IFNγ for 0.5h. (D-E) Saturated pixels are highlighted in red. (F) Immunoblot of IRF1, STAT1, and STAT2 expression in 2fTGH, STAT1-deficient (U3A) and STAT2-deficient (U6A) cells treated with IFNβ (500 IU/ml) or IFNγ for 4h. (G) Electromobility shift assay with nuclear extracts from IFNγ, IFNβ (125 IU/ml), IFNλ3 (500 ng/ml) treated PH5CH8 incubated with radiolabeled IRF1 probe. (H) Supershift EMSA for the identification of transcriptional regulators of IRF1. Nuclear extracts from IFNγ, IFNβ (125 IU/ml), IFNλ3 (500 ng/ml) treated PH5CH8 were co-incubated with radiolabeled IRF1 probe with indicated antibodies. (I) Immunoblot IRF1 and GAPDH analysis expression after treatment of PH5CH8 cells with IFNβ and/or IFNλ3 for 4h. (J) Immunoblot of IRF1 and β-Actin after treatment with IFNβ or increasing concentrations of IFNλ3 for 6h. Changes in mRNA expression are represented relative to mock-treated cells and normalized to HPRT. Unless otherwise indicated, cells were stimulated with 25 IU/ml of IFNβ, 100 ng/ml IFNλ3, or 5ng/ml IFNγ. Unless otherwise indicated, data is representative of 3 independent experiments. See also Figure S3.
To test if the activation of IRF1 was dependent on STAT1 activation, translocation, and binding to the IRF1 promoter, we co-incubated nuclear protein extracts from IFN-treated cells with a DNA probe derived from the IRF1 promoter STAT-binding site (Figure S3D). Only IFNβ and IFNγ-treated cell extracts induced a gel-shift band with the IRF1-promoter DNA (Figure 3G). To determine which STAT homo or heterodimer complexes were present, we co-incubated the DNA:protein complexes with antibodies against STAT proteins. Only incubation with antibodies against STAT1 led to a supershift (Figure 3H). These data demonstrate that activation of STAT1 and its homodimerization are essential for IRF1 induction, and these complexes are induced by type I and II, but not by type III IFN treatment. Since STAT1 is also essential for signal transduction downstream of IFNLR, we needed to exclude the possibility that IFNλ3 might inhibit IRF1 through an alternative pathway. To test this, we treated hepatocytes with IFNβ and IFNλ3 or IFNβ alone and did not observe changes in IRF1 protein expression (Figure 3I).
Current efforts to investigate the factors that govern IFNLR1 expression are limited due to the lack of specific antibodies to measure endogenous receptor density at the cell surface. Since increasing doses of IFNλ3 stimulation had no impact on the expression of IRF1 (Figure 3J), we hypothesized that the abundance of IFNLR1 could limit the activation of STAT1 and downstream induction of IRF1. To test this, we overexpressed IFNLR1 and measured downstream signaling activity. Overexpression of IFNLR1, significantly enhanced their sensitivity to IFNλ3 treatment without affecting the basal expression of STAT1 in cells (Figure 4A and B). We observed significant increase in STAT1 phosphorylation in IFNLR1 expressing cells in response to IFNλ3 at 0.5 to 4h post-treatment compared to EV expressing cells (Figure 4C, left). This increase in STAT1 activation correlated with a significant induction of IRF1 protein expression (4h post stimulation) following maximal STAT1 phosphorylation at 2h post stimulation and promoted the expression of CXCL10 and the ISG, MX1 (Figure 4D). We also confirmed the dependence of CXCL10 induction on IRF1 in IFNLR1 overexpressing cells, as CXCL10 expression was blunted in IRF1-deficient cells (Figure 4E).
Figure 4 – Expression levels of IFNLR1 dictate IRF1 inducibility by IFNλ, treatment.

(A) Immunoblot analysis of total STAT1 and ISG15 expression in IFNLR1 overexpressing Huh7 cells treated with IFNλ3 at the indicated doses for 24h. (B) Relative gene expression changes of ISG15 mRNA in IFNLR1 overexpressing cells treated with IFNλ3 at the indicated doses for 24h by qPCR. (C) Immunoblot analysis of STAT1 phosphorylated STAT1 (Y701), IRF1, and GAPDH in IFNLR1-overexpressing cells (IFNLR1; left) and control cells (Empty vector; right) stimulated with IFNβ or IFNλ3 for 0.5, 1.5, 2 and 4h. (D) Relative mean ± SD gene expression changes of CXCL10 and MX1 mRNA following 4h treatment with IFNβ or IFNλ3 in IFNLR1 overexpressing cells by qPCR. (E) Relative mean ± SD gene expression changes of CXCL10 and ISG15 mRNA in IFNLR1 overexpressing Wild-type (black) and IRF1-deficient (red) PH5CH8 cells treated with IFNλ3 for 4h by qPCR. (F) Average of the relative gene expression changes of IFNLR1 and CXCL10 mRNA in PH5CH8 cells stimulated with TNFα (10 ng/ml), poly I:C (2 μg/ml), or Sendai virus (SeV; 50 HAU/ml) for the indicated timepoints across 3 independent experiments mean ± SEM. Changes in mRNA expression are represented relative to mock-treated cells and normalized to HPRT. Unless otherwise indicated, cells were stimulated with 25 IU/ml of IFNβ or 100 ng/ml IFNλ3. Unless otherwise indicated, data is representative of 3 independent experiments. See also Figure S4.
Since IFNLR1 abundance dictated the strength of STAT1 signaling, we examined whether double-stranded RNA, viral infection, or inflammatory treatment induces IFNLR1 expression. Activation of the dsRNA sensors, TLR3 (pI:C) and RIG-I (SeV) or stimulation with TNFα did not induce IFNLR1 mRNA despite induction of CXCL10 (Figure 4F). Given the critical antiviral role of IFNΑ in curbing IAV dissemination, we tested whether IAV infection could alter the expression of IFNLR1. In vitro infection of A549 cells with IAV H1N1 (A/CA/04/2009) had no effect on IFNLR1 mRNA expression, despite inducing robust IFNB1 (Figure S4A). We observed a decrease in Ifnlr1 mRNA expression in whole lungs during in vivo infection with IAV H1N1 (A/PR/8/34) (Figure S4B). Based on these data, we conclude that IFNLR1 abundance is the limiting factor for STAT1-IRF1 axis and its downstream proinflammatory target genes. As viral sensing, innate immune, and proinflammatory responses fail to induce the expression of IFNLR1 in epithelial cells, we propose that low IFNLR1 expression could be an evolutionary adaptation to curtail unabated inflammation.
IRF1 regulates antiviral and inflammatory responses elicited by type I IFN.
We examined the contribution of IRF1 in the type I IFN-mediated transcriptional response by comparing WT and IRF1-deficient PH5CH8 cells. IRF1 was not necessary for the induction of ISG15 or IRF7 in either IFNβ or IFNλ3 treatments (Figure 5A); but it is required for the expression of a subset of ISGs, such as CXCL10 (Figure 5B), CIITA (Figure 5C), and TNFSF10 (Figure S5B). We asked whether differential induction of IRF1 is conserved across epithelial tissues, where distinct biological activities for type I and III IFNs have been previously reported (Figure S5A) (Davidson et al., 2016; Galani et al., 2017; Jilg et al., 2014; Marcello et al., 2006). In murine intestinal epithelial cells (IEC), IFNβ and IFNγ treatment resulted in the robust induction of IRF1 protein (Figure 5D) accompanied by the induction of Cxcl10 mRNA (Figure 5E, left), but only IFNβ or IFNλ3 induced significant expression of Isg15 (Figure 5E, right). Previous studies have suggested that cellular polarization enhances type III IFN responses in IECs (Bhushal et al., 2017). Thus, we studied primary small intestine organoid cultures from C57BL/6 mice where IRF1 induction was higher in IFNβ-treated organoids compared to a marginal increase in IRF1 expression induced by IFNλ3 (Figure 5F). The absence of IRF1 led to a significant dampening in the induction of Cxcl9 and Cxcl10 in IFNβ-treated organoids. No significant induction of these two genes was detectable in IFNλ3-treated organoids (Figure 5G). Similarly, the transcriptional induction of the IRF1-responsive gene, Ciita, was also impaired in Irf1−/− organoids (Figure S5C). While IFNΑ3-treated Irf1−/− organoids induced Isg15 gene expression, this response was enhanced in WT organoids (Figure S5C). Neither IRF1 expression (Figure S5D) nor proinflammatory treatment induced Ifnlr1 expression in primary IECs (Figure S5E). These results indicate that the preferential induction of IRF1-mediated inflammation by type I IFN is conserved across tissues.
Figure 5 – Central role of IRF1 in the response to IFN treatment.

(A) Immunoblot of IRF1, IRF7, ISG15, and Actin expression in WT and IRF1-deficient cells PH5CH8 treated with IFNβ or IFNλ3. (B-C) qPCR analysis of mean ± SD CXCL10 and CIITA mRNA expression in WT (circle) or IRF1-deficient cells (square) after IFNβ or IFNλ3 treatment for the indicated times. (B) Immunoblot analysis of IRF1 expression in immortalized murine small intestinal epithelial cells (IEC) treated with murine IFNβ (100 IU/ml), murine IFNλ3 (100 ng/ml) or murine IFNγ for 3 and 8h. (E) Relative gene expression changes of Cxcl10 and Isg15 mRNA in IEC treated with IFN as indicated above. (F) Immunoblot analysis of IRF1 expression in small intestine crypt-derived organoids stimulated with murine IFNβ (50 IU/ml) or murine IFNλ3 (200 ng/ml) for 4h. (G) Relative gene expression changes of Cxcl9 and Cxcl10 mRNA in IFN-treated organoids derived from WT or Irf1−/− mice, at 4h or 12h post stimulation. (H) Relative mean ± SD mRNA expression is normalized to Actin control. (H) Quantification of differentially expressed (DE) genes in PH5CH8 WT or IRF1-deficient PH5CH8 cells treated with IFNβ or IFNλ3 relative to genotype-matched untreated cells. DE cutoffs were set at a log2 fold change of |1| and a Benjamini-Hochberg adjusted p-value < 0.01. (I) Hierarchical clustering of 2402 DE genes following IFNβ treatment of WT or IRF1-deficient cells based on Euclidean distances. (J) Immunoblot analysis of USP18 protein IFNβ treatment of WT and IRF1-deficient PH5CH8 cells. (K) IFN-mediated protection against VSV-induced CPE (moi = 1) in WT and IRF1-deficient PH5CH8 cells pre-treated with IFNβ for 6, 12, or 24h prior to infection (top). Quantification of mean ± SEM dye uptake across 3 experiments 24h post infection (bottom). Black asterisks indicate significant differences between WT and IRF1-deficient cells and red asterisks indicate significant changes within the IRF1-deficient group. (L) Chemokines gene expression in our RNA-seq dataset. Genes highlighted in red are preferentially induced by IFNβ. Changes in mRNA expression are represented relative to mock-treated cells and normalized to HPRT. Unless otherwise indicated, cells were stimulated with 25 IU/ml of IFNβ or 100 ng/ml IFNλ3. Unless otherwise indicated, data is representative of 3 independent experiments. See also Figure S5 and Table S1.
To evaluate the differences in the transcriptional response to type I and III IFNs, we carried out next generation RNA sequencing. Genome-wide transcriptional profiling following IFN treatment of PH5CH8 cells corroborated the expression patterns observed with our targeted transcriptional screening of ISGs (Figure 1E). The abundance of differentially expressed (DE) genes in IFN-treated relative to mock-treated cells was lower in response to IFNλ3-stimulation relative to those observed with IFNβ at 12h post stimulation (Figure 5H). By 24h, significant changes in the transcriptional landscape were observed with either IFN treatment, with IFNλ3 promoting the expression of a higher number of ISGs. IRF1 loss led to a decrease in the overall number of differentially expressed (DE) transcripts by 24h. Specifically, IRF1 ablation led to the loss of CIITA along with genes associated with the activation of adaptive immune cells (Cluster IV, black) (Figure 5I and Table S1). There was an attenuation of genes involved in the regulation of coagulation pathways (Cluster II, yellow and V, blue) previously associated with enhanced immunopathogenic responses to viral infection. On the other hand, the loss of IRF1 sustained antiviral responses (Cluster III, skyblue and VI, cyan) (Figure S5F), likely through decreased expression of IFN-inducible IFNΑR inhibitors, such as USP18 (Figure 5J) (Speer et al., 2016; Zhang et al., 2015). This was consistent with the requirement for IRF1-mediated antiviral early upon IFNβ treatment, despite being dispensable at later timepoints (Figure 5K). Finally, IRF1 expression resulted in greater induction of chemokine genes, including CXCL10 and CXCL11, many of which were not induced in IFNλ3 treated cells (Figure 5L). These data suggest that IRF1 serves two crucial roles in the regulation of type I IFN responses: i) Promotes early induction of antiviral ISGs to curb viral spread and promote ligand-dependent IFNΑR downregulation, ii) Induces the expression of chemokines to recruit immune effector cells and promotes the activation of these cells.
Identifying molecular pathways uniquely induced by type III IFNs.
Our analysis revealed a unique set of DE genes after 24h of IFNλ3 treatment (Figure 6A). Through functional analysis of the distinct gene signatures induced by IFNs, we investigated the inferred activation state of kinases and observed an overlap in the enrichment of kinases involved in the regulation of IFN-mediated innate immune responses (JAK1, EIF2AK2, IKBKE) (Figure 6B). We observed an enrichment of the IFNλ-specific signature with MAPK kinase signaling (MAPK1 and MKNK1). These data corroborate previous work that demonstrated an IFNλ-specific dependency on MAPK signaling for the establishment of an antiviral state (Pervolaraki et al., 2017). Furthermore, we observed a significant enrichment of genes corresponding to the activation of the proto-oncogene tyrosine-protein kinase MER (MERTK), a member of the TAM receptor tyrosine kinase family highly associated with the resolution of inflammation. Using a similar approach, we investigated the subset of transcription factors activated or inhibited downstream of IFN-sensing and found an enrichment of classical antiviral regulators like IRFs and STAT1 that was greater in IFNβ at 12h and decreased by 24h (Figure 6C). Consistent with the delayed kinetics of IFNλ-mediated responses, the enrichment score for these factors increased by 24h post-treatment following IFNλ3 treatment. In IFNλ-treated cells, we uncovered the predicted activation of transcription factors involved in the regulation of hepatocyte differentiation (GATA4) (Enane et al., 2017), inflammation (Kang et al., 2015), and proliferation (NUPR1 and ID2) (Emma et al., 2016; Rodriguez et al., 2006). These results suggest that type I and III IFNs induce an overlapping antiviral response that is differentiated by the kinetic regulation of ISG expression. At later timepoints, unique kinase activity and transcription factors induced by IFNλ are likely involved in non-redundant cellular responses that could exert further antiviral activity and promote tissue reparative programs.
Figure 6 – Type I, but not type III IFNs promote immune cell recruitment into the lung.

(A) Intersection of IFNβ and IFNλ3 responsive genes in PH5CH8 WT cells. (B) Predicted activation state of kinases significantly associated transcriptional changes after IFNβ or IFNλ3 treatment using IPA. Color indicates predicted activation (purple)or predicted inhibition (green). (C) Predicted activation state of transcription factors found to be significantly associated with transcriptional changes after IFNβ or IFNλ3 treatment using IPA. Color indicates predicted activation (blue) or predicted inhibition (brown). (D) Bubble plot representation of significantly enriched biological functions in IFN-treated cells using IPA. Bubble color represents activation z-scores and bubble size represents the -log10 p-value of enrichment. Statistical significance was determined by an activation z-score > |2| and a -log10 p-value > 1.32, which correspond to a p-value of 0.05. Increases in -log10 p-value are indicative of increased statistical significance. (E) Immunoblot analysis of IRF1 in A549 cells treated with IFNβ (25 IU/ml) or IFNλ3 (100 ng/ml) over time. (F) Quantification of pulmonary expression of Cxcl10, Oas1a, and Isg15 mRNA following inoculation with murine IFNβ (2μg) or murine IFNΑ3 (4μg) relative to Actin control. (G) Quantification of immune cells in BAL of IFN-treated mice at 48h post-treatment. Unless otherwise indicated, data is representative of mean ± SEM of 3 independent experiments. See also Figure S6.
Differential recruitment of CXCR3+ cells by type I and III IFNs.
Analysis of the distinct immune biological functions associated with either type I or type III IFN treatment allowed us to capture the enrichment of genes involved in antiviral innate immune responses in both IFNβ and IFNλ-treated cells (Figure 6D). Under IFNλ treatment, we observed a diminished representation of genes associated with the quantity and activation of lymphocytes and leukocytes at both 12 and 24h. In contrast, these functions were enriched primarily after 12h of IFNλ treatment. Ablation of IRF1 led to a loss in the enrichment of genes associated with the activation of immune cells, but not those associated with antiviral functions. This is consistent with our previous data demonstrating that proximal and transient IFNβ-mediated induction of IRF1 leads to a burst in the induction of antiviral and pro-inflammatory genes.
Intranasal treatment of influenza A virus (IAV) infected mice with IFNα but not IFNλ induces the expression of inflammatory cytokines, immune cell infiltration, and activation (Davidson et al., 2016; Galani et al., 2017). Given that in human airway derived A549 cells, IRF1 induction was detected only after treatment with IFNβ and not IFNλ3 (Figure 6E), we tested whether murine intranasal treatment with IFNβ or IFNλ3, would be sufficient for the induction of ISGs and the recruitment of immune infiltrate in naive mice (Figure 6F). Consistent with our in vitro observations, we observed robust induction of Cxcl10 and other ISGs predominantly in IFNβ-treated lungs at 24h post-treatment that was resolved by 48h post-treatment. Treatment with IFNλ3 did not significantly induce Cxcl10 expression despite increases in Oas1a and Isg15. We also observed significant increase in the number of CD11c+ and CD11b+ macrophage/dendritic cells and Ly6G− and Ly6C+ monocytes in bronchoalveolar lavage (BAL) fluid of IFNβ-inoculated mice at 48h post treatment (Figure 6G and Figure S6A). In contrast, IFNλ3 inoculation did not induce any significant cellular infiltration compared to PBS treatment. Thus, type I IFNs are sufficient to promote the recruitment of inflammatory cellular mediators, whereas type III IFNs fail to promote such responses in vivo.
DISCUSSION
Here we identified unique transcriptional responses and biological consequences that differentiate type I and III IFNs. We found that the differences in the temporal ISG responses to type I or III IFN treatment were primarily regulated through transcription and not by translation. Gene expression in response to type I IFN stimulation (Type I IFN-ISG signature) was short-lived, whereas type III IFN-ISG signature was sustained over a longer time period following treatment. These differences were reflected by ISGF3 occupancy at ISG promoter regions observed at early and late time points post stimulation with IFNs. The temporal responses of type I IFN-ISG expression are partially explained by selective multifactorial ligand-dependent negative regulation of IFNΑR (Liu et al., 2008), but not the type III IFN receptor (Blumer et al., 2017). The temporal pattern of transcription supports a model in which the two IFN families have co-evolved to sustain an antiviral state against viral infections at barrier sites. The biological consequences of temporal regulation of ISGs were evident in the antiviral activity elicited by IFNs. IFNβ treatment led to early protection against virus-induced CPE while the protective effect of IFNλ3, while delayed, was sustained over longer periods of time. When we treated infected cells with both IFNs, we observed a sustained antiviral activity that suggested a non-redundant requirement of the two antiviral IFN systems.
One of the major differences observed between type I and III IFNs, was the differential induction of the pro-inflammatory transcription factor, IRF1. When we screened for the induction of signaling adaptors and transcription factors activated by type I and III IFNs, we found that type I, but not by type III IFNs induced IRF1. Differential IRF1 expression resulted in a distinct transcriptional signature, that included induction of IRF1-dependent CXCR3 ligands. This suggested a role for IRF1 in the diversification of ISGs that are inducible by type I and III IFNs. We then investigated the mechanisms that led to IRF1 responsiveness, we found that IRF1 induction by type I IFN was similar to IFNγ, where IRF1 transcription was induced by STAT1-homodimers (Leung et al., 1995). We showed that the phosphorylation of STAT1 was attenuated in IFNλ-treated cells, relative to that of cells treated with IFNβ. Thus, IFNλ-mediated STAT1 activation was insufficient to induce STAT1 homodimer formation required for IRF1 induction and downstream gene expression.
Since endogenous expression of IFNLR1 was insufficient to induce STAT1-IRF1 activation, we examined the effect of increasing IFNLR1 expression on IRF1 responsiveness. We observed robust STAT1 phosphorylation, IRF1 expression with concomitant CXCL10 gene induction, and the enhanced magnitude IFNλ responsive ISGs, such as MX1. These data suggested that the threshold for activation required for STAT1-dependent IRF1 expression was determined IFNLR1 abundance. We did not observe significant changes in IFNLR1 expression following pathogen sensing. As robust antibodies become available, follow up studies will determine whether potential regulatory mechanisms that control IFNRL1 surface expression impact the ability to promote STAT1-IRF1-dependent inflammation. These data suggested an evolutionary adaptation to control and maintain low levels of IFNLR1 expression to limit inflammatory responses, while sustaining robust antiviral activity.
IRF1 was dispensable for the innate antiviral activities of IFNs which are primarily carried out by the STAT1-STAT2-IRF9 transcriptional complex. Type I IFN-dependent IRF1 served as an amplifier of ISG and chemokine gene expression. Transcriptome analysis in IRF1-deficient cells supported our hypothesis that the distinct early induction of IRF1 by type I IFNs played an important role in enhancing antiviral responses and eliciting inflammation. We have shown that the IFNβ-IRF1 axis, and not IFNλ, induced various inflammatory chemokine genes, including the CXCR3 family ligands, and promoted a transcriptional profile consistent with the enrichment of genes that drive the activation and recruitment of immune effector cells. These ligands play an important role in the recruitment of CXCR3+ cells into sites of inflammation (Groom and Luster, 2011) and in coordinating adaptive immune responses. The temporal regulation of IFN-induced gene expression patterns was similar in IFNLR1 expressing non-hematopoietic compartments such as the lung and intestinal mucosal environments. Indeed, we observed the recruitment of CD11c+/CD11b+ and Ly6G−/Ly6C+ monocytes, macrophages, and dendritic cells into the lungs of IFNβ-inoculated mice. More importantly, these cells were not recruited following IFNλ3 treatment. These observations were consistent with previous studies that have reported that IFNλ, is the predominant IFN produced by the lung epithelium during low dose influenza A virus (IAV) infection (Galani et al., 2017). In these conditions, IFNλ, confers significant protection against IAV in the absence of the induction of chemokine and proinflammatory cytokine genes, and the production of type I IFN warrants higher infectious doses and enhances inflammation and tissue damage.
Although, type III IFNs are implicated in aiding tissue repair and regeneration, investigations into the signaling cascades that program such functions are not clear. Studies suggest a unique regulation of MAPK signaling pathway induced specifically by IFNλ,. Along with the MAPK pathway, our transcriptome data uncovered a gene signature that was consistent with the activation of the TAM receptor, MERTK. TAM receptors are expressed in macrophages, dendritic cells, and endothelial cells, and downregulate the expression of proinflammatory cytokine production following TLR activation and IFN treatment (Rothlin et al., 2007). We also identified novel transcription factors, induced by type III IFN, involved in cellular proliferation and differentiation. Given the involvement of these factors in the maintenance of cell-cell adhesion (Soini et al., 2018) and the regulation of inflammation (Kang et al., 2015), future investigation into how they regulate barrier integrity and the outcome of both acute and chronic infections is warranted.
Our study provides mechanistic insight into the distinct roles played by two IFN gene families that are evolutionarily conserved across vertebrates. We propose that STAT1-IRF1 axis is at the crossroads of these functions, serving as a branching point for both the antiviral and inflammatory IFN-mediated responses. Strong antiviral response to mucosal viruses, requires type I and III IFN signaling through shared signal transduction pathways. However, protracted inflammatory responses induced by IFNs decrease host fitness and contribute to the disruption of barrier integrity (Chiriac et al., 2017; Davidson et al., 2016; Forero et al., 2015; Lazear et al., 2015). Type III IFNs predominantly activate innate antiviral effectors in the local tissue without eliciting inflammation. The extent of IFNLR1 expression tunes the downstream IRF1 response which results in differences in the inflammatory responses observed after in vivo stimulation. We propose that receptor compartmentalization and regulation of cell surface abundance contributes to the differences in transcription factor activation that limits inflammation and confers a spatiotemporal division of labor. Thus, in sites of viral exposure the type I and III IFNs work in concert to control viral spread and limit tissue damage.
STAR METHODS
Cell culture conditions and Reagents
Immortalized human hepatocytes PH5CH8 (Kato et al., 1998), Huh7, hepatocyte-derived KO cells, 2fTGH cells (Pellegrini et al., 1989), and 2fTGH-derived U3A (STATl-deficient) (McKendry et al., 1991) and U6A (STAT2-deficient) cells (Leung et al., 1995) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 2mM Glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin and maintained at 37°C in 5% CO2. Huh7-derived IFNΑR1 and IFNLR1 deficient cells have been previously described (Hong et al., 2016; Jarret et al., 2016). PH5CH8-derived IRF1, STAT1 and STAT2deficient cells were derived using CRISPR editing technologies as previously described (Cuevas et al., 2016). Immortalized mouse small intestine epithelial cells (Schwerk et al., 2013) were maintained in complete muINTEPI medium (InSCREENeX). Human recombinant IFNβ (PBL Interferon Source), IFNΑ1, 2, and 3 (R&D Systems) and IFNα2, IFNγ (Shenandoah Biotechnology) were used at the indicated concentrations.
Plasmids
The pcDNA3.1/HisA/huIRF1 plasmid was used to overexpress IRF1 and has been previously described (Forero et al., 2014). The reporter plasmids pGL4/Full length (FL)-CXCL10 and ΔISRE CXCL10 firefly luciferase were a kind gift from Dr. David Proud (University of Calgary) have been previously reported (Clarke et al., 2010; Spurrell et al., 2005). The IRF1 and STAT1-targeting CRISPR/Cas9 plasmids were a kind gift from Veit Hornung (Ludwig-Maximilians-Universität München). For CRISPR/Cas9 targeting of STAT2, we generated the plasmid pRRL-MND-STAT2–2A-Puro by in-fusion cloning of the STAT2 gRNA sequence: 5’-AAAGGACGAAACACCGTGTGGACATTCGACAGTACTGTTTTAGAGCTAGAAATAGC AAG-3’ into pRRL-MND-STAT2–2A-Puro. The pGL2-IRF1-luc plasmid has been previously described (Deb et al., 2001). The eGFP expression plasmid, (pmaxGFP, Lonza), pGL3-Basic (Promega), human IFNλ Receptor Alpha expression plasmid, pUNO-hIL28RA (Invivogen) were obtained from commercial sources.
RNA isolation, reverse transcription, and quantification of gene expression.
Total RNA was extracted using the NucleoSpin RNA extraction kit (Macherey-Nagel). cDNA synthesis was performed using the QuantiTect RT kit (Qiagen) according to the manufacturer’s guidelines. qPCR was carried out using the ViiA7 qPCR system with TaqMan reagents (Life Technologies). Gene expression levels were normalized to Actin, HPRT or GAPDH as indicated. Probe-based assays utilized in this study were acquired from IDT or Life Technologies.
Metabolic labeling of nascent transcripts
Cells were treated with 500 μM of 4-thiouridine (4sU; Cayman Chemical) for 10 minutes before harvest. Total RNA was extracted using the NucleoSpin RNA extraction kit (Macherey-Nagel). Newly made RNAs (4sU-labeled) were fractionated following the protocol previously described (Garibaldi et al., 2017). Briefly, 100 μg of total RNA was biotinylated with biotin-HPDP (1 mg/mL in DMF; Thermo Fisher Scientific). RNA was extracted with chloroform:phenol:isoamyl alcohol two times (Thermo Fisher Scientific) and precipitated with ethanol. The biotinylated RNA was separated with Streptavidin M280 Dynabeads (Thermo Fisher Scientific). The beads were washed four times. The bound RNA was eluted with 100 mM DTT (Sigma-Aldrich) and recovered using the NucleoSpin RNA Clean-up kit (Macherey-Nagel).
Western blot analysis
Whole cell lysates were prepared from cells using RIPA buffer (10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented with Halt™ protease and phosphatase inhibitor cocktails (Pierce). Protein quantification and normalization was done using the BCA Protein Assay Kit (Pierce). 10–30 μg total protein were resolved by SDS-PAGE and transferred to PVDF membranes (Thermo Scientific). Membranes were probed overnight with antibodies diluted in 5% BSA in PBS-T (Phosphate-buffered saline/Tween 20), and species specific HRP conjugated antibodies. Chemiluminescent image acquisition was performed using a ChemiDoc XRS+ (BioRad).
Polysome Analysis
Polysome analysis was performed as described previously (Morita et al., 2013). Briefly, PH5CH8 cells were treated with 25IU/ml IFNβ or 100ng/μl IFNλ3 for the indicated times. Cells were treated with 100μg/ml cycloheximide (CHX) for 5 minutes to stall active translating ribosomes and then washed twice with ice-cold PBS containing 100μg/ml CHX prior to harvest. The cell pellet was resuspended in 425μl hypotonic buffer (5mM Tris-HCl; pH 7.5, 2.5mM MgCl2, 1.5mM KCl and 1X protease inhibitor cocktail) and transferred to pre-chilled microcentrifuge tubes. Each tube received 5μl of CHX (10mg/ml), 1μl of 1M DTT, 100U of RNasin Plus (Promega), 25 μl of 10% Triton X-100, and 25 μl of 10% Sodium Deoxycholate and samples were vortexed for 5 sec. Lysates were immediately centrifuged at 17,000 × g for 5 minutes at 4°C. The supernatants were transferred into fresh pre-chilled tubes and the OD260nm for each sample was measured using a Biotek Take3™ Micro-volume plate. Approximately OD260nm = 8–10 was loaded onto 10–50% sucrose gradient for each sample and 10% of the input was resuspended with Trizol (Thermo Fisher). Gradients were centrifuged at 35,000 r.p.m. at 4°C for 2 hours and then sampled using an Auto Densi-Flow Gradient Fractionator (Labconco) connected to an UA-6 absorbance monitor (OD245nm) and the Foxy R1 fraction collector. Approximately 14 1ml fractions were collected, in-vitro transcribed firefly luciferase RNA control was added to each fraction and an equal volume of Trizol was added to each fraction prior to total RNA extraction using the Direct-Zol 96-well kit (Zymo). Fractions corresponding to free ribonucleoprotein complexes (RNP), monosomes/light polysomes (2–3 ribosomes) and heavy polysomes (>4 ribosomes) were pooled after RNA extraction and cDNA synthesis was carried out as described above. Taqman gene expression card arrays were used to assess the expression of 41 ISGs across input, light polysomes and heavy polysomes (pooled samples and validated using gene specific Taqman gene expression assays.
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared from cell lines using the CellLytic NuCLEAR extraction kit (Sigma-Aldrich). Protein concentration was measured with a Bio-Rad protein assay, and samples were stored at −70°C until use. Double-stranded DNA oligonucleotide probes were synthesized (IDT) containing either the STAT-binding region of the IRF1 promoter (IRF-Prom: 5-GCCTGATTTCCCCGAAATGACGGCAC), or the IRF/STAT-binding region of the ISG15 promoter (ISG15-STAT: 5-GGCTTCAGTTTCGGTTTCCCTTTCCCGAGGCATGCC), Probes were labeled with α-[32P]deoxycytidine triphosphate (3000 Ci/mmol; PerkinElmer, Waltham, MA, USA) by fill-in using the Klenow fragment of DNA polymerase I (Invitrogen). [32P]-labeled double-stranded oligonucleotides were purified using mini Quick Spin Oligo Columns (Roche Diagnostics, Indianapolis, IN, USA). DNA-protein binding reactions were performed in a 10-μl mixture containing 5 μg nuclear protein and 1 μg poly[dI-dC] (Sigma-Aldrich) in 4% glycerol, 1 mM MgCl2, 0.5 mM ethylenediaminetetraacetic acid, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5). Nuclear extracts were incubated with 1 μl 32P-labeled oligonucleotide probe (10,000 cpm) either alone, in the presence of 2 μg of specific antibodies (Santa Cruz Biotechnology, Dallas, TX, USA), or with unlabeled consensus TF-binding oligos, incubated at room temperature for 20 min, and then loaded on a 5% polyacrylamide gel (37:5:1). Electrophoresis was performed in 0.5x TBE for 2 h at 130 V, and the gel was visualized by autoradiography.
Crystal Violet Uptake Assays
PH5CH8 cells control and PH5CH8-derived IRF1 deficient cells were plated in 24-well plates at a density of 2×105 cells/well. Cells were pre-treated with IFNβ (25 IU/ml) or IFNλ3 (100 ng/ml) in complete media for 6, 12, 24, 48, or 72h prior to infection. Cells were infected with VSV-GFP (Fredericksen and Gale, 2006) at a multiplicity of infection (moi) of 0.01 or moi 1 as indicated. Infections were carried out in serum-free media. Viral inoculum was removed following 1.5hrs of absorption at 37°C, cells were washed with PBS, and incubated under culture conditions described above. Following 24h of infection, the culture medium was removed, cells were fixed with 4% PFA in PBS, and stained with Crystal violet stain (3% w/v) in 50% ethanol. Plates were imaged using the ChemiDoc XRS+ (BioRad) imaging system and images were quantified using ImageJ.
Nuclear translocation assays
PH5CH8 cells (1×106 per treatment) were stimulated with 25 IU/ml IFNβ or 100 ng/ml of IFNλ3 for 30 mins. Cells were harvested by trypsinization, washed with PBS, and fixed in 1ml of 2% PFA/PBS for 30 minutes at room temperature. Cells were washed 2 times with PBS and permeabilized in 1ml of ice-cold methanol followed by incubation a 15–20min incubation at 4°C. Cells were washed twice with 1ml of FACS buffer and stained at 4°C for 30min with pSTAT1(Y701) antibody at a 1:400 followed by Alexa488 mouse anti-rabbit at a 1:200 dilution in FACS buffer (PBS+2% FBS+0.02% sodium azide). Cells were stained with 0.6nM DAPI solution for 15min at room temperature (RT) and washed twice with FACS buffer prior to image acquisition using the ImageStream imaging flow cytometer (Amnis). Single color controls were used to evaluate events corresponding to pSTAT1 nuclear translocation and data was analyzed using the IDEAS statistical analysis software (Amnis).
Chemokine secretion assays.
Chemokine secretion was measured using the LEGENDplex Human Proinflammatory Chemokine Panel (13-plex) kit following manufacturer’s guidelines. Samples were analyzed using a BD FACSCanto cell analyzer.
Luciferase reporter assay
Luciferase reporter assays were carried out in Huh7 cells. Briefly, cells were seeded in 6-well plates and transfected with 100 ng of pmax-eGFP and 600 ng of IRF1-luc, (FL)-CXCL10, or mut ISRE CXCL10 firefly luciferase reporter plasmids using the TransIT-X2 delivery system (Mirus) at a 3:1 TransIT-X2:DNA ratio. After 48h post-transfection, cells were stimulated with IFNβ, IFNλ3, or IFNγ at the indicated concentrations for 6h, for IRF1-luciferase reporter assays. Stimulations were carried out for 24h in CXCL10-luciferase reporter assays, prior to measurement of firefly luciferase activity. Luciferase activity was normalized to GFP expression.
RNA sequencing, data processing, and analysis
PH5CH8 control and IRF1 deficient cells were stimulated with 25 IU/ml IFNβ or 100 ng/ml of IFNλ3 for 12 or 24h (n=3 per treatment). Genotype-matched untreated cells were used as mock controls (n=3 per genotype). Total RNA was extracted as previously described. RNA integrity was assessed using the RNA 6000 Nano Kit and the 2100 Bioanalyzer (Agilent). Fluorometric quantification of total RNA was performed using the Qubit™ RNA BR assay kit (Invitrogen). cDNA libraries were synthesized using the TruSeq Stranded mRNA Library Prep Kit and sequenced using the Ilumina NextSeq 500 sequencer. Library preparation, QC, and sequencing was carried out by Seattle Genomics (www.seattlegenomics.com).
Both the genome sequence (fasta) and gene transfer files (gtf) for human were obtained using iGenomes (https://support.illumina.com/sequencing/sequencingsoftware/igenome.html). Raw RNAseq data (Fastq files) were demultiplexed and checked for quality (FastQC, version 0.11.3). Ribosomal RNA was digitally removed using Bowtie2 (version 2.3.4). Sufficient host reads (greater than twenty million per sample) were mapped to the human genome (GRCh37) using STAR (version 2.5.3a) and converted into gene counts with HTSeq (version 0.6.1). Prior to statistical analysis, gene counts were filtered with a cutoff of a mean of ten or greater across all samples using R statistical programming language (version 3.4.3) and ‘edgeR’ (version 3.20.9). Gene counts were normalized using voom and statistical analysis for differential expression was carried out in using ‘limma’ (version 3.34.8) in R. Functional analysis of transcriptional responses was performed using Ingenuity Pathway Analysis (IPA). Inferences in the activation state of upstream regulators was performed using IPA (Kramer et al., 2014).
In vivo responses to interferon treatment and influenza A virus infections.
Male C57BL/6 mice (6–8 weeks old) were anesthetized with ketamine/kylazine and administered 2 μg recombinant murine IFNβ (R&D) or 4 μg recombinant murine IFNλ3 (R&D) in 50 ml sterile PBS intranasally or given 50 ml sterile PBS as a control. Following 24h or 48h of treatment, bronchiole alveolar lavage (BAL) fluid was harvested by instilling PBS + 2mM EDTA intratracheally into the lung and collecting the lavage fluid. BAL cells were blocked 10 min, at RT in FACS buffer (PBS+2% HI FBS+0.02% sodium azide) with 2% rat serum for 10min, and then stained with antibodies for 30min on ice. Cells were subsequently resuspended in PBS, Absolute Count Beads (ThermoFisher) were added to the samples, and they were run on the LSRII (BD). Flow data was analyzed using FlowJo (TreeStar, Inc.), and total cell counts were graphed in Prism GraphPad. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparison tests. Flow cytometry data was generated from two independent experiments (n=4–6 mice/group). For infections, female C57BL/6 mice were anesthetized with ketamine/xylazine and administered 40 PFU A/PR/8/34 intranasally in sterile PBS. 24 or 48 h later, bronchiole alveolar lavage was performed, and lungs were subsequently harvested.
Whole lungs were harvested, incubated in RNALater at 4°C, placed into Precellys homogenization tubes containing TRI Reagent (ThermoFisher), and homogenized using a Precellys 24 tissue homogenizer (Bertin Industries). RNA was isolated from tissues using the RiboPure RNA Purification Kit (ThermoFisher) according the manufacturers protocol. cDNA was generated using an iScript cDNA Synthesis Kit (BioRad), and quantification of Isg15, Oas1a, Cxcl10, Ifnlr1 mRNA expression relative to Gapdh was determined by qRT-PCR. Quantification of viral RNA was performed by amplification of IAV NP relative to Chmp2a.
Evaluation of IFN responses in primary murine small intestine organoid cultures
Small intestinal crypt-derived organoids were grown as previously described (Nadjsombati et al., 2018). Crypts were harvested from 6–8 weeks old female and male C57BL/6 mice and Irf1−/−mice. Organoids were incubated for 4h with 50IU of recombinant murine IFNβ (R&D) or 200ng/ml IFNλ3 (R&D) in complete media. Organoids were harvested using Cell Recovery solution (Gibco), washed, and then lysed in RIPA buffer as described earlier for western blot analysis or RA1 buffer for quantification of gene expression as described above.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad software Inc. La Jolla, CA). Statistical significance of stimulation and time-dependent gene expression changes were analyzed using two-way ANOVA. Statistical significance in gene expression across stimulations were analyzed using one-way ANOVA. Statistical significance in median gene expression was determined using Mann-Whitney test. Across all experiments, p-values of < 0.05 were considered significant and are indicated by asterisks (*). * p-values of < 0.05; ** p-values of < 0.01; **** p-values of < 0.001; **** p-values of < 0.0001.
Data dissemination
The data generated in this study are available via the following accession identifiers on the NCBI-GEO database (GSE115198).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| IRF1 (D5E4) | Cell signaling | RRID: AB_10949108 |
| IRF1 (C-20 x) | Santa Cruz | RRID: AB_631838 |
| IRF3 (D83B9) | Cell signaling | RRID: AB_1904036 |
| IRF5 (10T1) | Abcam | RRID: AB_775785 |
| IRF7 | Cell signaling | RRID: AB_2127551 |
| IRF8 (C-19) | Santa Cruz | RRID: AB_649510 |
| IRF9 (C-20) | Santa Cruz | RRID: AB_2127709 |
| IRF9 (D2T8M) | Cell signaling | RRID: AB_2799885 |
| STAT1 (D1K9Y) | Cell signaling | RRID: AB_2737027 |
| pSTAT1 Y701 (58D6) | Cell signaling | RRID: AB_561284 |
| STAT2 (D9J7L) | Cell signaling | RRID: AB_2799824 |
| STAT2 (B-3) | Santa Cruz | RRID: AB_2810271 |
| pSTAT2 Y689 | Millipore | RRID: AB_2198439 |
| STAT3 (124H6) | Cell signaling | RRID: AB_331757 |
| STAT4 | Santa Cruz | RRID: AB_2810272 |
| STAT5 | Santa Cruz | RRID: AB_1129711 |
| USP18 (D4E7) | Cell signaling | RRID: AB_10614342 |
| ISG15 | Cell signaling | RRID: AB_2126201 |
| β-Actin-HRP (13E5) | Cell signaling | RRID: AB_1903890 |
| GAPDH (14C10) | Cell signaling | RRID: AB_561053 |
| mCXCR3-PE-Cy7 | Biolegend | RRID: AB_2086740 |
| mCD49b-PE | Biolegend | RRID: AB_313414 |
| mCD8a-BV421 | Biolegend | RRID: AB_10897101 |
| mCD4-APC-Cy7 | eBiosciences | RRID: AB_2534398 |
| mCD3-APC | eBiosciences | RRID: AB_11153519 |
| mCD19-BV785 | Biolegend | RRID: AB_11218994 |
| mCD45.2-PerCP-Cy5.5 | Biolegend | RRID: AB_893352 |
| mSiglecF-APC-Cy7 | BD | RRID: AB_2732831 |
| mCD11c-APC | eBiosciences | RRID: AB_469346 |
| mCD11b-PE | Biolegend | RRID: AB_312790 |
| mMHCII-A700 | eBiosciences | RRID: AB_494009 |
| mLy6C-BV421 | Biolegend | RRID: AB_2562177 |
| mLy6G-BV785 | Biolegend | RRID: AB_2566317 |
| Donkey Anti-Mouse IgG (HRP-Conjugated) | Jackson ImmunoResearch | RRID: AB_2340770 |
| Donkey Anti-Rabbit IgG (HRP-Conjugated) | Jackson ImmunoResearch | RRID: AB_10015282 |
| Bacterial and Virus Strains | ||
| VSV-GFP | Gale Lab | Fredericksen and Gale, 2006 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human IFNβ | PBL Assay Science | 111415–1 |
| Human IFNα2 | Shenandoah Biotechnology | 100–54-20ug |
| Human IFNλ1 | R&D Systems | 1598-IL-025 |
| Human IFNλ2 | R&D Systems | 8417-IL-025 |
| Human IFNλ3 | R&D Systems | 5259-IL-025 |
| Human IFNγ | Shenandoah Biotechnology | 100–77-20ug |
| Recombinant Mouse IFNβ | R&D Systems | 8234-MB/CF |
| Recombinant Mouse IFNλ3 | R&D Systems | 1789-ML/CF |
| Recombinant Mouse IFNγ | R&D Systems | 315–05 |
| muINTEPI medium | InSCREENeX | INS-ME-1005 |
| 4-Thiouridine (4sU) | Cayman Chemical Company | 16373 |
| Streptavidin M280 Dynabeads | Thermo Fisher Scientific | 11205D |
| Rnasin Plus | Fisher Scientific | PRN2615 |
| α-[32P] deoxycytidine triphosphate | Perkin Elmer | BLU513H |
| Klenow Fragment | Invitrogen | 18012021 |
| poly[dI-dC] | Sigma-Aldrich | 10108812001 |
| Mirus Bio TransIT-X2 | Fisher Scientific | MIR6000 |
| Mirus Bio TransIT-TKO | Fisher Scientific | MIR2150 |
| Cell Recovery Solution | Corning | 354253 |
| Critical Commercial Assays | ||
| QuantiTect RT kit | Qiagen | 205314 |
| TaqMan™ Universal Master Mix II, no UNG | Thermo Fisher Scientific | 4440048 |
| NucleoSpin® RNA Clean-up | Clontech | 740948.25 |
| Direct-Zol 96-well kit | Zymo Research | R2056 |
| CellLytic NuCLEAR | Sigma-Aldrich | NXTRACT-1KT |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | 20020594 |
| Qubit™ RNA BR Assay Kit | Thermo Fisher Scientific | Q10210 |
| NucleoSpin® RNA II | Clontech | 740955.25 |
| LEGENDplex Human Proinflammatory Chemokine Panel (13-plex) | Biolegend | 740007 |
| HiScribe™ T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S |
| RNA 6000 Nano Reagents | Agilent | 5067–1512 |
| Deposited Data | ||
| RNA sequencing Data | GEO | GSE115198 |
| Experimental Models: Cell Lines | ||
| PH5CH8 | RRID: CVCL_VL00 | |
| HuH7 | RRID: CVCL_0336 | |
| A549 | ATCC CCL-158 | RRID: CVCL_0023 |
| 2fTGH | Kind gift from George Stark | RRID: CVCL_0115 |
| U3A | Kind gift from George Stark | RRID: CVCL_9469 |
| U6A | Kind gift from George Stark | RRID: CVCL_D316 |
| muINTEPI | InSCREENeX | INS-CI-1007 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | RRID: IMSR_JAX:000664 |
| C57BL/6 Irf1 -/- | Michael S Diamond, Washington University, St. Louis, MO | |
| Oligonucleotides | ||
| EMSA IRF1-Prom 5'-GCCTGATTTCCCCGAAATGACGGCAC-3' |
Integrated DNA Technologies | |
| EMSA ISG15-STAT 5'-GGCTTCAGTTTCGGTTTCCCTTTCCCGAGGCATGCC-3' |
Integrated DNA Technologies | |
| STAT1 siRNA | Dharmacon | M-003543–01 |
| STAT2 siRNA | Thermo Fisher Scientific | s13530 |
| IRF9 siRNA | Dharmacon | M-020858–02 |
| IRF1 siRNA | Thermo Fisher Scientific | 115266 |
| Negative Control siRNA | Thermo Fisher Scientific | AM4613 |
| STAT2 gRNA sense 5'-AAAGGACGAAACACCGTGTGGACATTCGACAGTACTGTTTTAGAGCTAGAAATAGCAAG-3' |
Integrated DNA Technologies | This paper |
| STAT2 gRNA antisense 5'-CTTGCTATTTCTAGCTCTAAAACAGTACTGTCGAATGTCCACACGGTGTTTCGTCCTTT-3' |
Integrated DNA Technologies | This paper |
| ISG15 | Life Technologies | Hs01921425_s1 |
| MX1 | Life Technologies | Hs00895608_m1 |
| OAS1 | Life Technologies | Hs00973637_m1 |
| CXCL9 | Life Technologies | Hs00171065_m1 |
| CXCL10 | Life Technologies | Hs00171042_m1 |
| CXCL11 | Life Technologies | Hs00171138_m1 |
| IFNLR1 | Life Technologies | Hs00417120_m1 |
| IRF1 | Life Technologies | Hs00971959_m1 |
| STAT1 | Life Technologies | Hs01013996_m1 |
| STAT2 | Life Technologies | Hs01013123_m1 |
| IRF9 | Life Technologies | Hs00196051_m1 |
| CIITA | Life Technologies | Hs00172106_m1 |
| TNFSF10 | Life Technologies | Hs00921974_m1 |
| IL10R2 | Life Technologies | Hs001175123_m1 |
| HPRT1 | Integrated DNA Technologies | Hs.PT.58v.45621572 |
| Cxcl9 | Life Technologies | Mm00434946_m1 |
| Cxcl10 | Life Technologies | Hs00171042_m1 |
| Oas1a | Life Technologies | Mm00836412_m1 |
| Isg15 | Life Technologies | Mm01705338_s1 |
| Ciita | Life Technologies | Mm00482914_m1 |
| Ifnlr1 | Life Technologies | Mm00558035_m1 |
| Actin | Integrated DNA Technologies | Mm.PT.58.33257376.gs |
| Gapdh | Life Technologies | Mm99999915_g1 |
| mChmp2a Forward 5'-AGACGCCAGAGGAACTACTTC-3' |
Integrated DNA Technologies | |
| mChmp2a Reverse 5'-ACCAGGTCTTTTGCCATGATTC-3' |
Integrated DNA Technologies | |
| IAV NP Forward 5'-CGTTCTCCATCAGTCTCCATC-3' |
Integrated DNA Technologies | |
| IAV NP Reverse 5'-GAGTGACATCAAAATCATGGCG-3' |
Integrated DNA Technologies | |
| Recombinant DNA | ||
| pGL2-IRF1-luc | Deb et al., 2001 | |
| pGL4/Full length (FL)-CXCL10-luc | Clarke et al., 2010; Spurrell et al., 2005 | |
| pGL4/ISRECXCL10-luc | Clarke et al., 2010; Spurrell et al., 2005 | |
| pcDNA3.1/HisA/huIRF1 | Forero et al., 2014 | |
| IRF7–2D Flag | Gale Lab, University of Washington | |
| pcDNA-6xHis-p48 | Gale Lab, University of Washington | |
| Retro_STAT1_gRNA_Double BB_CMV | Gift from Veit Hornung | |
| Retro_IRF1_gRNA_Double BB_CMV | Gift from Veit Hornung | |
| pRRL-MND-STAT2–2A-Puro | This paper | |
| psPAX-2 | Gift from Didier Trono | RRID: Addgene_12259 |
| pCMV-VSV-G | Gift from Bob Weinberg | RRID: Addgene _8454 |
| pGL3-Basic | Promega | E1751 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | RRID:SCR_003070 |
| GraphPad Prism 8 | GraphPad Software, Inc | RRID: SCR_002798 |
| R Studio 1.1.442 | Rstudio, Inc | RRID: SCR_000432 |
| FlowJo Software | Threestar, Inc | RRID: SCR_008520 |
| Ingenuity Pathway Analysis | Qiagen | RRID: SCR_008653 |
| iGenomes | Illumina | |
| Other | ||
Highlights.
Temporal ISG induction by type I and III IFNs provide collaborative antiviral response.
Type I IFNs but not type III IFNs promote inflammation at the site of infection.
Type I IFNs induce a distinct IRF1-dependent inflammatory immune response.
IFNLR1 expression levels determine threshold of IRF1 induction.
Acknowledgements
This work was supported in part by R01AI108765 (RS), T32HL007312 (AF), T32AI007509 (MRH), R01 AI104002 (MG), U19 AI100625 (MG), R01 AI118916 (MG), R01 AI127463 (MG), P51 0D010425 (MG), 17P0ST33660907 (EAH), R01 AI118896 (SNS), and Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported in part by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, et al. (2008). An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol 180, 2474–2485. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, and Virgin HW (2017). Expression of Ifnlr1 on Intestinal Epithelial Cells Is Critical to the Antiviral Effects of Interferon Lambda against Norovirus and Reovirus. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushal S, Wolfsmuller M, Selvakumar TA, Kemper L, Wirth D, Hornef MW, Hauser H, and Koster M. (2017). Cell Polarization and Epigenetic Status Shape the Heterogeneous Response to Type III Interferons in Intestinal Epithelial Cells. Front Immunol 8, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau A, and Matthys P. (2009). Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev 20, 97–113. [DOI] [PubMed] [Google Scholar]
- Blumer T, Coto-Llerena M, Duong FHT, and Heim MH (2017). SOCS1 is an inducible negative regulator of interferon lambda (IFN-lambda)-induced gene expression in vivo. J Biol Chem 292, 17928–17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriac MT, Buchen B, Wandersee A, Hundorfean G, Gunther C, Bourjau Y, Doyle SE, Frey B, Ekici AB, Buttner C, et al. (2017). Activation of Epithelial Signal Transducer and Activator of Transcription 1 by Interleukin 28 Controls Mucosal Healing in Mice With Colitis and Is Increased in Mucosa of Patients With Inflammatory Bowel Disease. Gastroenterology 153, 123–138 e128. [DOI] [PubMed] [Google Scholar]
- Clarke DL, Clifford RL, Jindarat S, Proud D, Pang L, Belvisi M, and Knox AJ (2010). TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem 285, 29101–29110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas RA, Ghosh A, Wallerath C, Hornung V, Coyne CB, and Sarkar SN (2016). MOV10 Provides Antiviral Activity against RNA Viruses by Enhancing RIG-I-MAVS-Independent IFN Induction. J Immunol 196, 3877–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, McCabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, Hartmann R, and Wack A. (2016). IFNlambda is a potent anti-influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO Mol Med 8, 1099–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb A, Haque SJ, Mogensen T, Silverman RH, and Williams BR (2001). RNA-dependent protein kinase PKR is required for activation of NF-kappa B by IFN-gamma in a STAT1-independent pathway. J Immunol 166, 6170–6180. [DOI] [PubMed] [Google Scholar]
- Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, and Renauld JC (2004). Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem 279, 32269–32274. [DOI] [PubMed] [Google Scholar]
- Emma MR, Iovanna JL, Bachvarov D, Puleio R, Loria GR, Augello G, Candido S, Libra M, Gulino A, Cancila V, et al. (2016). NUPR1, a new target in liver cancer: implication in controlling cell growth, migration, invasion and sorafenib resistance. Cell Death Dis 7, e2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enane FO, Shuen WH, Gu X, Quteba E, Przychodzen B, Makishima H, Bodo J, Ng J, Chee CL, Ba R, et al. (2017). GATA4 loss of function in liver cancer impedes precursor to hepatocyte transition. J Clin Invest 127, 3527–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero A, Giacobbi NS, McCormick KD, Gjoerup OV, Bakkenist CJ, Pipas JM, and Sarkar SN (2014). Simian virus 40 large T antigen induces IFN-stimulated genes through ATR kinase. J Immunol 192, 5933–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero A, Tisoncik-Go J, Watanabe T, Zhong G, Hatta M, Tchitchek N, Selinger C, Chang J, Barker K, Morrison J, et al. (2015). The 1918 Influenza Virus PB2 Protein Enhances Virulence through the Disruption of Inflammatory and Wnt-Mediated Signaling in Mice. J Virol 90, 2240–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, and Gale M Jr. (2006). West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J Virol 80, 2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K, and Andreakos E. (2017). Interferon-lambda Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 46, 875–890 e876. [DOI] [PubMed] [Google Scholar]
- Garibaldi A, Carranza F, and Hertel KJ (2017). Isolation of Newly Transcribed RNA Using the Metabolic Label 4-Thiouridine. Methods Mol Biol 1648, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, et al. (2009). Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401. [DOI] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011). CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann EA, Gale M Jr., and Savan R. (2017). Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front Immunol 8, 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, and Taniguchi T. (2006). IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6, 644–658. [DOI] [PubMed] [Google Scholar]
- Hong M, Schwerk J, Lim C, Kell A, Jarret A, Pangallo J, Loo YM, Liu S, Hagedorn CH, Gale M Jr., and Savan R. (2016). Interferon lambda 4 expression is suppressed by the host during viral infection. J Exp Med 213, 2539–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, and Kola I. (1995). A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci U S A 92, 11284–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Negishi H, and Taniguchi T. (2013). The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 78, 105–116. [DOI] [PubMed] [Google Scholar]
- Jarret A, McFarland AP, Horner SM, Kell A, Schwerk J, Hong M, Badil S, Joslyn RC, Baker DP, Carrington M, et al. (2016). Hepatitis-C-virus-induced microRNAs dampen interferon-mediated antiviral signaling. Nat Med 22, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, and Durbin JE (2010). Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol 84, 11515–11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilg N, Lin W, Hong J, Schaefer EA, Wolski D, Meixong J, Goto K, Brisac C, Chusri P, Fusco DN, et al. (2014). Kinetic differences in the induction of interferon stimulated genes by interferon-alpha and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 59, 1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda N, Shimizu T, Tada Y, and Watanabe S. (2007). IL-18 enhances IFN-gamma-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol 37, 338–350. [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, and Elledge SJ (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Ikeda M, Sugiyama K, Mizutani T, Tanaka T, and Shimotohno K. (1998). Hepatitis C virus population dynamics in human lymphocytes and hepatocytes infected in vitro. J Gen Virol 79 (Pt 8), 1859–1869. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim MJ, Kim CH, Kang JW, Shin HK, Kim DY, Won TB, Han DH, Rhee CS, Yoon JH, and Kim HJ (2017). The Superiority of IFN-lambda as a Therapeutic Candidate to Control Acute Influenza Viral Lung Infection. Am J Respir Cell Mol Biol 56, 202–212. [DOI] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, and Donnelly RP (2003). IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4, 69–77. [DOI] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J Jr., and Tugendreich S. (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroczynska B, Mehrotra S, Arslan AD, Kaur S, and Platanias LC (2014). Regulation of interferon-dependent mRNA translation of target genes. J Interferon Cytokine Res 34, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, Gale M Jr., Klein RS, and Diamond MS (2015). Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med 7, 284ra259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Schoggins JW, and Diamond MS (2019). Shared and Distinct Functions of Type I and Type III Interferons. Immunity 50, 907–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung S, Qureshi SA, Kerr IM, Darnell JE Jr., and Stark GR (1995). Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol 15, 1312–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Plotnikov A, Banerjee A, Suresh Kumar KG, Ragimbeau J, Marijanovic Z, Baker DP, Pellegrini S, and Fuchs SY (2008). Ligand-independent pathway that controls stability of interferon alpha receptor. Biochem Biophys Res Commun 367, 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, and Rice CM (2006). Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898. [DOI] [PubMed] [Google Scholar]
- McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, Delker DA, Hagedorn CH, Carrington M, Gale M Jr., and Savan R. (2014). The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol 15, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKendry R, John J, Flavell D, Muller M, Kerr IM, and Stark GR (1991). High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc Natl Acad Sci U S A 88, 11455–11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, et al. (2013). mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 18, 698–711. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M. (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, Miller CN, Pollack JL, Nagana Gowda GA, Fontana MF, et al. (2018). Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 49, 33–41 e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, and Virgin HW (2015). Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini S, John J, Shearer M, Kerr IM, and Stark GR (1989). Use of a selectable marker regulated by alpha interferon to obtain mutations in the signaling pathway. Mol Cell Biol 9, 4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervolaraki K, Stanifer ML, Munchau S, Renn LA, Albrecht D, Kurzhals S, Senis E, Grimm D, Schroder-Braunstein J, Rabin RL, and Boulant S. (2017). Type I and Type III Interferons Display Different Dependency on Mitogen-Activated Protein Kinases to Mount an Antiviral State in the Human Gut. Front Immunol 8, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, Hergott D, Porter-Gill P, Mumy A, Kohaar I, et al. (2013). A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 45, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JL, Sandoval J, Serviddio G, Sastre J, Morante M, Perrelli MG, Martinez-Chantar ML, Vina J, Vina JR, Mato JM, et al. (2006). Id2 leaves the chromatin of the E2F4-p130-controlled c-myc promoter during hepatocyte priming for liver regeneration. Biochem J 398, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, and Lemke G. (2007). TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerk J, Koster M, Hauser H, Rohde M, Fulde M, Hornef MW, and May T. (2013). Generation of mouse small intestinal epithelial cell lines that allow the analysis of specific innate immune functions. PLoS One 8, e72700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, et al. (2003). IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4, 63–68. [DOI] [PubMed] [Google Scholar]
- Soini T, Eloranta K, Pihlajoki M, Kyronlahti A, Akinrinade O, Andersson N, Lohi J, Pakarinen MP, Wilson DB, and Heikinheimo M. (2018). Transcription factor GATA4 associates with mesenchymal-like gene expression in human hepatoblastoma cells. Tumour Biol 40, 1010428318785498. [DOI] [PubMed] [Google Scholar]
- Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, et al. (2016). ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun 7, 11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, and Proud D. (2005). Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 289, L85–95. [DOI] [PubMed] [Google Scholar]
- Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, Bassendine M, Spengler U, Dore GJ, Powell E, et al. (2009). IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 41, 1100–1104. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, et al. (2009). Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 41, 1105–1109. [DOI] [PubMed] [Google Scholar]
- Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, Kidd J, Kidd K, Khakoo SI, Alexander G, et al. (2009). Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461, 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. (2015). Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 517, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, and Hartmann R. (2007). Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol 81, 7749–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.