

Supplemental Digital Content is available in the text.
Keywords: brain; cell size; endothelium, vascular; models, animal; vascular disease
Abstract
Rationale:
We previously identified somatic activating mutations in the KRAS (Kirsten rat sarcoma viral oncogene homologue) gene in the endothelium of the majority of human sporadic brain arteriovenous malformations; a disorder characterized by direct connections between arteries and veins. However, whether this genetic abnormality alone is sufficient for lesion formation, as well as how active KRAS signaling contributes to arteriovenous malformations, remains unknown.
Objective:
To establish the first in vivo models of somatic KRAS gain of function in the endothelium in both mice and zebrafish to directly observe the phenotypic consequences of constitutive KRAS activity at a cellular level in vivo, and to test potential therapeutic interventions for arteriovenous malformations.
Methods and Results:
Using both postnatal and adult mice, as well as embryonic zebrafish, we demonstrate that endothelial-specific gain of function mutations in Kras (G12D or G12V) are sufficient to induce brain arteriovenous malformations. Active KRAS signaling leads to altered endothelial cell morphogenesis and increased cell size, ectopic sprouting, expanded vessel lumen diameter, and direct connections between arteries and veins. Furthermore, we show that these lesions are not associated with altered endothelial growth dynamics or a lack of proper arteriovenous identity but instead seem to feature exuberant angiogenic signaling. Finally, we demonstrate that KRAS-dependent arteriovenous malformations in zebrafish are refractory to inhibition of the downstream effector PI3K but instead require active MEK (mitogen-activated protein kinase kinase 1) signaling.
Conclusions:
We demonstrate that active KRAS expression in the endothelium is sufficient for brain arteriovenous malformations, even in the setting of uninjured adult vasculature. Furthermore, the finding that KRAS-dependent lesions are reversible in zebrafish suggests that MEK inhibition may represent a promising therapeutic treatment for arteriovenous malformation patients.
Editorial, see p 744
In This Issue, see p 703
Meet the First Author, see p 704
Brain arteriovenous malformations (bAVMs) are abnormal direct connections between arteries and veins that bypass a capillary network.1 Because blood flow and pressure are not dissipated, the vein can become distended and enlarged. Over time, altered flow dynamics may lead to extensive remodeling of the conduit vessels into a tangled nidus; a hallmark of bAVMs.2 Because of their propensity to leak or rupture, these vascular malformations are the leading cause of hemorrhagic stroke in children and young adults.1 Current therapy is limited to surgical resection, endovascular embolization or stereotactic radiosurgery, but this carries significant risk and is not always possible depending on location. No pharmacological interventions have been developed, which is due to our rudimentary understanding of the biology underlying the origin and progression of sporadic bAVMs. By performing exome sequencing on resected patient bAVM tissues and matching peripheral blood as a germline control, our group found that somatic activating mutations in the gene KRAS (Kirsten rat sarcoma viral oncogene homologue)—which encodes RAS GTPase (guanosine triphosphate hydrolase)—occurred in the endothelium of ≈60% of all sporadic bAVMs.3 This finding of somatic KRAS mutations has since been confirmed by others.4–7 However, no preclinical studies have been performed to test whether active KRAS mutations are sufficient to drive the formation of arteriovenous malformations (AVMs).
KRAS activating mutations have been observed in both codon 12 (G12V, G12D, G12C) and in codon 61 (Q61H).3–7 These same mutations are highly prevalent in cancer, where they constitutively activate KRAS by preventing GTP hydrolysis.8 Activating mutations have also been identified in BRAF and MAP2K1/MEK (mitogen-activated protein kinase kinase 1) in AVMs.6,7 Each of these mutated genes function in the MEK/ERK (extracellular signal-regulated kinase) kinase cascade, suggesting this pathway plays a key role in AVM pathogenesis. Indeed, we observed robust MEK/ERK activity in all bAVM tissues that we examined, even those that did not contain detectable KRAS mutations, indicating that excessive activity of this pathway is a central feature in the pathogenesis of these lesions.3
Interestingly, somatic mutations in the endothelium have been observed in several other vascular malformations, including capillary malformations and venous malformations, but the mutations and signaling pathways in these diseases are distinct.9–13 Based on this information, targeted therapies for venous malformations have been tested in slow-flow lesions in patients, with some success.14–16 MEK signaling has been implicated in artery development17–19 and angiogenesis,20,21 and we previously showed that inhibition of the MEK pathway could suppress abnormal endothelial cell (EC) gene expression networks and morphology in active KRAS-transfected cells in vitro.3 These findings suggest that exploration of MEK/ERK pathway inhibition may be warranted in sporadic bAVM patients. This is intriguing given the recent approval of several MEK inhibitors for treating cancer.22 However, the failure to demonstrate that KRAS activation definitively leads to bAVMs in animal models remains a significant hurdle to testing these, and other novel therapies in a preclinical setting.
Thus, we set out to create mouse and zebrafish models of KRAS-induced, sporadic bAVMs. We found that pan-endothelial expression of constitutively active KRAS led to early postnatal lethality in mice, with focal cranial hemorrhage. However, both central nervous system-specific postnatal endothelial gain of KRAS activity, as well as later pan-endothelial induction in adults, bypassed this early lethality and led to dilated vessels and bAVMs. Similar phenotypes were observed upon endothelial-specific expression of mutant KRAS in the developing zebrafish, as embryos demonstrated overt hemorrhage and clear AV shunts, with accompanying alterations in vascular morphogenesis, including increased EC size, ectopic sprouting, and expanded vascular lumens. Notably, brain hemorrhage and existing AV shunts were partially rescued by pharmacological inhibition of MEK activity. Collectively, these results establish novel animal models for the future study of KRAS-mediated bAVM progression. Furthermore, this study indicates that MEK inhibition is a promising therapeutic strategy for treating bAVMs in humans.
Methods
The authors declare that all supporting data are available within the article and its Data Supplement. RNA-seq data has been deposited at the Gene Expression Omnibus, our informatics RStudio pipeline is contained within the Data Supplement, and all plasmids will be deposited at Addgene. Reasonable requests for fish or mice will be fulfilled by the corresponding authors.
A complete description of Methods is included in the Data Supplement. Please see the Major Resources Table in the Data Supplement.
Results
Early Postnatal Expression of KRASG12D in the Murine Endothelium Results in Cerebral Hemorrhage and Death
To explore the cellular and molecular mechanisms that underlie bAVMs in patients harboring somatic KRAS gain of function mutations, we generated compound transgenic mice that harbor a tamoxifen-inducible, EC-specific Cre recombinase (Cdh5(PAC)-CreERT2, which we will refer to hereafter as Cdh5-CreER)23 and a conditional, Cre-activatable mutant Kras allele.24 In this lox-stop-lox mutant Kras conditional mouse strain (referred to as KrasG12D), expression of the constitutively active KrasG12D mutant transcript requires Cre-dependent removal of a transcriptional stop cassette (Online Figure IA). This conditional mutant allele is targeted to the endogenous Kras locus, which ensures that physiological levels of constitutively active KrasG12D transcript are only expressed from the native promoter following removal of the stop cassette. Of note, activation of the Cdh5-CreER driver at P1 led to uniform expression of a recombined Rosa26RFP lineage reporter throughout the entire brain vasculature at P14 (Online Figure II). Kaplan-Meier survival analysis determined that administration of tamoxifen at P1 to Cdh5-CreER; KrasG12D (hereafter referred to as inducible EC-specific KrasG12D, or iEC-KrasG12D) mice on a variety of fluorescent reporter backgrounds (see Methods in the Data Supplement) resulted in significant lethality beginning around P12 (Online Figure IA and IB; Control, n=48; iEC-KrasG12D, n=138). Gross morphological analyses of surviving animals at P21 revealed an incompletely penetrant phenotype of focal intracranial hemorrhage (Control, n=0/116; iEC-KrasG12D, n=11/32), which was confirmed via 3D imaging of the cortical cerebrovasculature via light sheet microscopy (Online Figure IC through IF). However, cortical AVMs were not observed. In contrast to other established models of bAVMs, such as Notch gain of function,25 the cerebral vessels did not appear dilated, but instead appeared thin and fragile (Online Figure IF; Online Movies I and II). Vessel density within the brain at P21 was indistinguishable between Kras mutants (n=5) and control animals (n=7; Online Figure III), suggesting that KRAS gain of function may not stimulate EC proliferation, in agreement with our previous in vitro studies.3 Analysis of iEC-KrasG12D mice on a pure Rosa26RFP reporter background recapitulated the early postnatal lethality, but mutants failed to show frequent cranial hemorrhage (n=1/6) or cortical bAVMs (n=0/6) at P14 (Online Figure IVA through IVD). The lethality of iEC-KrasG12D pups was not due to a failure to thrive, as they were of comparable size and weight to their littermate controls (Online Figure IVG and IVH). Analysis of visceral organs likewise did not reveal obvious hemorrhage in the intestines, lung, or liver at P14 (Online Figure IVE and IVF). Assessment of cardiac structure and function at P14 and P21 failed to reveal any consistent alterations in iEC-KrasG12D mice (Online Figure V). As we only analyzed surviving animals at P14 and P21, and these animals did not feature either bAVMs or organ hemorrhage, and showed mostly normal cardiac performance, the cause of this early postnatal lethality remains unclear. Nevertheless, these results led us to posit that more time and perhaps hemodynamic feedback are required to form bAVMs, and that early lethality in this model precludes lesion formation.
Postnatal Expression of KRASG12D in the Murine Central Nervous System Endothelium Bypasses Early Lethality and Produces bAVMs
To more specifically assess the role of elevated KRAS activity in the cerebrovasculature, we utilized the Slc1o1c1(BAC)-CreER26 driver line to restrict mutant KRAS expression to the brain endothelium. This approach has the added benefit of more closely mimicking the location of somatic gain of KRAS function mutations observed in human patients.3 Females harboring a Cre-dependent Rosa26RFP reporter were crossed to males containing the Slc1o1c1-CreER allele. RFP signal (a visual readout for CreER activity) was observed in the endothelium of the brain and retina, likely reflecting a general central nervous system-wide endothelial activity of the transgene (Online Figure VIA). In contrast to previous reports using the same reporter,26 we observed CreER activity in the absence of tamoxifen (Online Figure VIB and VIC). With this leakiness in mind, we maintained the KrasG12D allele in Rosa26RFP/RFP dams, separate from KrasWT/WT; Slc1o1c1-CreER driver sires, then treated all offspring with tamoxifen at P1. Notably, immunohistochemical analysis of 8-week-old brains revealed robust and uniform labeling of the cerebrovasculature throughout the brain, as well as expression in nonvascular tissues, as previously described26 (Online Figure VII).
Initiation of mutant KrasG12D expression using the inducible brain EC driver Slco1c1-CreER (hereafter referred to as ibEC-KrasG12D) at P1 did not affect survival up to 8 weeks of age (Control, n=0/47 dead; ibEC-KrasG12D, n=0/28 dead; Figure 1A and 1B). Unlike the iEC-KrasG12D pups at P21, ibEC-KrasG12D brains lacked overt signs of hemorrhage at P21 (Control n=0/14; ibEC-KrasG12D n=0/7; Figure 1C). This was also the case in 8-week-old adult animals, where we only rarely observed intracranial hemorrhage in the Kras mutants compared with their control littermates (Control, n=0/47; ibEC-KrasG12D, n=1/28; Figure 1D and 1E). Notably, perfusion with fluorescent-labeled lectin, followed by epifluorescence examination, revealed the presence of bAVMs in ibEC-KrasG12D adults at 8 weeks of age (n=11/21; Figure 1F and 1G). These abnormalities, which occurred in the cortex, just anterior to the cerebellum, as well as near the olfactory bulb, were not detected in control animals at 8 weeks of age (n=0/32; Figure 1F and 1G). Optical clearing and light sheet confocal microscopy revealed that the vasculature surrounding these bAVMs was abnormal, as the vessels appeared tortuous and dilated, similar to the niduses associated with these lesions in human patients, whereas these vessels appeared normal in control animals (Figure 1F, far right, upper and lower panels; also see Online Movies III and IV).
Figure 1.
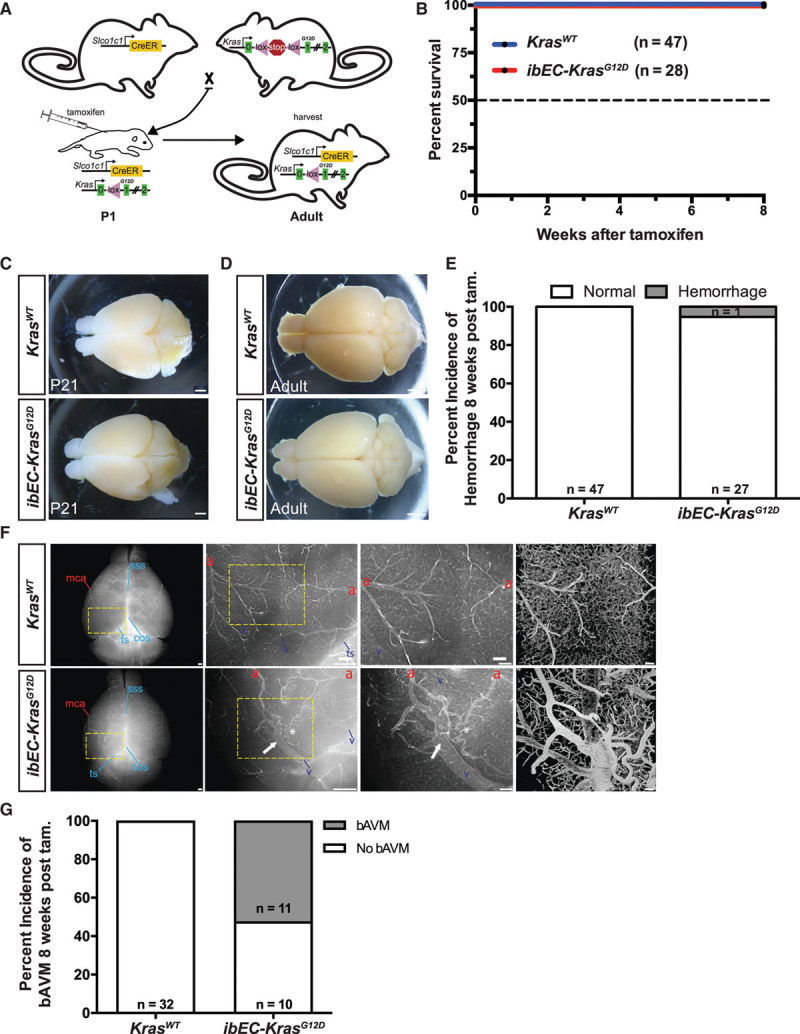
Postnatal expression of active KRAS (Kirsten rat sarcoma viral oncogene homologue) in the murine central nervous system endothelium bypasses early lethality and produces brain arteriovenous malformations. A, Breeding scheme for generating inducible brain endothelial cell KRAS mutant mice (ibEC-KrasG12D). Tamoxifen is delivered to pups at P1 and tissues are harvested at 8 wk of age. B, No survival defects were observed in ibEC-KrasG12D mice or control KrasWT littermates. C, Representative phase microscopy images of the dorsal surface of the brain at P21 show no cerebral hemorrhages in either ibEC-KrasG12D mice (n=0/7) or control littermates (n=0/14; quantification not shown). Scale bar=500 µm. D, Representative phase microscopy images of the dorsal surface of the brain at 8 wks. Scale bar=500 µm. E, Quantification of the incidence of hemorrhage at 8 wk of age in both control (n=0/35) and ibEC-KrasG12D mice (n=1/19). Fisher exact test; P=0.373. F, Representative dorsal surface view, olfactory bulb at the top and cerebellum at the bottom, via direct fluorescence microscopy of an 8-week-old adult mouse brain following perfusion with fluorescent lectin. Arteriovenous shunts, or fusions, between cerebral arteries (red letter a) and veins (blue letter v), as well as venous dilation (white arrow) and tortuosity (asterisk) are evident in ibEC-KrasG12D animals but not the control littermates at 8 wk of age. Magnified areas (yellow boxes) are shown in the panels to the right. Far right panels are flattened reconstructions of volume rendered images of the cortical vasculature following CLARITY clearing and lightsheet confocal microscopy. Scale bar=500 µm for first 2 (left to right) upper and lower panels for wild-type and mutant brain images (with yellow dashed boxes), and = 100 µm for upper and lower far right magnified images. G, Quantification of the incidence of brain arteriovenous malformation (bAVM) at 8 wk of age. Fisher exact test; P=4.6×10−6. COS indicates confluence of sinus veins; MCA, middle cerebral artery; SSS, superior sagittal sinus vein; and TS, transverse sinus vein.
Pan-Endothelial Expression of KRASG12D in Adult Mice Bypasses Lethality and Produces bAVMs
We next sought to determine whether Kras gain of function throughout the adult murine endothelium was sufficient to produce bAVMs. Interestingly, in endothelial-specific loss of function mouse models of Hereditary Hemorrhagic Telangiectasia, such as endothelial-specific loss of Alk1 or Eng, central nervous system AVMs arise if loss of function is induced in early embryonic stages27–30; however, they only appear in adult stages with the addition of an exogenous angiogenic stimulus.28,31 Similarly, gain of constitutive Notch1 or 4 activity must be initiated during the early postnatal period to produce bAVMs.29 Treatment of iEC-KrasG12D and control littermates with tamoxifen between 2 and 4 months of age did not affect overall survival up to 8 weeks later (Control, n=0/23; iEC-KrasG12D, n=0/19; Figure 2A and 2B). In a pilot cohort, no differences in viability were detected up to 36 weeks later (Control, n=0/4; iEC-KrasG12D, n=0/3; Figure 2A and 2B). In contrast to our early postnatal experiments, pan-endothelial induction of mutant KRAS expression in adult mice failed to induce hemorrhage after 8 weeks (Control, n=0/23; iEC-KrasG12D, n=0/19; Figure 2C and 2D). However, this induction regime was sufficient to induce bAVMs in iEC-KrasG12D animals within 8 weeks of treatment in more than half of all animals examined (Control, n=0/15; iEC-KrasG12D, n=6/11; Figure 2E and 2F). A similar phenotypic penetrance was observed 36 weeks after induction in a small cohort with a limited sample size (Control, n=0/4; iEC-KrasG12D, n=2/3; Figure 2G). Thus, expression of KrasG12D in the endothelium is sufficient to induce bAVMs in both postnatal and adult mice.
Figure 2.

Pan-endothelial expression of active KRAS (Kirsten rat sarcoma viral oncogene homologue) in adult mice induces brain arteriovenous malformations. A, Representative model for induction of pan-endothelial mutant KRAS activity in adult mice. B, No survival defects were observed in KRAS mutant mice or control animals up to 8 and 36 wk following induction. C, No evidence of hemorrhage was detected at 8 wk post-treatment in adult KRAS mutant or control animal brains. Representative phase microscopy images of the dorsal and ventral surfaces of the brain are shown. Scale bar=500 µm. D, Quantification of the incidence of cranial hemorrhage 8 wk after initiating tamoxifen treatment at 2 to 4 mo of age. E, Representative whole mount epifluorescent views following perfusion of fluorescent-conjugated tomato lectin reveal the presence of brain arteriovenous malformations (bAVMs; white arrow) just distal to the olfactory bulb (i) and in the cortex just proximal to the cerebellum (Eii and Eiii). a=artery, v=vein. Scale bar=500 µm for far-left panels, and Ei and Eii, and scale bar=100 µm for Eiii. F, Quantification of bAVM incidence 8 wk post tamoxifen induction. Fisher exact test; P=0.002. G, Quantification of the incidence of bAVM at 36 wk post–tamoxifen induction. The sample size was too small for statistical analysis.
Expression of Mutant KRAS in the Endothelium of Embryonic Zebrafish Alters Vascular Morphology
To assess the consequences of expressing active KRAS in the endothelium at a cellular level, we utilized embryonic zebrafish. Tol2-transposable element-flanked transgenes, with expression of either fluorescently-tagged wild-type (WT) or mutant human KRAS under the control of an endothelial-specific kdrl promoter, were injected along with transposase mRNA at the 1 to 4 cell stage to generate mosaic embryos (Figure 3A). Importantly, the mosaic nature of KRAS expression in this transient transgenic system mimics the somatic nature of KRAS mutations in human patients. Expression of N-terminal EYFP-tagged human KRASWT in the endothelium did not have an appreciable effect on the phenotype of ECs in the intersomitic vessels (ISVs) or in the dorsal aorta (DA) (Figure 3B). In contrast, expression of N-terminal EGFP-tagged human KRASG12V dramatically altered EC morphology (Figure 3C through 3E). Ectopic sprouting defects were apparent in 39.0±6.7% of ISVs imaged at 48 hpf that contained KRASG12V-expressing ECs (Figure 3C[i and ii]; arrows and Figure 3F). At earlier stages (ie, 36 hpf), ectopic sprouts containing numerous filopodia could be visualized in Tg(kdrl:LifeAct-GFP) embryos injected with kdrl:mScarlet-KRASG12V (Figure 3D). Additionally, 54.1±7.0% of ISVs expressing KRASG12V had a noticeably larger diameter, and of these ≈86% had lamellipodia-like protrusions that resulted in misshapen, bulbous vessels (Figure 3C[ii and iii]; arrowheads, Figure 3E, Figure 3G). The lumen of KRASG12V-expressing ISVs was expanded, as evidenced by an abundance of gata1:dsRed-expressing red blood cells within the ISV lumen (Figure 3E). Abnormal morphology was also noted when KRASG12V-expressing cells were located in the DA or posterior cardinal vein (Figure 3C[i and ii]; arrowheads). Similar phenotypes were evident in ISVs expressing BFP-tagged KRASG12D in the endothelium (Online Figure VIIIA and VIIIB). The ectopic sprouting and expanded vessel diameter phenotypes were confined to ISVs containing mutant KRAS-expressing ECs, as transgene-negative ISVs were phenotypically normal (Figure 3F and 3G). However, ectopic sprouts were observed occasionally in nontransgenic ECs adjacent to mutant KRAS-expressing cells (Online Figure VIIIA), suggestive of noncell autonomous effects.
Figure 3.
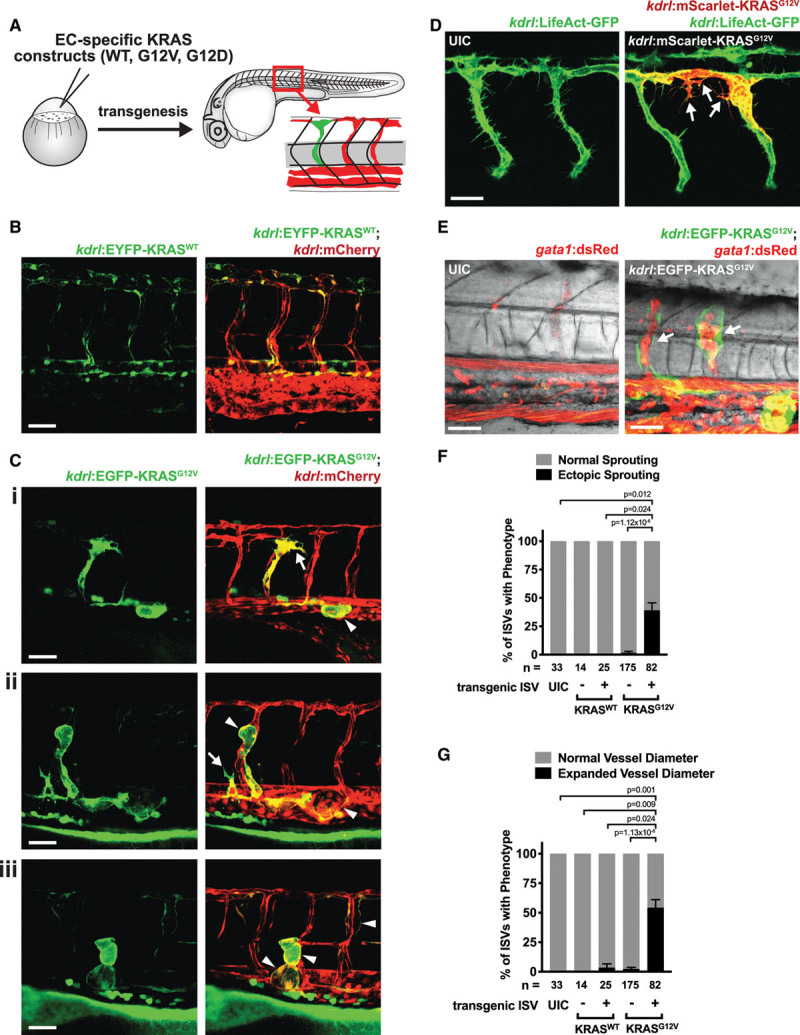
Expression of active KRAS (Kirsten rat sarcoma viral oncogene homologue) in the endothelium of embryonic zebrafish alters cellular morphology. A, Schematic of mosaic analysis of KRAS expression in the endothelium. Wild-type KRAS (KRASWT) or mutant KRAS (KRASG12V or KRASG12D) were expressed under the control of an endothelial-specific promoter, kdrl. B, Representative image of a Tg(kdrl:mCherry) embryo injected with a kdrl:EYFP-KRASWT construct. No changes in cellular phenotype were noted. 52 h post-fertilization (hpf). C, Representative images of Tg(kdrl:mCherry) embryos injected with a kdrl:EGFP-KRASG12V construct. Arrows indicate ectopic sprouting, while arrowheads indicate vessels with enlarged vessel diameter. Ci; 48 hpf, Cii and Ciii; 52 hpf. D, Representative images of Tg(kdrl:LifeAct-GFP) embryos injected with a kdrl:EGFP-KRASG12V construct or an uninjected control (UIC). Arrows indicate ectopic sprouts. 36 hpf. E, Representative image of a Tg(gata1:dsRed) embryo injected with a kdrl:EGFP-KRASG12V construct or an UIC. Arrows indicate expanded, blood-filled lumens in intersomitic vessels (ISVs) expressing KRASG12V. 64 hpf. Quantification of ectopic sprouting (F) or expanded vessel diameter (G) phenotypes in ISVs containing cells expressing the kdrl:KRASWT or kdrl:KRASG12V transgene (+) or in transgene-negative (−) ISVs or in ISVs in UIC embryos at 48 hpf. Shown is the mean±SEM of the percentage of embryos with the indicated phenotype across multiple experiments. The total number of ISVs assessed is shown below. Kruskal-Wallis test with Dunn multiple comparisons test. Scale bar=50 μm for B, C; 30 μm for D, E.
We next determined whether EC hypertrophy (eg, enhanced cell size) or hyperplasia (eg, increased cell number) were responsible for the wider ISVs. Individual ECs expressing active KRAS had highly abnormal cellular shapes (Figure 4A) and KRASG12V-expressing ISVs had significantly (≈2.1-fold) increased ISV diameters (Figure 4B) and expanded blood volumes (Figures 3E and 4A). Of note, the effect on ISV diameter was cell autonomous as regions of ISVs that lacked transgene expression were of normal diameter (Figure 4A). Quantification of the number of ECs per ISV revealed that ISV dilation was not likely due to an augmented number of cells, as there was only a modest, nonsignificant (≈1.17-fold) increase in the number of ECs (Figure 4C), but the average size of ECs was significantly increased by ≈1.7-fold (Figure 4D). To further assess the ability of active KRAS signaling to alter cell size, we quantified cell area in human umbilical vein ECs electroporated with control or KRASG12V constructs. This revealed a ≈1.9-fold increase in cell size in cells expressing mutant KRAS (Figure 4E and 4F), consistent with our previous observations.3
Figure 4.
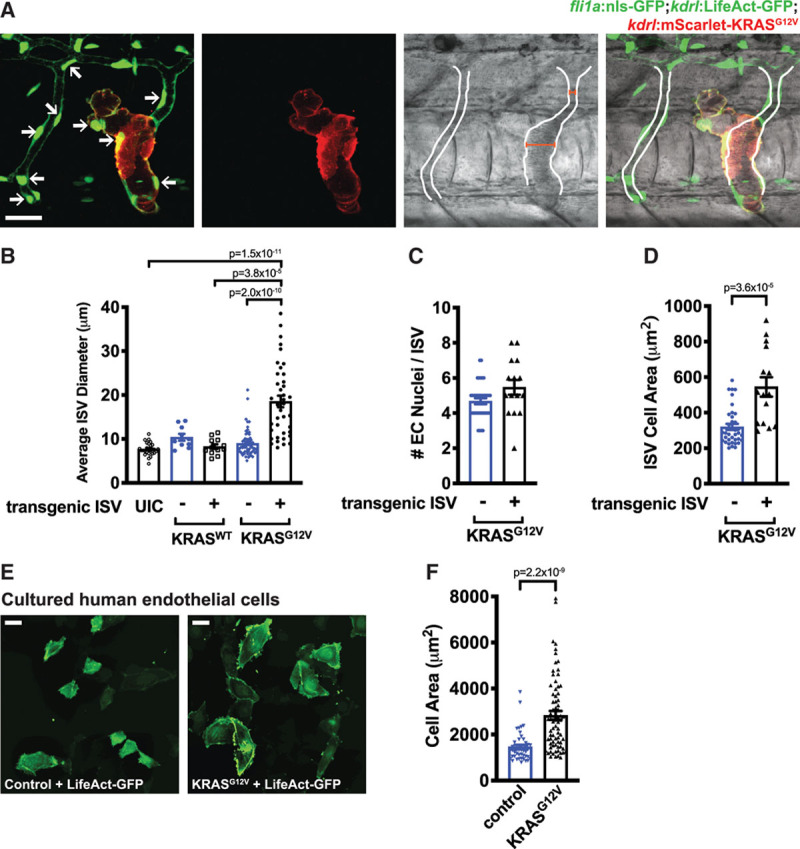
Active KRAS (Kirsten rat sarcoma viral oncogene homologue) expression increases the size of endothelial cells and expands vessel diameter. A, Representative composite brightfield/fluorescent images of a Tg (fli1:nls-GFP; kdrl:LifeActGFP) embryo injected with kdrl:Scarlet-KRASG12V showing altered vessel morphology and expanded lumen (outlined from brightfield image of blood flow). Endothelial cell (EC) nuclei are indicated by arrows. Widths of intersomitic vessels (ISVs) with nontransgenic and KRASG12V-positive cells are indicated with red bars. 48 hpf. Scale bar=20 µm. B, Quantification of average diameter of ISVs containing cells expressing the kdrl:KRASWT or kdrl:KRASG12V transgene (+) or in transgene-negative (−) ISVs or in ISVs in uninjected control (UIC) embryos. Kruskal-Wallis test with Dunn multiple comparisons test. C, Quantification of the number of EC nuclei in ISVs in Tg(fli1a:nls-GFP) embryos containing cells expressing the kdrl:KRASG12V transgene (+) or in transgene-negative (−) ISVs. Unpaired Mann-Whitney U test; P=0.055. D, Quantification of ISV cell area (ie, total ISV area divided by the number of fli1a:nls-GFP nuclei) in ISVs containing cells expressing the kdrl:KRASG12V transgene (+) or in transgene-negative (−) ISVs. Unpaired Mann-Whitney U test. Representative images (E) and quantification (F) of human umbilical vein endothelial cells electroporated with control or KRASG12V constructs, together with a LifeAct-GFP construct. Scale bar=40 µm. Unpaired Mann-Whitney U test.
Activated KRAS Expression Leads to Arteriovenous Shunting and is Associated With Cranial Hemorrhages
Observing blood flow by bright field microscopy or visualizing fluorescently labeled red blood cells in Tg(gata1:dsRed) embryos revealed that KRASG12V expression in the endothelium resulted in AV shunts between the DA and the CV (Figure 5A and 5B). Quantification revealed that 47±4.5% of embryos injected with kdrl:EGFP-KRASG12V developed shunts in the distal axial vasculature at 48 hpf (Figure 5C). In contrast, embryos injected with kdrl:EYFP-KRASWT did not have a significant increase in the number of shunts (Figure 5C; Online Figure VIIIC). AV shunts were also prevalent in Tg(kdrl:LifeAct-GFP) embryos expressing BFP-KRASG12D under the control of kdrl regulatory elements, and this was accompanied by a breakdown of the normal separation between the DA and posterior cardinal vein, and a disruption of actin polymerization (Figure 5D).
Figure 5.
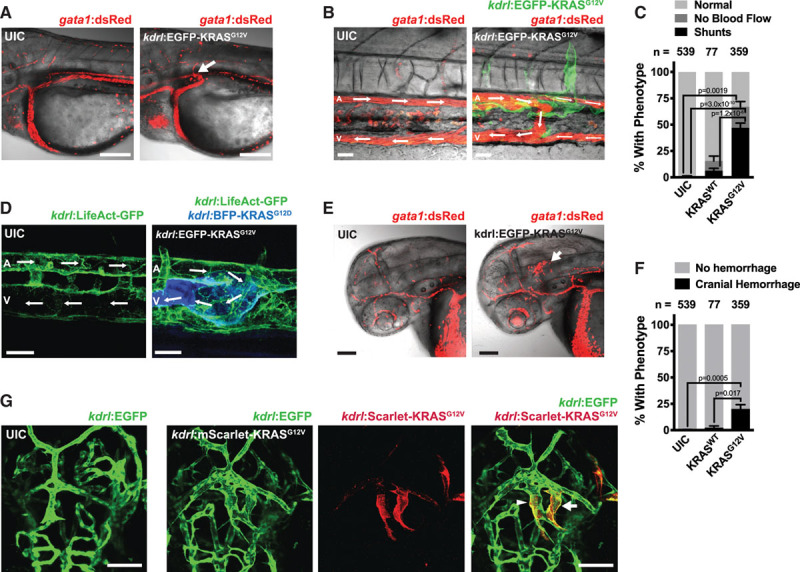
Active KRAS (Kirsten rat sarcoma viral oncogene homologue) expression in the endothelium drives the formation of arteriovenous shunts and cranial hemorrhage. A, Representative image of an arteriovenous (AV) shunt (indicated by arrow) between the dorsal aorta (DA) and the cardinal vein (CV) in a Tg(gata1:dsRed) zebrafish embryo injected with kdrl:EGFP-KRASG12V, but not in uninjected controls (UIC). Scale bar=100 μm. 60 hpf. B, Representative image of an AV shunt between the DA (A) and posterior CV (V) in the trunk of a Tg(gata1:dsRed) embryo injected with kdrl:EGFP-KRASG12V, but not in UIC. Blood flow is indicated by arrows. Note that EGFP-KRASG12V-expressing cells are present at the location of the shunt. Scale bar=30 μm. 64 hpf. C, Quantification of AV shunts and no blood flow phenotypes in the trunk of embryos injected with kdrl:EYFP-KRASWT, kdrl:EGFP-KRASG12V or UIC, as assessed under bright field imaging at 48 hpf. Data are mean±SEM of the percentages of phenotypes from multiple independent experiments. The total number of embryos analyzed are indicated above. One-way ANOVA with Tukey’s multiple comparisons test. D, Representative images of Tg(kdrl:LifeAct-GFP) embryos injected with a kdrl:BFP-KRASG12D construct or UIC demonstrating an AV shunt and altered actin polymerization. Scale bar=20 μm. 72 hpf. E, Representative image of a cranial hemorrhage in a Tg(gata1:dsRed) embryo injected with kdrl:EGFP-KRASG12V but not in UIC. Scale bar=90 μm. 48 hpf. F, Quantification of cranial hemorrhages in embryos injected with kdrl:EYFP-KRASWT, kdrl:EGFP-KRASG12V or UIC, as assessed under bright field at 48 hpf. Data are mean±SEM of the percentages of phenotypes from multiple independent experiments. The total number of embryos analyzed are indicated above. One-way ANOVA with Tukey’s multiple comparisons test. G, Representative images of cranial blood vessels in Tg(kdrl:EGFP) embryos injected with kdrl:mScarlet-KRASG12V and in UIC. The arrow indicates an abnormal vascular connection with large lumen and the arrowhead indicates a vessel with an expanded lumen. Scale bar=50 μm. 54 hpf.
Endothelial-specific expression of KRASG12V in zebrafish was associated with cranial hemorrhages in 19.7±4.6% of embryos, as visualized by cranial blood pooling by bright field imaging (not shown) or confocal microscopy in Tg(gata1:dsRed) embryos (Figure 5E and 5F). In contrast, expression of KRASWT did not induce this phenotype (Figure 5F; Online Figure VIIIC). Examination of vascular morphology revealed that KRASG12V expression led to a malformed cerebral vasculature, including larger diameter vessels and abnormal connections between vessels (Figure 5G).
KRAS Signaling Does Not Alter AV Identity, but Activates Angiogenic Signaling
To determine whether expression of mutant KRAS affected AV identity, we examined the activity of fluorescent arterial reporters in vivo. The Tg(Dll4:GFP) reporter line has high activity in arterial ISVs and the DA of the developing embryo. We did not observe an increase in the already robust reporter activity in ISVs or the DA in embryos expressing KRASG12D in the endothelium (Online Figure IXA). However, we found that Dll4 reporter activity was elevated in shunts between the DA and posterior cardinal vein and expression was observed in some KRASG12D-expressing venous cells (Online Figure IXA). However, when we examined Notch activity—a critical determinant of AV identity—using the transgenic Notch reporter line Tg(Tp1:GFP), only modest levels of Notch signaling were evident in shunts and no expression was detected in KRASG12V-expressing venous cells (Online Figure IXB). This suggests that expression of active KRAS may not dramatically alter AV identity. As we previously demonstrated that the Dll4:GFP reporter is regulated by VEGF signaling and requires a single, intact ETS transcription factor DNA binding sequence, and is largely Notch-independent, we assessed whether ETS reporter activity was altered by KRAS expression. To do so, we generated a novel ETS reporter line by concatemerizing the ETS B-site of the murine Dll4 intron 3 enhancer (8X).19 Reporter activity was detected in ECs of some of the sprouting ISVs and the DA at 24 hpf. At later developmental stages (ie, 48 and 72 hpf), low levels of reporter activity remained confined to the DA and select ISVs (Online Figure IXC). ETS reporter activity was induced in KRASG12V-expressing cells, including within shunts and in venous cells, and was also elevated in neighboring, transgene-negative cells (Online Figure IXD).
Activated KRAS Enhances EC Permeability In Vitro in a MEK-Dependent Manner
Our previous study revealed that KRAS-driven downregulation of VE-Cadherin at cell-cell junctions in cultured ECs required MEK activity.3 To further explore the role of KRAS in EC permeability and integrity, we performed a transwell leak assay in cultured human umbilical vein ECs. KRASG12V expression enhanced EC permeability by ≈2-fold compared with ECs electroporated with a control plasmid (Figure 6A). Importantly, permeability was reduced by treatment of KRASG12V-expressing cells with 2 different MEK inhibitors, SL327 or U0126, for 18 hours before the addition of the FITC tracer, but was not affected by addition of the PI3K inhibitor (PI3Ki), LY294002 (Figure 6A), despite significant inhibition of pAKT levels (Online Figure XA). To further determine the requirement for PI3K and MEK kinase activity downstream of precocious KRAS activation in ECs, we performed transcriptional profiling in human umbilical vein ECs electroporated with empty vector or KRASG12V, followed by treatment with LY294002, U0126 or vehicle control. In total, KRASG12V expression upregulated 460 genes and downregulated 236 genes (log2-fold change >0.5 or <−0.5 and adjusted P-value <0.1; Figure 6B). Gene ontology analysis showed that KRASG12V expression upregulated processes such as angiogenesis, cell cycle, regulation of MAP kinase activity, and RNA processing, among others (Figure 6B and 6C). Conversely, KRASG12V expression led to downregulation of such processes as cytokine production, response to stress, and immune responses (Figure 6B and 6C). Further analysis of KRAS-expressing cells treated with PI3K inhibitors or MEK inhibitors identified 147 KRAS-induced, MEK-dependent targets that fell into such categories as blood vessel development, regulation of cell migration, and RNA processing (Figure 6Dand 6E). Critically, KRAS-induced processes that required MEK activity—such as angiogenesis and cell migration—were not affected by PI3Ki (Figure 6F and 6G). Remarkably, PI3Ki only downregulated 21 unique targets, with just a single GO term emerging (Figure 6E). Additional analyses of this dataset can be found in Online Figures XI through XIII. Collectively, these results demonstrate that a subset of specific phenotypic and molecular changes induced by KRASG12V expression in ECs require MEK, but not PI3K.
Figure 6.
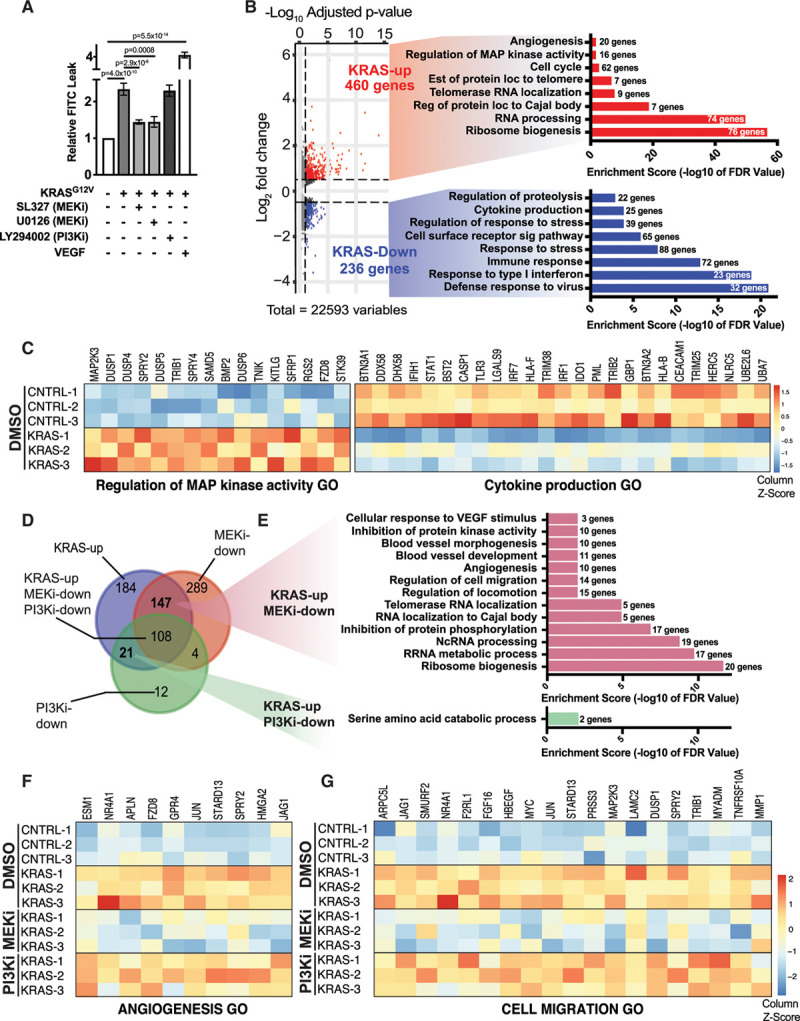
KRASG12V (Kirsten rat sarcoma viral oncogene homologue)-signaling disrupts endothelial cell (EC) barrier and alters gene regulatory networks in a MEK (mitogen-activated protein kinase kinase 1)-dependent manner. A, EC barrier was assessed using a transwell leak assay with a FITC-Dextran tracer in HUVECs expressing KRASG12V±MEK inhibition (MEKi) with SL327 or U0126 or PI3K inhibition (PI3Ki) with LY294002 or control cells (ie, empty vector). VEGF treatment was included as a positive control for EC permeability. n=9 for control, KRASG12V and KRASG12V+SL327; n=3 for KRASG12V+U0126 and KRASG12V+LY294002; n=6 for VEGF. One-way ANOVA with Tukey’s multiple comparisons test. For simplicity, not all significant comparisons are indicated. PI3Ki was not able to rescue KRAS-dependent leak; P=0.9999. B, Volcano-plot of differentially expressed genes in HUVECs expressing KRASG12V compared with empty vector control. Associated select GO terms (with enrichment score below and number of genes to the right) for genes up-regulated by KRASG12V are shown in red, while those downregulated are shown in blue. C, Heat maps of differentially expressed transcripts detected by RNA-seq in 2 representative GO categories are shown (n=3 biological replicates). D, A Venn diagram shows the overlap between genes that are differentially up-regulated in HUVECs expressing KRASG12V compared with empty vector control following treatment with vehicle (DMSO), PI3Ki (LY294002), or MEKi (U01236). A small subset of KRASG12V upregulated targets (460 total genes) are uniquely affected by PI3Ki (21 genes), although MEKi affects a larger cohort of these KRAS-induced transcripts (147 genes). Significant genes in the Venn diagram were included if they displayed a Benjamini–Hochberg adjusted P-value of <0.1 and a log2 (fold change) >0.5. E, Associated select GO terms of MEKi- and PI3Ki-sensitive genes. F and G, Heat maps of differentially expressed transcripts detected by RNA-seq in 2 representative GO categories. KRASG12V-induced transcripts in these GO categories are not downregulated following PI3Ki but are decreased following MEKi. See Online Table II for gene list of KRAS-induced, MEK-dependent and PI3K-dependent transcripts, along with a full list of linked Gene Ontology categories.
Inhibition of MEK Activity, but Not PI3K Signaling, Rescues Vascular Defects in KRASG12D-Expressing Embryos
To assess the importance of the MEK signaling pathway for shunt formation, embryos were treated with 1 μM of the MEK inhibitor, SL327, or an equivalent amount of vehicle (ie, DMSO) at 28 hpf, before the formation of observable shunts, and the prevalence of AV shunts was later assessed at 48 hpf. Importantly, this dose of SL327 does not impair ISV formation (data not shown) but does lower pERK levels (Online Figure XB). We found that SL327 treatment reduced the proportion of KRASG12V-injected embryos that had shunts between the DA and posterior cardinal vein, demonstrating that KRAS-mediated shunt formation requires MEK activity (Online Figure XC).
Next, we sought to determine whether MEK activity was required to maintain established AV shunts. Zebrafish injected with kdrl:EGFP-KRASG12V were screened for the presence of AV shunts at ≈36 hpf and then embryos were treated with 1 μM SL327 or an equivalent amount of vehicle control (ie, DMSO), and the presence of shunts was assessed 24 hours later (≈60 hpf). While ≈19% of embryos exposed to DMSO for 24 hours had reversal of established shunts, treatment with SL327 rescued this defect in ≈66% of embryos (Figure 7A and 7B). To determine the specificity of this response, we treated embryos with established KRASG12V-induced shunts at 36 hpf with the PI3Ki, LY294002, at a dose (10 µM) that inhibits pAKT levels but does not affect blood flow (Online Figure XD and XE), and assessed shunts 24 hours later (Online Figure XF; Figure 7B). No change in shunt frequency was observed with this treatment.
Figure 7.
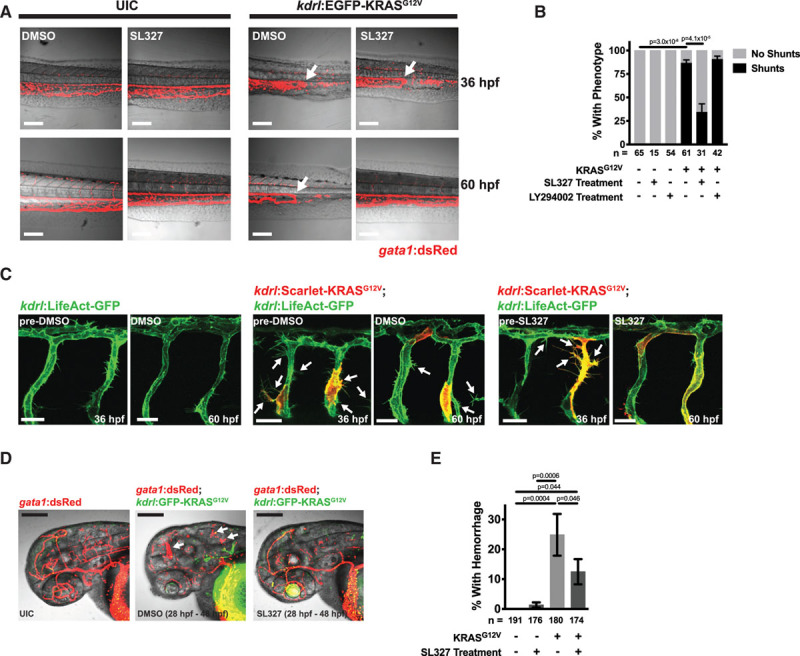
MEK (mitogen-activated protein kinase kinase 1) inhibition can rescue phenotypes caused by active KRAS (Kirsten rat sarcoma viral oncogene homologue) signaling in endothelial cells. A, Representative images of Tg(gata1:dsRed) embryos, that were either uninjected (ie, UIC) or were injected with kdrl:EGFP-KRASG12V. Embryos were imaged at 36 hpf to identify embryos with AV shunts, at which point these embryos were exposed to either DMSO or 1 μM of the MEK inhibitor, SL327. The same embryos were imaged at 60 hpf. Scale bar=40 μm. B, Quantification of phenotypes after 24 h of SL327 (1 μM), LY294002 (PI3K inhibitor, 10 μM), or vehicle (ie, DMSO) treatment. All KRASG12V embryos had shunts at the beginning of treatment. Data are mean±SEM of the percentages of phenotypes from 5 (SL327) or 4 (LY294002) independent experiments. The SL327 and LY294002 treatments were performed in different experiments and data are combined in one graph for visualization purposes, but analysis was done separately. The total number of embryos analyzed are indicated below. One-way ANOVA with Tukey’s multiple comparison test. For simplicity, not all significant comparisons are indicated. PI3K was not able to rescue established shunts; P=0.816. See Online Figure X for more data on LY294002 treatment. C, Representative images of Tg(kdrl:LifeAct-GFP) embryos injected with kdrl:mScarlet-KRASG12V or UIC. Images were taken at 36 hpf and embryos were treated with 1 μM SL327 or DMSO control and imaging was then repeated on the same embryos at 60 hpf. kdrl:mScarlet-KRASG12V-injected embryos had ectopic sprouts (indicated by arrows) and these continued to be present at 60 hpf in embryos exposed to DMSO, but this phenotype was normalized by treatment with SL327. Scale bar=20 μm. D, Hemorrhage phenotype in Tg(gata1:dsRed) embryos treated with DMSO or 1 μM SL327 from 28 to 48 hpf. Hemorrhages are indicated by arrows. Scale bar=200 μm. E, Quantification of the percentage of embryos with cranial hemorrhage (mean±SEM) from 6 independent experiments. The total number of embryos analyzed are indicated below. Statistical significance was determined by one-way ANOVA with Tukey multiple comparison test.
We next determined if MEK inhibition could also reverse KRAS-induced morphological changes in ISVs. Embryos carrying a Tg(kdrl:LifeAct-GFP) reporter that were injected with kdrl:mScarlet-KRASG12V displayed enhanced ectopic EC sprouting compared with uninjected controls at ≈36 hpf (Figure 7C). Embryos were then treated with DMSO or SL327 and the same embryos were imaged again later at ≈60 hpf. Ectopic sprouts were still present in kdrl:KRASG12V-expressing embryos treated with DMSO, while vascular morphology appeared to be normalized in SL327-treated embryos (Figure 7C).
Finally, we sought to determine whether inhibition of MEK activity could prevent the formation of cranial hemorrhages in kdrl:KRASG12V-injected embryos. Indeed, addition of SL327 at ≈28 hpf reduced the percentage of embryos that developed hemorrhage by 48 hpf compared with control embryos (Figure 7D and 7E).
Discussion
AVMs in the brain vasculature are a significant source of morbidity and mortality in children and young adults.32 New therapies, particularly pharmacological approaches, are desperately needed as existing surgical interventions pose significant risks to patients.33 It is now apparent that somatic mutations that lead to constitutive activation of the KRAS-MEK pathway are the most common known genetic cause of sporadic bAVMs and occur in the majority of such lesions.3–7 Yet, the mechanisms whereby this signaling pathway can promote abnormal connections between arteries and veins is poorly understood. Here, we have developed the first animal models of this disease and find that expression of active KRAS (G12V or G12D) in the endothelium is sufficient to drive the formation of bAVMs in mice and zebrafish. Furthermore, we demonstrate in zebrafish that inhibition of MEK can block and even reverse AV shunts. These models will serve as valuable platforms to uncover the molecular mechanisms involved in sporadic bAVM pathogenesis and to develop novel therapies. Critically, the lesions observed in our animal models are analogous to human bAVM pathology, providing strong evidence that activating KRAS mutations are causative of the disease and that MEK inhibition may be a feasible approach for treatment of patients with bAVMs.
While these same KRAS mutations promote proliferation of tumorigenic cells, we demonstrate that expression of activated KRAS in the endothelium does not affect proliferation but instead leads to striking changes in EC phenotype. These phenotypes include ectopic sprouting, increased cell size, disorganization of cell shape, expansion of lumen diameter, and abnormal connections between arteries and veins. While we observed AV shunts, establishment of AV identity did not appear to be altered. Instead, we find increased activation of ETS reporter activity in KRAS-expressing cells and in neighboring ECs in vivo. Given the critical role of ETS factors in regulating angiogenesis, coupled with our previous observation of a VEGF-like transcriptional signature in KRAS expressing cells,3 and our finding of ectopic sprouting in vivo upon KRAS expression, we suggest that alterations in protrusive and migratory behavior plays a significant, or even causative role in the formation of abnormal connections between arteries and veins. In contrast to other AVM mouse models (eg, Hereditary Hemorrhagic Telangiectasia),31,34–36 injury or external angiogenic stimuli are not required for AVM formation in adult vessels as KRAS signaling may provide the angiogenic and remodeling cues. Stochastic aberrant connections between arteries and veins or additional physiological cues that remain to be uncovered may contribute to the focal nature of bAVMs in our mouse model (despite widespread expression of mutant KRAS in all ECs).
A characteristic feature of AVMs are their expanded vascular lumens. Of note, a recent immunohistochemistry study demonstrated that KRASG12D tended to be expressed in ECs of dilated vessels in bAVMs.4 This matches our observation in zebrafish and mouse models that active KRAS-expressing vessels feature a distinctly large diameter. The mechanisms responsible for the expanded lumen are not yet clear, but we provide evidence that active KRAS signaling leads to enhanced EC size. Identification of the pathways activated downstream of KRAS would help determine the mechanisms underlying KRAS-induced alterations in cell shape and lumen diameter. Given the involvement of blood flow in mediating morphogenic defects in distinct types of vascular malformations,37,38 it will also be of interest to determine how KRAS-dependent increases in lumen diameter are involved in disease pathogenesis and whether KRAS-mutant cells have abnormal responses to flow.
Although multiple RAS family members are expressed in ECs (ie, KRAS, NRAS, HRAS), it remains unclear why sporadic bAVMs involve activating mutations exclusively in KRAS. Furthermore, mutations in negative regulators of RAS signaling (eg, RASA1/p120-RasGAP) are associated with capillary-malformation arteriovenous malformation (CM-AVM1) in humans39 and a subset of these patients present with intracranial AVMs,40 and while mouse models present with vascular and lymphatic defects,41–45 they did not feature bAVMs. Interestingly, activating NRAS variants have been observed in a highly aggressive vascular malformation of the lymphatics, known as lymphangiomatosis,46,47 and HRAS gain of function variants have been observed in ECs isolated from a patient with an extracranial AVM.48 A recent transgenic mouse model in which active HRAS was expressed in the postnatal endothelium showed that the abnormal and sustained activation of the RAS pathway can drive vascular defects, including dilation of capillary vessels and diffuse hemorrhage in the brain, but not other organs.49 It is notable, however, that these phenotypes seem to be distinct from those observed following KRAS activation in the endothelium, as no bAVMs were noted. This points to unique roles for RAS proteins in vessel morphogenesis and homeostasis, which requires further exploration.
As KRAS is difficult to target pharmacologically, most therapies for treating KRAS-mutant cancers have focused on targeting downstream signaling molecules, including MEK.50–54 We are encouraged by our finding that inhibition of MEK signaling can reverse already-established AV shunts in KRAS-expressing zebrafish. It will be important to determine whether advanced lesions in the mouse brain—which feature a tangled nidus—can be reversed through inhibiting effectors that act downstream of KRAS signaling. If the abnormal vessel architecture that we observed requires continual KRAS activity and MEK/ERK signaling, similar to addiction to KRAS signaling in cancer,55 then pharmacological blockade of these downstream effectors may offer hope to patients suffering from sporadic bAVM. Indeed, recent studies using animal models of venous and lymphatic malformations and small pilot studies in humans have shown that pharmacological blockade of aberrant signaling pathways may be a feasible therapeutic approach for managing vascular malformations.16,56,57 However, whether MEK inhibition can reverse advanced cerebrovascular lesions in humans, which may have been present for decades and have undergone extensive remodeling due to chronic responses to altered blood flow and inflammation, remains to be seen. Notably, a recent case report of efficacy of MEK inhibitor therapy (trametinib) in reducing the size and vessel caliber of a skin AVM that contained activating MAP2K1 mutations, provides some optimism.58
Taken together, we demonstrate that KRAS gain of function mutations in ECs can drive abnormal vascular morphology and AVMs in mouse and zebrafish models and that these lesions can be reversed by inhibiting MEK signaling. Furthermore, unlike other genetic animal models, expression of active KRAS in adult ECs is sufficient to induce bAVM formation. These findings provide new insight into sporadic bAVMs and offer hope that therapeutic approaches can be utilized to negate KRAS-mediated alterations in vessel morphology.
Acknowledgments
We thank Kate Wythe for the model graphics in Figures 1A, 2A, and 3A, and the graphic abstract. The authors also thank the Optical Imaging and Vital Microscopy core, run by Dr Mary Dickinson and Mr Jason Kirk, for assistance with both normal confocal and lightsheet imaging, data analysis, and 3-dimensional image reconstruction. J.E. Fish and J.D. Wythe conceptualized the study; J.E. Fish, C.P. Flores Suarez, E. Boudreau, A.M. Herman, and J.D. Wythe designed experiments; C.P. Flores Suarez, E. Boudreau, A.M. Herman, M.C. Gutierrez and P.V. DiStefano conducted the majority of experiments with help from D. Gustafson, M. Cui, Z. Chen, T.S. Schexnayder, C.S. Ward, and K.B. De Ruiz; M.C. Gutierrez and J.D. Wythe performed the bioinformatic analysis; all authors acquired data; J.E. Fish, C.P. Flores Suarez, E. Boudreau, A.M. Herman, M.C. Gutierrez, C.S. Ward, and J.D. Wythe analyzed the data; J.E. Fish and J.D. Wythe wrote the original draft of the manuscript; J.E. Fish, A.M. Herman, P.V. DiStefano, M.C. Gutierrez, D. Gustafson, C.S. Ward, I. Radovanovic, and J.D. Wythe secured funding; all authors edited and approved the manuscript.
Sources of Funding
T.S. Schexnayder and C.S. Ward in the Mouse Metabolism and Phenotyping Core at Baylor College of Medicine were supported with funding from National Institutes of Health (UM1HG006348, R01DK114356, R01HL130249). J.E. Fish is supported by a Canada Research Chair from the Canadian Institutes of Health Research (CIHR) and his lab received infrastructure funding from the Canada Foundation for Innovation, the John R. Evans Leaders Fund and the Ontario Research Fund. Research in the Fish laboratory is supported by a Project Grant from CIHR (PJT 155922), the US Department of Defense (W81XWH-18-1-0351) and a Medicine by Design Team Project Award, which receives funding from the Canada First Research Excellence Fund. A.M. Herman was supported by the National Institutes of Health (5T32HL007676-27, 5T32HL007676-28) and American Heart Association Postdoctoral Award (19POST34430008). P.V. DiStefano was supported by a post-doctoral fellowship from the Toronto General Hospital Research Institute. D. Gustafson is supported by a Canada Graduate Scholarship from CIHR. M.C. Gutierrez was supported by an American Heart Association Predoctoral Award (19PRE34410104). J.D. Wythe is supported by institutional startup funds from the Cardiovascular Research Institute at Baylor College of Medicine, the Caroline Wiess Law Fund for Research in Molecular Medicine, the Curtis and Doris K. Hankamer Foundation Basic Research Fund, and the ARCO Foundation Young Teacher-Investigator Award. J.D. Wythe and work within the Wythe laboratory is supported by the Department of Defense (W81XWH-18-1-0350) and CIHR (PJT 155922).
Disclosures
None.
Supplemental Materials
Detailed Methods
Online Figures I–XIII
Uncropped Western Blots
Online Tables I–V
Online Videos I–IV
Online File I
References59–83
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronymns
- AVM
- arteriovenous malformation
- bAVM
- brain arteriovenous malformation
- DA
- dorsal aorta
- EC
- endothelial cell
- ISV
- intersomitic vessel
J.E.F., C.P.F.S., and E.B. contributed equally to this article.
For Sources of Funding and Disclosures, see page 741.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.119.316500.
Novelty and Significance
What Is Known?
Somatic mutations in the KRAS (Kirsten rat sarcoma viral oncogene homologue) gene are detected within the endothelial cells (ECs) lining the vessels of brain arteriovenous malformations (bAVMs) in the majority of patients with sporadic lesions.
Histological analysis shows that these activating mutations in KRAS lead to the constitutive activation of the downstream MAPK/MEK (mitogen-activated protein kinase kinase 1)/ERK (extracellular signal-regulated kinase) signaling pathway.
In cultured ECs, expression of active KRAS increases migration and alters cell shape and actin dynamics without affecting proliferation.
What New Information Does This Article Contribute?
In both mouse and zebrafish models, expression of mutant KRAS in the endothelium is sufficient to induce bAVMs in vivo.
In vivo, ECs expressing active KRAS have increased cell size and increased sprouting behavior, and vessels are tortuous with dilated lumens, and abnormal connections occur between arteries and veins. Critically, these ECs do not show increased proliferation.
Inhibition of MEK/ERK signaling, but not PI3K activity can suppress the KRAS-induced gene signature in cultured ECs, and can rescue lesion formation and reverse established arteriovenous shunts.
Arteriovenous malformations are the leading cause of brain hemorrhage in children and young adults. Most bAVMs are sporadic (ie, no familial history of inheritance). We recently showed that somatic activating KRAS mutations occur in the endothelium of the majority of these lesions, and that this leads to activation of the MAPK/MEK/ERK signaling pathway in cultured ECs. This suggested that pharmacological treatments for brain arteriovenous malformations may be possible. However, whether somatic KRAS mutations actually cause these vascular lesions, and the requirement of the MAPK/MEK/ERK pathway in maintaining these malformations, were not known. Furthermore, the mechanisms that lead to sporadic arteriovenous malformation have not been deciphered. Here, we establish the first animal models of KRAS-dependent bAVMs in both mice and zebrafish. We demonstrate that expression of active KRAS in the endothelium of the brain is sufficient to produce bAVMs. At the cellular level, blood vessels expressing active KRAS have expanded lumens, ectopic sprouting, and abnormal connections between arteries and veins. At the molecular level, we identify MEK-dependent angiogenic gene networks that are activated in KRAS-expressing ECs. Importantly, inhibition of MEK in our zebrafish model reveals that established arteriovenous shunts can be reversed, giving hope that such approaches may be useful in a clinical setting.
References
- 1.Solomon RA, Connolly ES., Jr. Arteriovenous malformations of the brain. N Engl J Med 20173761859–1866doi: 10.1056/NEJMra1607407 [DOI] [PubMed] [Google Scholar]
- 2.Yun JH, Kwon DH, Lee EJ, Lee DH, Ahn JS, Kwun BD. New nidus formation adjacent to the target site of an arteriovenous malformation treated by Gamma Knife surgery. J Neurosurg 2012117suppl120–125doi: 10.3171/2012.8.GKS12994 [DOI] [PubMed] [Google Scholar]
- 3.Nikolaev SI, Vetiska S, Bonilla X, Boudreau E, Jauhiainen S, Rezai Jahromi B, Khyzha N, DiStefano PV, Suutarinen S, Kiehl TR, et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med 2018378250–261doi: 10.1056/NEJMoa1709449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oka M, Kushamae M, Aoki T, Yamaguchi T, Kitazato K, Abekura Y, Kawamata T, Mizutani T, Miyamoto S, Takagi Y. KRAS G12D or G12V mutation in human brain arteriovenous malformations. World Neurosurg 2019126e1365–e1373doi: 10.1016/j.wneu.2019.03.105 [DOI] [PubMed] [Google Scholar]
- 5.Priemer DS, Vortmeyer AO, Zhang S, Chang HY, Curless KL, Cheng L. Activating KRAS mutations in arteriovenous malformations of the brain: frequency and clinicopathologic correlation. Hum Pathol 20198933–39doi: 10.1016/j.humpath.2019.04.004 [DOI] [PubMed] [Google Scholar]
- 6.Hong T, Yan Y, Li J, Radovanovic I, Ma X, Shao YW, Yu J, Ma Y, Zhang P, Ling F, et al. High prevalence of KRAS/BRAF somatic mutations in brain and spinal cord arteriovenous malformations. Brain 201914223–34doi: 10.1093/brain/awy307 [DOI] [PubMed] [Google Scholar]
- 7.Al-Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, Knox R, Pittman A, Clark G, Baird W, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest 20181281496–1508doi: 10.1172/JCI98589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serebriiskii IG, Connelly C, Frampton G, Newberg J, Cooke M, Miller V, Ali S, Ross JS, Handorf E, Arora S, et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun. 2019;10:3722. doi: 10.1038/s41467-019-11530-0. doi: 10.1038/s41467-019-11530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lapinski PE, Doosti A, Salato V, North P, Burrows PE, King PD. Somatic second hit mutation of RASA1 in vascular endothelial cells in capillary malformation-arteriovenous malformation. Eur J Med Genet 20186111–16doi: 10.1016/j.ejmg.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, Eklund L, Boon LM, Vikkula M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet 200941118–124doi: 10.1038/ng.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limaye N, Kangas J, Mendola A, Godfraind C, Schlögel MJ, Helaers R, Eklund L, Boon LM, Vikkula M. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet 201597914–921doi: 10.1016/j.ajhg.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 20133681971–1979doi: 10.1056/NEJMoa1213507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L, Couto JA, Pinto A, Alexandrescu S, Madsen JR, Greene AK, Sahin M, Bischoff J. Somatic GNAQ mutation is enriched in brain endothelial cells in Sturge-Weber syndrome. Pediatr Neurol 20176759–63doi: 10.1016/j.pediatrneurol.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, et al. Sirolimus is efficacious in treatment for extensive and/or complex slow-flow vascular malformations: a monocentric prospective phase II study. Orphanet J Rare Dis. 2018;13:191. doi: 10.1186/s13023-018-0934-z. doi: 10.1186/s13023-018-0934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Cai Y, Goines J, Pastura P, Brichta L, Lane A, Le Cras TD, Boscolo E. Ponatinib combined with rapamycin causes regression of murine venous malformation. Arterioscler Thromb Vasc Biol 201939496–512doi: 10.1161/ATVBAHA.118.312315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boscolo E, Limaye N, Huang L, Kang KT, Soblet J, Uebelhoer M, Mendola A, Natynki M, Seront E, Dupont S, et al. Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects. J Clin Invest 20151253491–3504doi: 10.1172/JCI76004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong CC, Peterson QP, Hong JY, Peterson RT. Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and MAP kinase/ERK signaling. Curr Biol 2006161366–1372doi: 10.1016/j.cub.2006.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng Y, Larrivée B, Zhuang ZW, Atri D, Moraes F, Prahst C, Eichmann A, Simons M. Endothelial RAF1/ERK activation regulates arterial morphogenesis. Blood 20131213988–96, S1doi: 10.1182/blood-2012-12-474601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wythe JD, Dang LT, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DY, Oettgen P, Black BL, et al. ETS factors regulate Vegf-dependent arterial specification. Dev Cell 20132645–58doi: 10.1016/j.devcel.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin M, Beane TJ, Quillien A, Male I, Zhu LJ, Lawson ND. Vegfa signals through ERK to promote angiogenesis, but not artery differentiation. Development 20161433796–3805doi: 10.1242/dev.137919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fish JE, Cantu Gutierrez M, Dang LT, Khyzha N, Chen Z, Veitch S, Cheng HS, Khor M, Antounians L, Njock MS, et al. Dynamic regulation of VEGF-inducible genes by an ERK/ERG/p300 transcriptional network. Development 20171442428–2444doi: 10.1242/dev.146050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yaeger R, Corcoran RB. Targeting alterations in the RAF-MEK pathway. Cancer Discov 20199329–341doi: 10.1158/2159-8290.CD-18-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010465483–486doi: 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- 24.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001153243–3248doi: 10.1101/gad.943001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy PA, Lam MT, Wu X, Kim TN, Vartanian SM, Bollen AW, Carlson TR, Wang RA. Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. Proc Natl Acad Sci U S A 200810510901–10906doi: 10.1073/pnas.0802743105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridder DA, Lang MF, Salinin S, Röderer JP, Struss M, Maser-Gluth C, Schwaninger M. TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med 20112082615–2623doi: 10.1084/jem.20110398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M, Arthur HM. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res 20101061425–1433doi: 10.1161/CIRCRESAHA.109.211037 [DOI] [PubMed] [Google Scholar]
- 28.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M, Arthur HM. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One. 2014;9:e98646. doi: 10.1371/journal.pone.0098646. doi: 10.1371/journal.pone.0098646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen CM, Huang L, Murphy PA, Lawton MT, Wang RA. Mouse Models of Cerebral Arteriovenous Malformation. Stroke 201647293–300doi: 10.1161/STROKEAHA.115.002869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest 20091193487–3496doi: 10.1172/JCI39482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen W, Sun Z, Han Z, Jun K, Camus M, Wankhede M, Mao L, Arnold T, Young WL, Su H. De novo cerebrovascular malformation in the adult mouse after endothelial Alk1 deletion and angiogenic stimulation. Stroke 201445900–902doi: 10.1161/STROKEAHA.113.003655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leblanc GG, Golanov E, Awad IA, Young WL; Biology of Vascular Malformations of the Brain NINDS Workshop Collaborators Biology of vascular malformations of the brain. Stroke 200940e694–e702doi: 10.1161/STROKEAHA.109.563692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawton MT, Rutledge WC, Kim H, Stapf C, Whitehead KJ, Li DY, Krings T, terBrugge K, Kondziolka D, Morgan MK, et al. Brain arteriovenous malformations. Nat Rev Dis Primers. 2015;1:15008. doi: 10.1038/nrdp.2015.8. doi: 10.1038/nrdp.2015.8. [DOI] [PubMed] [Google Scholar]
- 34.Xu B, Wu YQ, Huey M, Arthur HM, Marchuk DA, Hashimoto T, Young WL, Yang GY. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J Cereb Blood Flow Metab 200424237–244doi: 10.1097/01.WCB.0000107730.66603.51 [DOI] [PubMed] [Google Scholar]
- 35.Hao Q, Zhu Y, Su H, Shen F, Yang GY, Kim H, Young WL. VEGF induces more severe cerebrovascular dysplasia in endoglin than in Alk1 mice. Transl Stroke Res 20101197–201doi: 10.1007/s12975-010-0020-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi EJ, Walker EJ, Shen F, Oh SP, Arthur HM, Young WL, Su H. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc Dis 201233540–547doi: 10.1159/000337762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugden WW, Meissner R, Aegerter-Wilmsen T, Tsaryk R, Leonard EV, Bussmann J, Hamm MJ, Herzog W, Jin Y, Jakobsson L, et al. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat Cell Biol 201719653–665doi: 10.1038/ncb3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rochon ER, Menon PG, Roman BL. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development 20161432593–2602doi: 10.1242/dev.135392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 2003731240–1249doi: 10.1086/379793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, Clapuyt P, Hammer F, Dubois J, Baselga E, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat 200829959–965doi: 10.1002/humu.20746 [DOI] [PubMed] [Google Scholar]
- 41.Chen D, Teng JM, North PE, Lapinski PE, King PD. RASA1-dependent cellular export of collagen IV controls blood and lymphatic vascular development. J Clin Invest 20191293545–3561doi: 10.1172/JCI124917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 1995377695–701doi: 10.1038/377695a0 [DOI] [PubMed] [Google Scholar]
- 43.Lapinski PE, Kwon S, Lubeck BA, Wilkinson JE, Srinivasan RS, Sevick-Muraca E, King PD. RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J Clin Invest 2012122733–747doi: 10.1172/JCI46116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lapinski PE, Lubeck BA, Chen D, Doosti A, Zawieja SD, Davis MJ, King PD. RASA1 regulates the function of lymphatic vessel valves in mice. J Clin Invest 20171272569–2585doi: 10.1172/JCI89607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lubeck BA, Lapinski PE, Bauler TJ, Oliver JA, Hughes ED, Saunders TL, King PD. Blood vascular abnormalities in Rasa1(R780Q) knockin mice: implications for the pathogenesis of capillary malformation-arteriovenous malformation. Am J Pathol 20141843163–3169doi: 10.1016/j.ajpath.2014.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barclay SF, Inman KW, Luks VL, McIntyre JB, Al-Ibraheemi A, Church AJ, Perez-Atayde AR, Mangray S, Jeng M, Kreimer SR, et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet Med 2019211517–1524doi: 10.1038/s41436-018-0390-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manevitz-Mendelson E, Leichner GS, Barel O, Davidi-Avrahami I, Ziv-Strasser L, Eyal E, Pessach I, Rimon U, Barzilai A, Hirshberg A, et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis 201821287–298doi: 10.1007/s10456-018-9595-8 [DOI] [PubMed] [Google Scholar]
- 48.Konczyk DJ, Goss JA, Smits PJ, Huang AY, Al-Ibraheemi A, Sudduth CL, Warman ML, Greene AK. Arteriovenous malformation associated with a HRAS mutation. Hum Genet 20191381419–1421doi: 10.1007/s00439-019-02072-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li QF, Decker-Rockefeller B, Bajaj A, Pumiglia K. Activation of Ras in the vascular endothelium induces brain vascular malformations and hemorrhagic stroke. Cell Rep 2018242869–2882doi: 10.1016/j.celrep.2018.08.025 [DOI] [PubMed] [Google Scholar]
- 50.Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev 20175361–69doi: 10.1016/j.ctrv.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 51.Merchant M, Moffat J, Schaefer G, Chan J, Wang X, Orr C, Cheng J, Hunsaker T, Shao L, Wang SJ, et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS One. 2017;12:e0185862. doi: 10.1371/journal.pone.0185862. doi: 10.1371/journal.pone.0185862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim MO, Dombi E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest 2013123340–347doi: 10.1172/JCI60578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang MT, Fer N, Galeas J, Collisson EA, Kim SE, Sharib J, McCormick F. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat Commun. 2019;10:3055. doi: 10.1038/s41467-019-11044-9. doi: 10.1038/s41467-019-11044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tetsu O, McCormick F. ETS-targeted therapy: can it substitute for MEK inhibitors? Clin Transl Med. 2017;6:16. doi: 10.1186/s40169-017-0147-4. doi: 10.1186/s40169-017-0147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009136823–837doi: 10.1016/j.cell.2009.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li D, March ME, Gutierrez-Uzquiza A, Kao C, Seiler C, Pinto E, Matsuoka LS, Battig MR, Bhoj EJ, Wenger TL, et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat Med 2019251116–1122doi: 10.1038/s41591-019-0479-2 [DOI] [PubMed] [Google Scholar]
- 57.Triana P, Dore M, Cerezo VN, Cervantes M, Sánchez AV, Ferrero MM, González MD, Lopez-Gutierrez JC. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 20172786–90doi: 10.1055/s-0036-1593383 [DOI] [PubMed] [Google Scholar]
- 58.Lekwuttikarn R, Lim YH, Admani S, Choate KA, Teng JMC. Genotype-guided medical treatment of an arteriovenous malformation in a child. JAMA Dermatol 2019155256–257doi: 10.1001/jamadermatol.2018.4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villefranc JA, Amigo J, Lawson ND. Gateway compatible vectors for analysis of gene function in the zebrafish. Dev Dyn 20072363077–3087doi: 10.1002/dvdy.21354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Y, Cashman TJ, Nevis KR, Obregon P, Carney SA, Liu Y, Gu A, Mosimann C, Sondalle S, Peterson RE, et al. Latent TGF-β binding protein 3 identifies a second heart field in zebrafish. Nature 2011474645–648doi: 10.1038/nature10094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20022087–90doi: 10.1038/nbt0102-87 [DOI] [PubMed] [Google Scholar]
- 62.Zhao L, Borikova AL, Ben-Yair R, Guner-Ataman B, MacRae CA, Lee RT, Burns CG, Burns CE. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc Natl Acad Sci U S A 20141111403–1408doi: 10.1073/pnas.1311705111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 20072363088–3099doi: 10.1002/dvdy.21343 [DOI] [PubMed] [Google Scholar]
- 64.Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res 2012221059–1068doi: 10.1101/gr.133546.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 2015861369–1384doi: 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, Grgic I, Kumar S, Humphreys BD, Humphreys B, et al. Cell-specific translational profiling in acute kidney injury. J Clin Invest 20141241242–1254doi: 10.1172/JCI72126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 201013133–140doi: 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 2002244305–318doi: 10.1006/dbio.2002.0597 [DOI] [PubMed] [Google Scholar]
- 69.Zudaire E, Gambardella L, Kurcz C, Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One. 2011;6:e27385. doi: 10.1371/journal.pone.0027385. doi: 10.1371/journal.pone.0027385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Respress JL, Wehrens XH. Transthoracic echocardiography in mice. J Vis Exp. 2010:1738. doi: 10.3791/1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 20051325199–5209doi: 10.1242/dev.02087 [DOI] [PubMed] [Google Scholar]
- 72.Traver D, Paw BH, Poss KD, Penberthy WT, Lin S, Zon LI. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat Immunol 200341238–1246doi: 10.1038/ni1007 [DOI] [PubMed] [Google Scholar]
- 73.Proulx K, Lu A, Sumanas S. Cranial vasculature in zebrafish forms by angioblast cluster-derived angiogenesis. Dev Biol 201034834–46doi: 10.1016/j.ydbio.2010.08.036 [DOI] [PubMed] [Google Scholar]
- 74.Parsons MJ, Pisharath H, Yusuff S, Moore JC, Siekmann AF, Lawson N, Leach SD. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev 2009126898–912doi: 10.1016/j.mod.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, Maquet E, Gauquier A, Cabochette P, Fukuhara S, Mochizuki N, et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/beta-catenin pathway during brain angiogenesis. Elife. 2015;4:e06489. doi: 10.7554/eLife.06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roman BL, Pham VN, Lawson ND, Kulik M, Childs S, Lekven AC, Garrity DM, Moon RT, Fishman MC, Lechleider RJ, et al. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development 20021293009–3019 [DOI] [PubMed] [Google Scholar]
- 77.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 201714417–419doi: 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blighe K, Rana S, Lewis M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 152. https://githubcom/kevinblighe/EnhancedVolcano. 2020.
- 81. http://bioinformaticspsbugentbe/webtools/Venn/
- 82.Kolde R. pheatmap: Pretty Heatmaps. R package version 1012. https://CRANR-projectorg/package=pheatmap. 2019.
- 83.Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020362628–2629doi: 10.1093/bioinformatics/btz931 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.