

The SLC12 gene family encodes electroneutral cation-coupled chloride cotransporters. McNeill et al. identify de novo variants in SLC12A2 that are associated with a neurodevelopmental disorder or with non-syndromic sensorineural deafness, and provide further evidence for a role of SLC12A2 in neurodevelopment.
Keywords: corticogenesis, neurodevelopmental disorder, de novo mutation, exome, brain
Abstract
The SLC12 gene family consists of SLC12A1–SLC12A9, encoding electroneutral cation-coupled chloride co-transporters. SCL12A2 has been shown to play a role in corticogenesis and therefore represents a strong candidate neurodevelopmental disorder gene. Through trio exome sequencing we identified de novo mutations in SLC12A2 in six children with neurodevelopmental disorders. All had developmental delay or intellectual disability ranging from mild to severe. Two had sensorineural deafness. We also identified SLC12A2 variants in three individuals with non-syndromic bilateral sensorineural hearing loss and vestibular areflexia. The SLC12A2 de novo mutation rate was demonstrated to be significantly elevated in the deciphering developmental disorders cohort. All tested variants were shown to reduce co-transporter function in Xenopus laevis oocytes. Analysis of SLC12A2 expression in foetal brain at 16–18 weeks post-conception revealed high expression in radial glial cells, compatible with a role in neurogenesis. Gene co-expression analysis in cells robustly expressing SLC12A2 at 16–18 weeks post-conception identified a transcriptomic programme associated with active neurogenesis. We identify SLC12A2 de novo mutations as the cause of a novel neurodevelopmental disorder and bilateral non-syndromic sensorineural hearing loss and provide further data supporting a role for this gene in human neurodevelopment.
Introduction
Neurodevelopmental disorders (NDD) affect 1–5% of the population and demonstrate clinical and aetiological heterogeneity (Wright et al., 2015). De novo mutations (DNM) are associated with NDD in ∼25% of cases (McRae et al., 2017). Corticogenesis is the process by which neuronal progenitors proliferate and migrate to form the cerebral cortex (Urbán and Guillemot, 2014). Corticogenesis is tightly regulated by transcriptional programmes, with temporal and spatial regulation of gene expression. In humans, corticogenesis begins around embryonic Day 42 and is largely completed by birth (Urbán and Guillemot, 2014). Many of the genes affected by DNM in NDD play a role in corticogenesis (McRae et al., 2017; Zawerton et al., 2019), and corticogenesis genes are strong candidates for NDD. The SLC12 gene family consists of SLC12A1–SLC12A9, encoding electroneutral cation-coupled chloride transporters (Arroyo et al., 2013). Several of these genes are known to cause human disease (Supplementary Table 1). There is no clear gene–disease relationship for SLC12A2 (NKCC1). However, SCL12A2 has been shown to play a role in corticogenesis (Young et al., 2012) and the excitatory-inhibitory GABA switch during brain development (He et al., 2014). SLC12A2 represents a good candidate gene for NDD.
Study of an Slc12a2/NKCC1 null mouse demonstrated that the transporter is a key mechanism in the accumulation of the K+-rich endolymph in the inner ear (Delpire et al., 1999). Absence of NKCC1 causes sensorineural deafness and balance deficits. Little is known about a brain phenotype in the NKCC1 knockout mouse. However, NKCC1 expression in central neurons is developmentally regulated in rodents (Plotkin et al., 1997). Expression is likely highest when the neurons are born in the subventricular zone at a time when the intracellular chloride (Cl−) concentration is highest (Ben-Ari, 2012). High Cl− facilitates the development of GABA-mediated giant synaptic potentials, by which GABA excites developing neurons to promote growth and synapse formation when glutamatergic inputs have not yet developed (Ben-Ari, 2012). Disruption of NKCC1 expression during development is likely to have consequences for brain development.
SLC12A2 undergoes alternative splicing, with eight isoforms identified by the Genotype-Tissue Expression Project (GTEx) (Supplementary Fig. 1) (Stranger et al., 2017). The full-length isoform is 27 exons, with exon 21 being spliced out in the other major isoform. In mouse cochlea, only the exon 21-containing isoform is expressed (Dixon et al., 1999). Deafness in the sy mouse is due to an exon 21 frameshift in Slc12a2 (Dixon et al., 1999). In the brain, both exon 21-containing and exon 21-deleted isoforms are expressed (Morita et al., 2014). Here, we describe six patients with DNM in SLC12A2 associated with an NDD and three with bilateral sensorineural hearing loss (BLSNHL) and exon 21 variants.
Materials and methods
Ascertainment of individuals with SCL12A2 variants
Probands with protein altering single nucleotide variants (SNVs) in SLC12A2 were identified in Patients 1–5 by exome sequencing in the Deciphering Developmental Disorders study (DDD) (Wright et al., 2015). Subjects 6–9 were identified via Genematcher (Sobreira et al., 2015). Written consent was obtained from parents/guardians.
In silico assessment of pathogenicity of SCL12A2 variants
The effects of SLC12A2 variants were assessed using multiple in silico tools (Schwarz et al., 2010; Shihab et al., 2013). The excess in occurrence of de novo SLC12A2 protein-altering variants in DDD was assessed using DenovolyzeR (Ware et al., 2015), which compares the observed: expected number DNMs to identify an elevated de novo mutation rate for a given gene. Spatial clustering of missense SLC12A2 DNMs was assessed using Denovonear, which calculates the probability of the observed spatial pattern of DNM arising by chance. We used MuPIT, which maps the effect of SNVs onto 3D-protein structures, to visualize the structural effects of DNMs (Niknafs et al., 2013).
Transcriptomic study of SLC12A2 expression in the developing human brain
Variations in SLC12A2 transcript levels in the human brain among different developmental stages and anatomical regions were investigated using data from the BrainSpan Atlas of the Developing Human Brain (Miller et al., 2014). Single cell RNA (scRNA) sequencing data from the human brain at 16–18 weeks gestation were obtained from Pollen et al. (2015). NetworkAnalyst, which uses protein-protein interaction networks to analyse gene expression studies, was used to explore transcriptomic differences in scRNA data between SLC12A2 expressing and non-expressing cells (Xia et al., 2015).
In silico study of expression of SLC12A2 splice isoforms
RNA-seq data from the BrainSpan Atlas of the Developing Human Brain were used to analyse levels of SLC12A2 exon expression during brain development. RNA-seq data from developing mouse cochlea were examined for alternative splicing of SLC12A2 during cochlear development (Ranum et al., 2019).
In vitro assessment of NKCC1 ion transporter function
NKCC1 function was assessed through K+ influx measurements into Xenopus laevis oocytes injected with wild-type or mutant NKCC1 cRNA. Detailed protocols have been published and summarized in the Supplementary material (Delpire et al., 2011).
Data availability
Exome sequencing data from the DDD study are available at the European Genome-Phenome Archive (https://ega-archive.org/studies/EGAS00001000775).
Results
Identification of SLC12A2 variants in children with neurodevelopmental disorders
Through trio exome sequencing, we identified six children with NDD and DNM in SLC12A2. Detailed clinical reports are provided in the Supplementary material and Table 1. All had intellectual disability or developmental delay varying from mild to severe. Three had an autistic spectrum disorder. Patient 1 had cerebral cortical dysplasia on brain MRI. Patient 5 had agenesis of the corpus callosum. Two had BLSNHL. Patient 4 had spastic diplegia and Patient 5 had spastic quadriparesis. In addition we identified three individuals from two unrelated families with congenital non-syndromic BLSNHL and an SCL12A2 variant (Table 1). There was no shared facial dysmorphology. None had a pathogenic copy number variant, any additional candidate DNM (Supplementary Table 2) or causal variant in a deafness gene.
Table 1.
Summary of clinical and genomic data for SLC12A2 variant carriers
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | |
|---|---|---|---|---|---|---|---|---|---|
| Age, years; sex | 1; Male | 9; Male | 15; Female | 3; F | 6; F | 21; Male | 2; Male | 44; Male | 5; Male |
| Variant (Genome build GRCh-37/hg19) | g.5127450305C>T | g.5127469897G>A | g.5127503511G>A | g.5127466837A>T | g.5127420201dup | g.5127466845delinsCT | g.5127512805G>A | g.5127512802G>A | g.5127512802G>A |
| c.DNA variantNM_001046.3 | c.980C>T | c.1229G>A | c.2675G>A | c.1127A>T | c.555dupG | c.1135_1136delGCinsCT | c.2938G>A | c.2935G>A | c.2935G>A |
| ACMG criteria | 4 | 4 | 5 | 4 | 4 | 4 | 4 | 4 | 4 |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | Unknown | Inherited from Case 8 |
| Amino acid change | p.(Ala327Val) | p.(Arg410Gln) | p.(Trp892*) | p.(Asn376Ile) | p.(His186AlafsTer17) | p.(Ala379Leu) | p.(Glu980Lys) | p.(Glu979Lys) | p.(Glu979Lys) |
| Exon (of 27) | 4 | 6 | 18 | 5 | 1 | 5 | 21 | 21 | 21 |
| gnomAD frequency | 0 | 0.0000070 (2 cases) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CADD-Phred score | 26.0 | 31 | 40 | 27.3 | 33 | 28 | 23 | 23 | 23 |
| GERP | 5.36 | 4.94 | 4.8 | 4.92 | 3.97 | 4.92 | 5.11 | 5.11 | 5.11 |
| MutationTaster | Disease causing (1) | Disease causing (1) | Disease causing (1) | Disease causing (1) | N/A | Disease causing (1) | Disease causing (1) | Disease causing (1) | Disease causing (1) |
| FATHMM | Damaging (−5.14) | Damaging (−2.17) | Damaging (0.977) (FATHMM-MKL) | Damaging (−5.18) | N/A | Damaging (−5.03) | Damaging (−1.91) | Damaging (−1.9) | Damaging (−1.9) |
| PolyPhen-2 | Probably damaging (1) | Probably damaging (1) | N/A | Probably damaging (1) | N/A | Probably damaging (1) | Possibly damaging (0.682) | Possibly damaging (0.799) | Possibly damaging (0.799) |
In silico assessment of SLC12A2 variants supports pathogenicity
Seven of the eight identified SLC12A2 variants were de novo and all had Combined Annotation Dependent Depletion (CADD) scores >20 (Table 1 and Fig. 1A). The missense variants affected evolutionarily conserved residues, conserved between SLC12 gene family members (Supplementary Fig. 2). We assessed whether the DNM rate in the DDD cohort was elevated. We collected all eight SLC12A2 protein-altering DNMs (the five pathogenic variants plus three variants predicted to be benign) in DDD (Supplementary material). Using DenovolyzeR, we showed a 9.62-fold DNM enrichment in SLC12A2 (P = 2.71 × 10−6, after correction for multiple testing P = 0.05).
Figure 1.
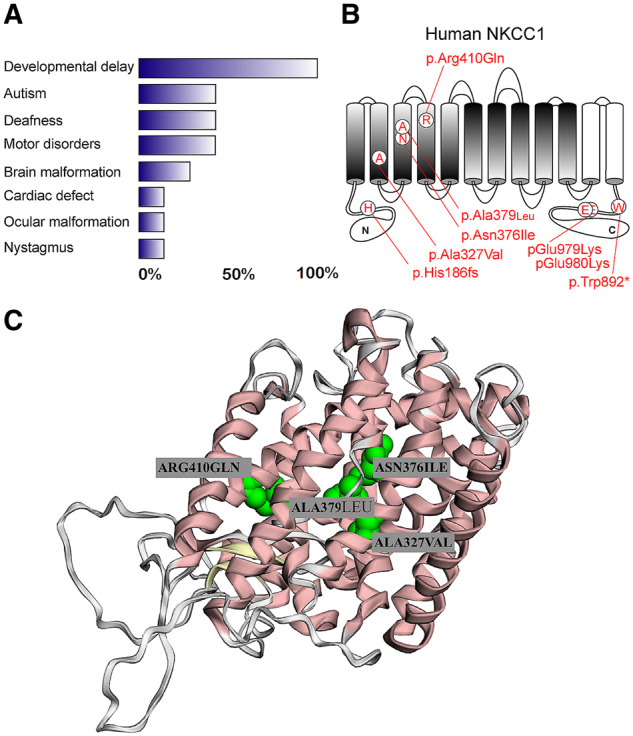
SLC12A2 missense variants. (A) Chart summarizing frequency of clinical features in children with SLC12A2 de novo mutations and an NDD. (B) Schematic representation of NKCC1 with 12 transmembrane domains: five inverted transmembrane domains + five symmetry transmembrane domains (shaded), followed by TM11 and TM12 (white transmembrane domains). The position of patient mutations is indicated in red. (C) Three-dimensional structure of NKCC1 demonstrating location of missense variants at the core of the protein, with high potential to disrupt protein structure.
SLC12A2 sequence data from the gnomAD database demonstrated significant constraint for protein-truncating variants in SLC12A2 [58.7 expected versus 11 observed, pLI = 0.96, observed: expected 0.19 (0.12–0.31)], but not for missense SNVs [594 observed versus 430 expected, Z = 2.4, observed: expected 0.72 (0.67–0.78)]. We then used gnomAD data to examine the spatial distribution of missense SNVs in SLC12A2, to look for differential prevalence of missense variants in protein domains (implying regional constraint). There was no difference in the percentage of residues with a synonymous variant in the functionally important domains (transmembrane domains) (Chen et al., 2019) and domains of no known function (19% versus 22%, P = 0.13). In the functionally significant domains, 35% of residues had a missense variant compared to 43% of residues in the domains of no known functional significance (chi-squared, P = 0.0037). In addition, constraint data from Samocha et al. (2017) indicate that amino acids 1–836 are depleted of missense variants (observed 173, expected 294.8, ratio 0.58, chi-squared 50.3) while the remainder of the protein is not. This suggests constraint of missense variation in the functionally significant domains of SLC12A2.
Pathogenic SLC12A2 variants alter NKCC1 activity
To assess the functional significance of the mutations, we injected wild-type NKCC1 and mutant NKCC1 cRNAs in Xenopus laevis oocytes and performed standard K+ influx measurements under isotonic (basal) and hypertonic (stimulated) conditions. We used the mouse cDNA, which is 95% conserved compared to human SLC12A2. For every mutation tested, Supplementary Fig. 2 demonstrates homology of the mutated and surrounding residues within the Slc12a transporter family and among NKCC1 proteins from six species (sea urchin and five vertebrates). All tested mutations demonstrated significant reduction in K+ influx (Fig. 2 and detailed results in the Supplementary material).
Figure 2.
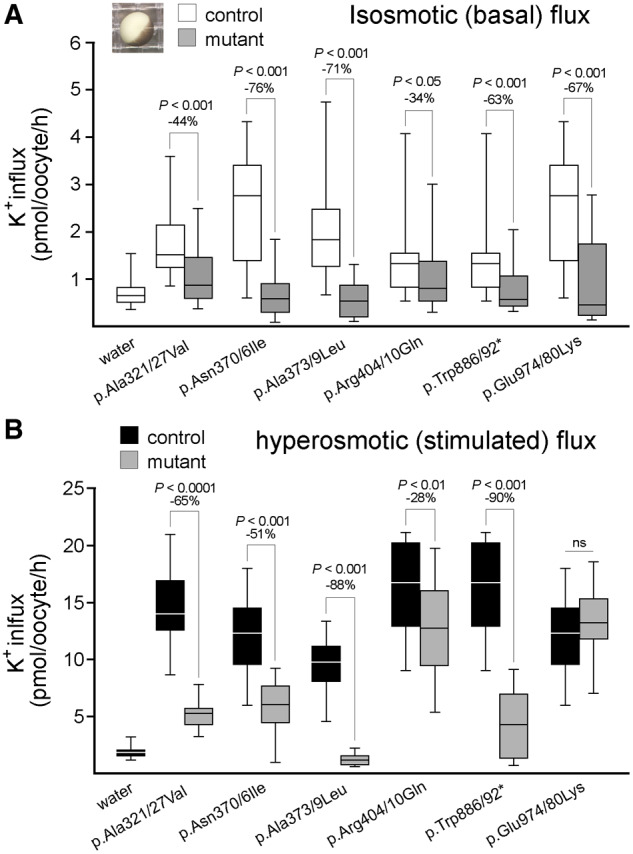
Functional analysis of NKCC1 mutation in Xenopus laevis oocytes. (A and B) Box plots demonstrating K+ influx measured in oocytes injected with water (negative control), water containing 15 ng wild-type (control) or mutant mouse Slc12a2/NKCC1 cRNA. Horizontal line represents median, extent of box demonstrates interquartile range and whiskers minimum and maximum. K+ fluxes were measured under basal isosmotic (200 mOsM) or stimulated hyperosmotic (270 mOsM) conditions and are expressed in picomoles K+/oocyte/hour. As the mutants were tested in different experiments on different oocytes, each mutant flux is accompanied by its own control flux. Statistical differences were determined using one-way ANOVA. Each experiment represents 20–25 oocytes per mutation. Note that the mouse residue numbers are slightly different from human.
Spatial proximity of SLC12A2 missense de novo mutations altering NKCC1 activity
We wondered whether DNMs [p.(Ala327Val), p.(Arg410Gln), p.(Asn376Ile), p.(Ala379Pro), p.(Glu980Lys)] clustered within the protein. Clustering analysis with Denovonear demonstrated a greater spatial proximity within the protein than expected by chance (P = 0.027), with the majority of DNMs falling into transmembrane domains (Fig. 1B). We then used MuPIT to map these DNMs onto NKCC1 3D protein structure, showing all DNMs embedded within the transmembrane core of the protein (Fig. 1C).
Expression of SLC12A2 in developing human brain supports a role in neurogenesis
We next examined SLC12A2 expression in developing human brain using microarray data from the BrainSpan atlas. SLC12A2 expression was significantly higher (Mann-Whitney U-test, Z = −8.36 P < 0.001) in neuroanatomical regions with high neurogenesis (ventricular zone, subventricular zone) compared with regions of less active neurogenesis (cortical plate, subplate) at 15–16 weeks gestation (Fig. 3A). For a range of brain expressed genes with no known role in neurogenesis there was no differential expression in neurogenic niches (data not shown).
Figure 3.
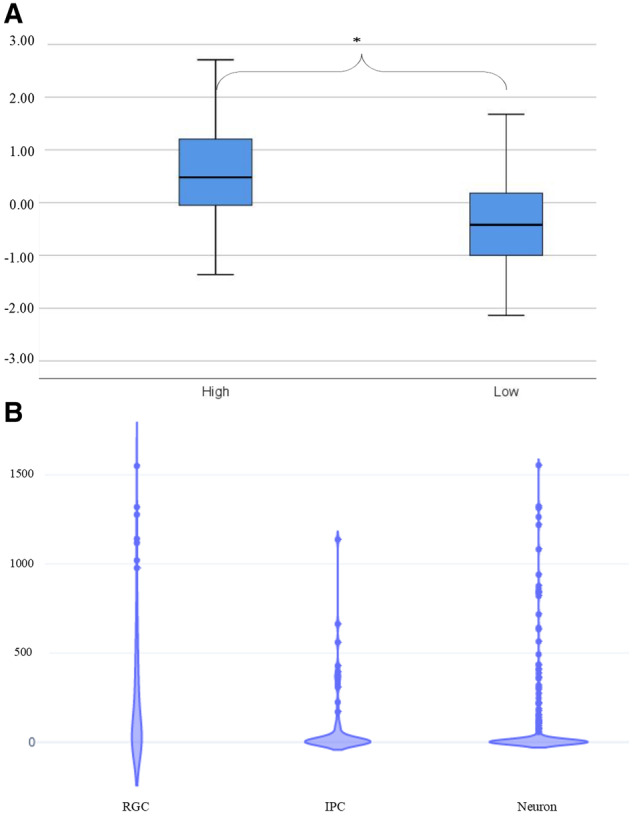
Transcriptomic analysis of SCL12A2 expression in developing human brain. (A) Box plots of microarray data comparing SLC12A2 expression in neuroanatomical regions of high and less active neurogenesis at 15 weeks gestation. Horizontal lines represent median, extent of box interquartile range and whiskers minimum and maximum. Expression is significantly higher in areas of active neurogenesis (asterisk). (B) Single cell RNA sequencing data demonstrating significantly higher expression of SLC12A2 in radial glia cells (RGC) compared to intermediate progenitor cells (IPC) (Mann-Whitney U-test, Z = −3.3, P = 0.001) and neurons (z = −5.2, P < 0.001). Units are counts per million reads (CPM).
Single-cell RNA sequencing data from 16–18 weeks gestation foetal brain were then used to identify populations of cells that express SLC12A2 during development. The marker genes defined by Pollen et al. (2015) were used to identify radial glia (e.g. PAX6, SOX2, VIM), intermediate progenitors (e.g. EOMES, RBFOX1) and neurons (e.g. MYT1L, NEUROD1, SATB2). SLC12A2 expression was significantly higher in radial glia than in intermediate progenitors (Mann-Whitney U-test, Z = −3.3, P = 0.001) or neurons (z = −5.2, P < 0.001), but did not differ between intermediate progenitors and neurons (z = −1.06, P = 0.28, Fig. 3B). In radial glial there was no expression of other SLC12 gene family members known to have a role in neuronal development or function. This suggests other SLC12A2 family members cannot compensate for reduction in SLC12A2 function associated with SLC12A2 DNM.
In silico study of expression of SLC12A2 splice isoforms
RNA-seq data from BrainSpan quantifies levels of exon expression. Supplementary Fig. 3A demonstrates an excess of transcripts for exon 1–28 compared to exon 21 in human foetal brain. SLC12A2 isoforms lacking exon 21 therefore exist in developing human brain. RNA-seq of developing mouse cochlea demonstrated no alternative splicing of exon 21; the exon 21-containing isoform is the only transcript expressed (Supplementary Fig. 3B).
SLC12A2 expressing cells display transcriptomic profiles of active neurogenesis
To investigate the functional properties of SLC12A2 expressing cells we used the single cell RNA sequencing data from Pollen et al. (2015) to select cells robustly expressing SLC12A2 (>100 counts per million) and those with no SLC12A2 expression. This was done agnostic to cell type. We then used NetworkAnalyst to identify differentially expressed genes in SLC12A2 expressing cells. This identified 589 differentially expressed genes (corrected P-value < 0.05 with > 2-fold difference in expression). Images of networks generated and a full list of enrichment terms is provided in Supplementary Fig. 4.
Given that we were analysing scRNA data from foetal brain, we used tissue-specific (brain cortex) co-expression network analysis to form a network from differentially expressed genes. NetworkAnalyst created a gene co-expression network (Supplementary material). This network was enriched for Reactome terms [e.g. L1CAM interaction and axon guidance (both FDR corrected P = 0.00085)] and Gene Ontology (GO) biological process terms [e.g. axonogenesis (P = 0.0027), neurodevelopment (P = 0.0036)] relevant to neurodevelopment. We then used the label propagation algorithm to identify five modules within the network (Supplementary material). Details of the enrichment analysis of all five modules are provided in the Supplementary material. Module 1 (coloured blue) was the largest (435 genes, Supplementary Fig. 4A). It was enriched for Reactome term ‘L1CAM interaction’ (P = 0.010) and GO biological process terms ‘cytoskeleton dependent intracellular transport’ (P = 0.00005) and ‘synaptic transmission’ (P = 0.00072) as well as Panther biological process term ‘nervous system development’ (P = 0.017). Module 2 (coloured red, 104 genes, Fig. 4B) was enriched for the Reactome term ‘notch-HLH transcription pathway’ (P = 0.0016) and GO biological process terms for ‘neuron formation’ (neuron differentiation, generation of neurons, neurogenesis, nervous system development, all P = 0.01). Modules 3 (coloured white, 123 genes), 4 (coloured green, 117 genes) and 5 (coloured yellow, 123 genes) were not enriched for neurogenesis terms. SCL12A2 expressing cells manifest a transcriptomic programme suggesting an active role in neurogenesis.
Discussion
We describe six individuals with a pleiotropic NDD associated with DNM in SCL12A2. All had developmental delay, ranging from mild to severe. Three had autistic spectrum disorder. In addition, we identified three patients with BLSNHL and no NDD. Two individuals with likely pathogenic variants in SLC12A2 have previously been reported. We described a female patient with a protein-truncating variant in exon 22 of SLC12A2 (Delpire et al., 2016). She presented with lung, gastrointestinal tract, endocrine and exocrine gland deficits with seizure-like episodes and EEG abnormalities, but no hearing impairment. Recently, a 5-year-old male with uniparental disomy for chromosome 5 with a 22 kb deletion of SLC12A2 was described with bilateral sensorineural deafness, global developmental delay and failure to thrive (Macnamara et al., 2019). Our report confirms that SLC12A2 variants are associated with a pleiotropic NDD and exon 21 variants with BLSNHL.
Pleiotropy is well recognized in NDD (McRae et al., 2017). The explanation for pleiotropy associated with SLC12A2 is unclear. The severe phenotype in the boy reported by MacNamara et al. (2019) is likely related to the presence of a homozygous variant in SLC12A2, combined with the effects of uniparental disomy for chromosome 5. Patients 3 and 5 in our series had protein truncating variants and their phenotypes were subjectively more severe than those of the patients with missense variants. A larger series of SLC12A2 variants will be required to confirm any genotype-phenotype correlation. Patient 7 was 47, XYY. The contribution of this is not clear. XYY syndrome has been associated with a mild reduction in IQ (Green et al., 2019). There is no clear association between BLSNHL or tongue fasciculations and XYY syndrome. The cochlea expresses only the SLC12A2 isoform containing exon 21, suggesting exon 21 plays a critical developmental role in cochlea (Dixon et al., 1999) and explaining why exon 21 variants cause BLSNHL. Notably, deafness in the sy mouse is due to an exon 21 Slc12a2 mutation (Dixon et al., 1999). In human foetal brain, significant amounts of the SLC12A2 isoform lacking exon 21 are expressed (Morita et al., 2014). This may compensate for deleterious effects of exon 21 variants and explain why exon 21 variants do not cause an NDD.
In both murine models (Magalhães and Rivera, 2016) and cell systems (Young et al., 2012), loss of Slc12a2/NKCC1 has been shown to inhibit neurogenesis. SLC12A2 is highly expressed in areas of active neurogenesis and SLC12A2 expression is higher in radial glia than in intermediate progenitor cells or neurons at 16–18 weeks gestation. Transcriptomic profiling indicates that SLC12A2 expressing cells (at 16–18 weeks gestation) manifest a transcriptomic programme reflecting active neurogenesis. Reduced function of SLC12A2 may result in an NDD by altering the delicate process of corticogenesis, and/or by dysregulating the excitatory-inhibitory GABA switch.
The phenotype of humans with SLC12A2 variants has similarities to animal models. An slc12a2 null zebrafish displays collapse of the otic vesicle with reduced endolymph (Abbas and Whitfield, 2009). This paper did not describe the brain phenotype. The otic malformation in the zebrafish model has relevance to the hearing loss in SLC12A2 variant carriers. Several Slc12A2 murine models exist. We initially reported an Slc12A2 null mouse with cochlear malformations, loss of hair cells and hearing impairment (Delpire et al., 1999). The brain phenotype in this mouse has not been studied extensively. Reduced neuronal proliferation has been demonstrated in the lateral ganglionic eminence of the null mouse (Magalhães and Rivera, 2016), and NKCC1 knockdown by short-hairpin RNA reduced neuronal proliferation in the murine subventricular zone (Young et al., 2012). The similarities between animal models and the human phenotype of SCL12A2, the role of SLC12A2 in neuronal development, the results of in silico analyses and the effects of the SNVs on NKCC1 biochemical function provide strong support for a causal role of SLC12A2 variants in NDD and BSNHL.
Web resources
https://gnomad.broadinstitute.org/
https://www.networkanalyst.ca/
https://github.com/jeremymcrae/denovonear
https://mupit.icm.jhu.edu/MuPIT_Interactive
Funding
Funding for the project was provided by the Wellcome Trust. We declare that those who collected data and deposited it in the DECIPHER database bear no responsibility for its use and interpretation in the current work. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). Department of Health’s National Institute for Health Research Biomedical Research Centres funding scheme. E.D. is funded by National Institutes of Health grants GM118944, DK093501 and R01DK110375.
Competing interests
The authors report no competing interests.
Supplementary Material
Glossary
- BLSNHL = bilateral sensorineural hearing loss;
- DDD =
deciphering developmental disorders;
- DNM =
de novo mutations;
- NDD =
neurodevelopmental disorders
References
- Abbas L, Whitfield TT.. Nkcc1 (Slc12a2) is required for the regulation of endolymph volume in the otic vesicle and swim bladder volume in the zebrafish larva. Development 2009; 136: 2837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JP, Kahle KT, Gamba G.. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Aspects Med 2013; 34: 288–98. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. The yin and yen of GABA in brain development and operation in health and disease. Front Cell Neurosci 2012; 6: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TA, Orlando BJ, Zhang J, Latorraca NR, Wang A, Hollingsworth SA, et al. Structure and mechanism of the cation-chloride cotransporter NKCC1. Nature 2019; 572: 488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Gagnon KB, Ledford JJ, Wallace JM.. Housing and husbandry of Xenopus laevis affect the quality of oocytes for heterologous expression studies. J Am Assoc Lab Anim Sci 2011; 50: 46–53. [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Lu J, England R, Dull C, Thorne T.. Deafness and imbalance associated with inactivation of the secretory Na- K-2Cl co-transporter. Nat Genet 1999; 22: 192–5. [DOI] [PubMed] [Google Scholar]
- Delpire E, Wolfe L, Flores B, Koumangoye R, Schornak CC, Omer S, et al. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb Mol Case Stud 2016; 2: a001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP.. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet 1999; 8: 1579–84. [DOI] [PubMed] [Google Scholar]
- Green T, Flash S, Reiss AL.. Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. Neuropsychopharmacology 2019; 44: 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Nomura T, Xu J, Contractor A.. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci 2014; 34: 446–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnamara EF, Koehler AE, D’Souza P, Estwick T, Lee P, Vezina G, et al. Kilquist syndrome: a novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum Mutat 2019; 40: 532–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães AC, Rivera C.. NKCC1-deficiency results in abnormal proliferation of neural progenitor cells of the lateral ganglionic eminence. Front Cell Neurosci 2016; 10: 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017; 542: 433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature 2014; 508: 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Callicott JH, Testa LR, Mighdoll MI, Dickinson D, Chen Q, et al. Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and Schizophrenia. J Neurosci 2014; 34: 4929–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niknafs N, Kim D, Kim R, Diekhans M, Ryan M, Stenson PD, et al. MuPIT interactive: webserver for mapping variant positions to annotated, interactive 3D structures. Hum Genet 2013; 132: 1235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E.. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol 1997; 33: 781–95. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, et al. Molecular identity of human outer radial glia during cortical development. Cell 2015; 163: 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum PT, Goodwin AT, Yoshimura H, Kolbe DL, Walls WD, Koh JY, et al. Insights into the biology of hearing and deafness revealed by single-cell RNA sequencing. Cell Rep 2019; 26: 3160–71.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv 2017: 148353. doi.org/10.1101/148353.
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D.. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–6. [DOI] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 2013; 34: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A.. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015; 36: 928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, et al. Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat Genet 2017; 49: 1664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbán N, Guillemot F.. Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci 2014; 8: 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware JS, Samocha KE, Homsy J, Daly MJ.. Interpreting de novo Variation in Human Disease Using denovolyzeR. Curr Protoc Hum Genet 2015; 87: 7.25.1–.25.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, Van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015; 385: 1305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Gill EE, Hancock R.. Networkanalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc 2015; 10: 823–44. [DOI] [PubMed] [Google Scholar]
- Young SZ, Morgan Taylor M, Wu S, Ikeda-Matsuo Y, Kubera C, Bordey A.. NKCC1 knockdown decreases neuron production through GABAA-regulated neural progenitor proliferation and delays dendrite development. J Neurosci 2012; 32: 13630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawerton A, Yao B, Yeager JP, Pippucci T, Haseeb A, Smith JD, et al. De Novo SOX4 variants cause a neurodevelopmental disease associated with mild dysmorphism. Am J Hum Genet 2019; 104: 246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Exome sequencing data from the DDD study are available at the European Genome-Phenome Archive (https://ega-archive.org/studies/EGAS00001000775).