

Abstract
Objective:
To identify clinically relevant predictive biomarkers of trastuzumab resistance.
Material and methods:
MTT, FACS assays, immunoblotting and immunocytochemistry were used to phenotypically characterize drug responses of two cell models BT474R and SKBR3R. Student’s t-test and Spearman’s correlation were applied for statistic analysis.
Results:
The activity of a downstream effector of the HER2 pathway phosphorylated ribosomal protein S6 (p-rpS6), was suppressed by trastuzumab in the parental cell lines yet remained unchanged in the resistant cells following treatment. The level of p-rpS6 was inversely correlated to the drug induced growth inhibition of trastuzumab-resistant cells when they are treated with selected HER2 targeting drugs.
Conclusion:
p-rpS6 is a robust post-treatment indicator of HER2 pathway-targeted therapy resistance.
Keywords: Biomarker, breast cancer, cell model, drug response, trastuzumab resistance
Introduction
Human epidermal growth factor receptor-2 (HER2) is a member of the epidermal growth factor receptor family and is encoded by the proto-oncogene HER2/neu. It is amplified or overexpressed in 15–20% of patients with breast cancer (Chung et al., 2013). This group of patients represents a subcategory of more aggressive tumour phenotypes with higher rates of disease recurrence and worse prognosis (Gonzalez-Angulo et al., 2009; Slamon et al., 1987, 1989; Tripathy, 2009). The development and adoption of the humanized monoclonal antibody trastuzumab (Herceptin) has changed the natural history of HER2+breast cancer and can improve overall and disease free survival, particularly when used in combination with conventional chemotherapy agents (Cobleigh et al., 1999; Pernas et al., 2012; Vogel et al., 2002).
However, the clinical benefit of trastuzumab is limited by de novo and/or acquired resistance which inevitably occurs in the advanced setting (Gianni et al., 2011; Gonzalez-Angulo et al., 2009; Sawaki et al., 2004). Known drivers of resistance include HER2 truncation, up-regulation of HER2 downstream signalling pathways and escape of antibody dependent cellular cytotoxicity (ADCC) (Chung et al., 2013; Nahta, 2012), which has led to the development of additional therapeutics targeting HER2 and its downstream signalling molecules (Lauring et al., 2013). A majority of these drugs are still in developmental phases or undergoing clinical trials, while only a few (lapatinib, everoliums, temsirolimus) have gained FDA approval for clinical use (Parker et al., 2012). Besides limited clinical benefits (Alvarez et al., 2011; Cameron et al., 2010) and toxicity (Sendur et al., 2013), a lack of reliable predictive markers to measure and/or track a patient’s response is one of the major clinical obstacles in breast cancer treatment today (Gonzalez-Angulo & Blumenschein, 2012; La Thangue & Kerr, 2011; Tan et al., 2009).
In breast cancer, several baseline (pre-treatment) biomarkers are used as predictive indices and help guide the choice of treatment. For example, ER/PR receptor status is used to determine whether hormonal therapy should be administered, whereas HER2 expression is used to decide whether trastuzumab or lapatinib-based regimens should be applied. However, for measuring a patient’s response to targeted therapies, we still rely on clinical end points, such as overall survival (OS), progression free survival (PFS) and pathological complete response (pCR) (Ellis et al., 2008).
One widely accepted prognostic marker in breast cancer is Ki67, a nuclear antigen expressed in all phases of the cell cycle except G0. Elevated Ki67 expression of a specimen represents a higher cell proliferation score, which can indicate worse outcome (Dowsett et al., 2011). However, Ki67 expression is not always concordant with clinical outcomes when used as an end point to predict patients’ responses to treatment (Bottini et al., 2000; Jalava et al., 2006; Van Diest et al., 2004; Yoshioka et al., 2013) due to issues such as tissue handling, staining and lack of standardization in scoring (Jalava et al., 2006). Therefore, identifying more accurate and specific indicators of response to HER2-targeted agents is imperative for improving breast cancer treatment outcome. Moreover, as more drugs are being developed and tested for other growth factor pathways, and resistance mechanisms bypassing these pathways are being identified, a more distal biomarker that predicts growth inhibition in preclinical models and clinical benefit in patients is of high priority.
In HER2+breast cancer, overexpression of HER2 often leads to hyper activation of several downstream signalling pathways that regulates cell growth, proliferation and survival. The ribosomal protein S6 (rpS6) is a downstream effector of the HER2 signalling pathway. Its phosphorylation has been shown to associate with several intracellular processes such as protein synthesis, cellular growth and proliferation (Ruvinsky et al., 2005), and plays an important role in cancer malignancy (Chaisuparat et al., 2012; Molinolo et al., 2007).
We hypothesize that the more distal components of the HER2 signalling pathway would correlate most tightly with growth inhibition in trastuzumab-sensitive and resistant cell lines since mechanisms of resistance can bypass the more proximal pathway segments. Therefore, we systematically assessed HER2 signalling pathway mediators in relationship to growth inhibition using trastuzumab-sensitive and resistant cell lines before and after treatment with trastuzumab and other pathway modulating agents. In this study, we identify and characterize phosphor-rpS6 (p-rpS6) expression as a marker of trastuzumab resistance in HER2-overexpressing breast cancer cell models. Specifically, we demonstrate a correlation between p-rpS6 expression levels and response to several targeted agents against HER2 and downstream signalling molecules. Following additional in vivo and clinical validation, p-rpS6 could be used as a potential molecular marker to predict an individual patient’s responsiveness to therapies targeting the HER2 signalling pathway. Ultimately, p-rpS6 activity could help guide the course of treatment and improve outcome in targeted breast cancer therapies for HER2-overexpressing breast cancer.
Material and methods
Cell culture and resistance sublines generation
Two trastuzumab-sensitive HER2+breast cancer cell lines SKBR3 and BT474 obtained from American Type Culture Collections (ATCC, Manassas, VA) were maintained in McCoy-5A and Hybri-Care (ATCC, Manassas, VA) growth medium plus 10% fetal bovine serine (Invitrogen, Carlsbad, CA), 50 units/mL penicillin and 50 μg/mL streptomycin in a 37 °C humidified incubator with 5% CO2. Subcultures were carried out when the cells were ~90% confluent.
To select resistant sublines, SKBR3 and BT474 cells were cultured in the presence of 200 μg/ml of trastuzumab (Genentech, San Francisco, CA) for over 12 months with medium refreshed every 3–4 days to allow the formation of resistant colonies.
Trastuzumab susceptibility and IC50 value determination
During the process of establishing and maintaining trastuzumab-resistant sublines, trastuzumab sensitivity (of selected clones and parental cells) was routinely monitored by MTT (Thiazolyl Blue Tetrazolium Bromide, Sigma, Sigma-Aldrich Corp., St. Louis, MO) assay. Briefly, log phase growing cells were seeded at 3000 cells/well in 96-well culture plates in quadruplicate for 24 h, then exposed to serial dilutions of trastuzumab (0.025–400 μg/ml) for 7 days, followed by an incubation with 5 mg/ml of MTT (Sigma-Aldrich, St. Louis, MO) in growth medium for 2 h. The optical densities (ODs) at 570 nm were detected with a microplate reader (BioTek Instruments, Winooski, VM), background was subtracted using the 690 nm OD readings. IC50 values were calculated using prism 5.0 (GraphPad Software, La Jolla, CA). A resistant subline was defined when its IC50 value surpassed 100 μg/ml (the IC50 values of the parental lines are ~1 μg/ml). Resistant sublines (BT474R and SKBR3R) were maintained in 20 μg/ml trastuzumab and were cultured in the absence of trastuzumab for two passages before each characterization experiment.
Cell proliferation assay
Cell proliferation was analysed by EdU (5-ethynyl-2′-deoxyuridine) incorporation following manufacture’s instruction (Life technologies, Carlsbad, CA). Briefly, cells were seeded onto chamber slides and were treated or untreated with trastuzumab for 24 h, then incubated with EdU for 60 min, fixed, and the Click-it cocktail was added. EdU incorporated cells were counted and recorded with a fluorescent microscope (Nikon, Melville, NY). Experiments were repeated at least three times. For each treatment, three fields were randomly chosen and 300 cells/field were counted. Percentages of EdU+cells were compared among different treatments.
Cell cycle analysis
G0/G1 arrest was examined by cell cycle analysis. Briefly, cells were treated or untreated with trastuzumab for 24 h, followed by trypsinization, cold 70% ethanol fixation, propidium iodide plus RNaseA staining (Sigma-Aldrich), and then read on a FACSCalibur cytometer (BD Biosciences, San Jose, CA). Quantitative analysis of the percentage of cells in the G0/G1, S or G2/M phases of the cell cycle was carried out using the Cell Quest Software (BD Biosciences) at the University of Southern California Flow Cytometry Core.
Immunoblotting
Immunoblotting was carried out to investigate key gene expression alterations of HER2 and downstream signalling pathways. Pelleted cells were washed with cold PBS and lysed with RIPA buffer with proteinase and phosphatase inhibitors (Thermo Scientific Pierce, Rockford, IL). Protein concentrations were determined by Bradford Dye Reagent (BioRad, Hercules, CA). About 10–20 μg/lane of whole cell lysates were separated by a gradient Tris–glycine gel (Life Technologies Corp, Grand Island, NY). Bands were transferred to a nitrocellulose film (BioRad), blocked and probed with targeted primary antibodies (Supplementary Table S1). After binding with secondary antibodies (LiCOR Biosciences), bands were visualized with an Odyssey CLx system and analysed by Image Studio version 2.0 (LiCOR Biosciences).
Immunocytochemistry-immunofluorescence (ICC-IF)
To study the expressions of p-rpS6 and Ki67, ICC-IF was used. Primary antibodies p-rpS6 (cell signalling, Cat# 2211, 1:500 dilution) and Ki67 (Santa Cruz, Cat# sc23900, 1:200 dilution) were incubated with the cells on slides overnight at 4 °C. Stained cells were visualized with a fluorescent microscope (Nikon) and fluorescence intensities were quantified by ImageJ (imagej.nih.gov).
Statistical analysis
Student’s t-test was used to compare the differences of trastuzumab treated and untreated cells in cell proliferation and cell cycle studies. The association between the expression of key effectors in HER2 signalling pathway and HER2-targeted drugs leading to growth inhibition was examined using a Spearman’s correlation. A p value <0.05 was considered statistically significant.
Results
Trastuzumab-resistant breast cancer sublines BT474R and SKBR3R tolerated higher concentrations of trastuzumab treatment compared to parental cells
Two trastuzumab-resistant sublines, designated as BT474R and SKBR3R were derived from the HER+breast cancer cell lines BT474 and SKBR3, respectively, after 1 year of exposure. These cell lines were selected in our study to represent either ER/PR positive (BT474) or negative (SKBR3) breast cancers.
The resistant sublines were first compared to their parental cell counterparts for growth inhibitory response to trastuzumab. Figure 1(a) displays the dose response curves of BT474R, SKBR3R and their parental cell lines, BT474 and SKBR3, to trastuzumab after 7 days of exposure to trastuzumab. BT474R, SKBR3R demonstrated resistance to increasing concentrations of trastuzumab treatments relative to BT474 and SKBR3. The IC50 values for the resistant lines (BT474R=905±105 μg/ml; SKBR3R=1412±206 μg/ml) are >100-fold higher than their corresponding parental lines (BT474=1±0.5 μg/ml; SKBR3=2±0.8 μg/ml) (Figure 1b, Table 1).
Figure 1.
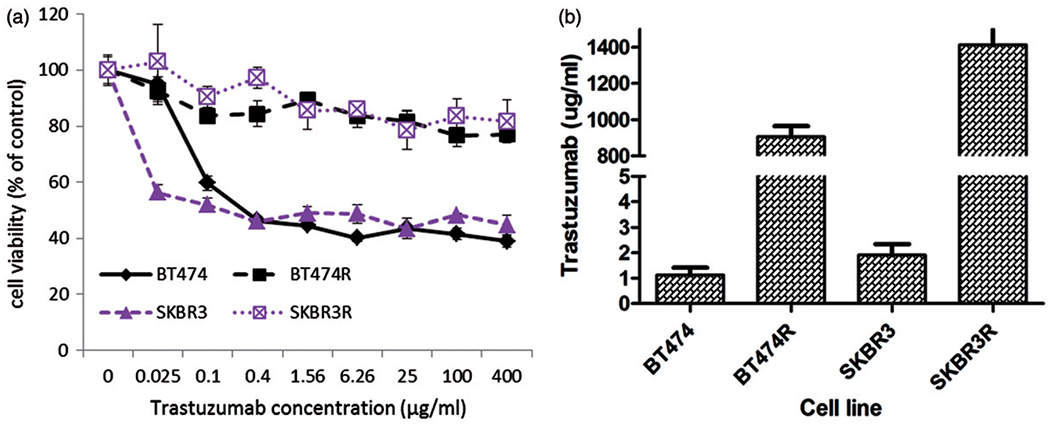
Trastuzumab sensitivities of BT474R, SKBR3R and their parental cell lines BT474 and SKBR3. (a) Dose response curves of the four cell lines after 7 days trastuzumab treatments. Cell viability is the percentage of viable cells with respect to cells untreated with trastuzumab. (b) GraphPad Prism 5 modelled IC50 values of the resistant sublines BT474R and SKBR3R and their parental cells BT474 and SKBR3 to trastuzumab (μg/ml). Bars are means of three independent experiments and standard deviations.
Table 1.
IC50 values of trastuzumab-sensitive and resistant cells to trastuzumab, rapamycin and lapatinib.
| Cell lines | IC50–T (μg/ml) | IC50–R (nM) | IC50–L (nM) |
|---|---|---|---|
| BT474 | 1.1±0.5 | 3.5±1.2 | 48.5±1.1 |
| BT474R | 904.7±104.6 | 5.5±1.2 | 68.0±1.1 |
| SKBR3 | 1.9±0.8 | 4.5±1.2 | 75.1±1.2 |
| SKBR3R | 1412.7±206.1 | 4.6±1.3 | 79.4±1.1 |
T: trastuzumab; R: rapamycin; L: lapatinib.
To determine whether the development of resistance in BT474R and SKBR3R is specific to trastuzumab, we treated these cell lines with two common small molecule inhibitors targeting the HER2 pathway, rapamycin (mTOR/TORC1 inhibitor) and lapatinib (dual EGFR1 and HER2 kinase inhibitor). The dose response curves showed that the resistant cells responded to lapatinib and rapamycin in a near identical manner as the parental cells (Supplementary Figure S1) and had similar IC50 values (Table 1).
We show that the BT474R and SKBR3R sublines acquired trastuzumab resistance after a year under selective pressure from trastuzumab treatment. It has been reported that patients with HER2+breast cancers that initially responded to trastuzumab eventually develop clinical resistance or progression after a medium duration of 7–12 months (Hayashi et al., 2012; Nahta & O’Regan, 2010). Therefore, the time frame in which we used to generate the in vitro trastuzumab resistance models is concordant with what has been clinically reported for the development of trastuzumab resistance.
Increased percentage of proliferating cells and an unaltered cell cycle in the resistant sublines after trastuzumab treatment
When cancer recurs or progresses, the clinical course is usually more aggressive. We therefore assessed the proliferation rate of the resistant cell lines using EdU DNA incorporation. BT474R and SKBR3R cells exhibited an increased DNA synthesis rate compared to their parental BT474 and SKBR3 counterparts (Figure 2a and b). Following treatment with trastuzumab, the parental lines showed a significant reduction in cell proliferation that was not seen in the resistant lines.
Figure 2.
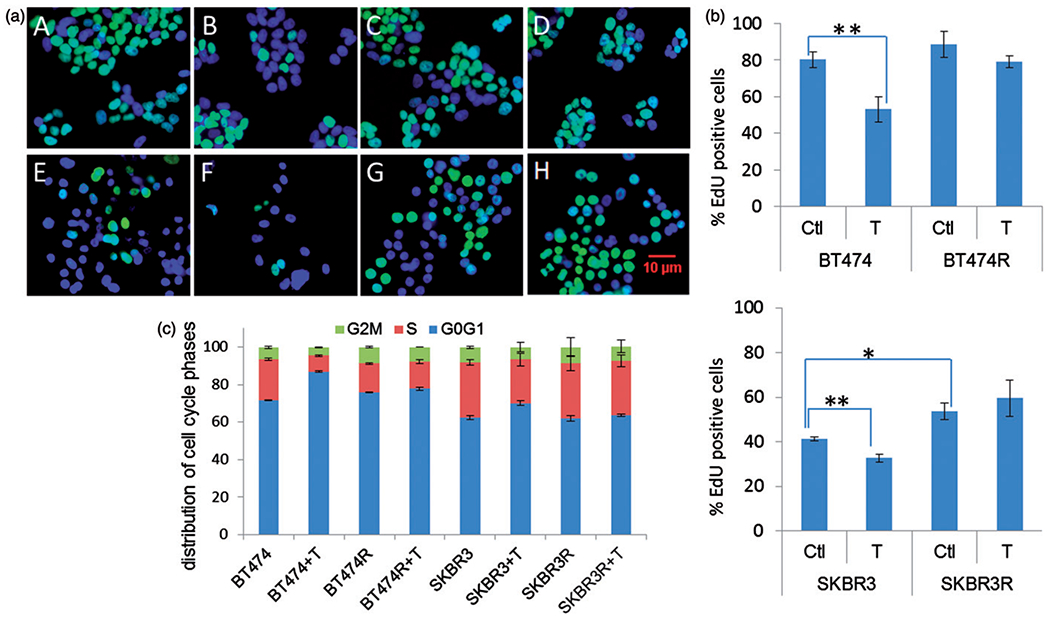
Trastuzumab-resistant cells BT474R and SKBR3R expressed more aggressive phenotypes than parental BT474 and SKBR3. (a) Cell proliferation measured by EdU incorporation. EdU incorporated cells are shown in green. Cell nuclei were stained blue (DAPI). A: BT474, B: BT474+T, C: BT474R, D: BT474R+T, E: SKBR3, F: SKBR3+T, G: SKBR3R, H: SKBR3R+T. (b) Quantification of the data from a Student’s t-test were used to process the data. [* significant (t<0.05); ** most significant (t<0.01)]. Experiments were repeated at least three times. Bars represent mean and standard deviation values. Ctl: control untreated; T: trastuzumab treated. (c) FACS data demonstrates the G0/G1 fraction of the cell cycle was increased and S-phase was decreased after exposing to trastuzumab in BT474 and SKBR3 cells; in the trastuzumab-resistant BT474R and SKBR3R cells, neither G0/G1 or S-phase was affected by trastuzumab exposure.
One of the proposed mechanisms of trastuzumab activity is through inhibition of signal transduction that ultimately leads to G0/G1 arrest and prevents entry into S-phase. Figure 2(c) displays flow-cytometry data showing the cell cycle distribution of trastuzumab-resistant sublines with no significant changes observed in G0/G1 or S fractions after 24 h of trastuzumab treatment (G0/G1 and S, after versus before: 77.8 and 14.6% versus 76.0 and 15.4% in BT474R, 63.6 and 29.0 % versus 62.0 and 29.5% in SKBR3R). In contrast, when the parental BT474 and SKBR3 cells were treated with trastuzumab, the G0/G1 fractions increased (from 71.7 to 87.0% in BT474 and 62.5 to70.2% in SKBR3) and the S1 fractions decreased (from 21.8 to 8.5% in BT474 and 29.4 to 23.3% in SKBR3) (Figure 2c).
Phosphorylated (p-rpS6) was constitutively expressed in BT474R and SKBR3R cells regardless of exposure to trastuzumab
Preclinical and clinical investigations have suggested that deregulation of the HER2 downstream signalling pathways is a principle mechanism of trastuzumab resistance in breast cancers (Chung et al., 2013; Nahta & O’Regan, 2010; Mohd Sharial et al., 2012). In our preclinical cell models, we observed that key proteins in the Akt/mTOR branch of HER2 signalling were differentially expressed (Figure 3a, Supplementary Figure S2). SKBR3 cells expressed less phospho-HER2 (pHER2) than BT474 cells yet the amount of total HER2 was relatively consistent between the lines (Figure 3a). There was no difference in total Akt between the resistant and the parental lines, but phosphorylated Akt (pAkt) expression decreased after 24 h treatment with trastuzumab in parental and resistant cells (Figure 3a), suggesting that pAkt or upstream mediators may not be major effectors in trastuzumab resistance in this model.
Figure 3.
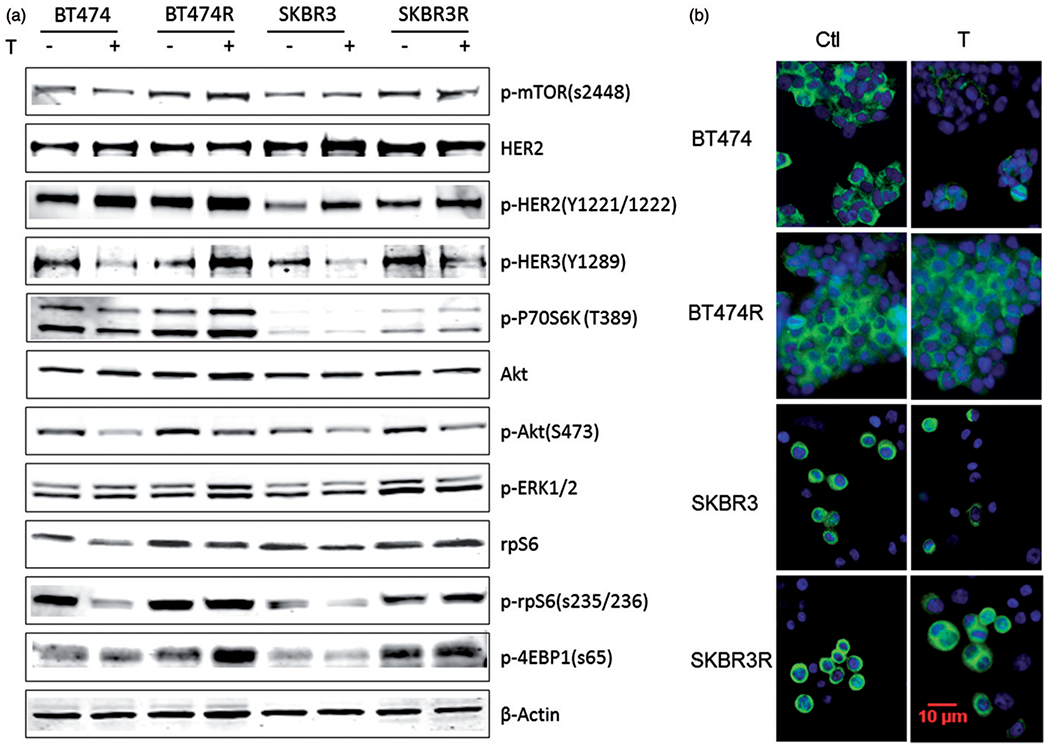
p-rpS6 expression was not suppressed by trastuzumab treatment in trastuzumab-resistant BT474R and SKBR3R cells, but it was decreased in parental BT474 and SKBR3 cells. (a) Immunoblotting of whole cell lysate of key effectors in HER2 signalling pathway demonstrating altered expression of some markers in response to trastuzumab treatment. (b) Immunocytochemistry staining of p-rpS6 in BT474, SKBR3 and trastuzumab-resistant BT474R and SKBR3R cells. P-rpS6 was stained green in the cytoplasm, cell nuclei were stained with DAPI (blue lighter).
rpS6 is one of the downstream effectors of HER2 signalling. It serves as a distal mediator of regulating protein synthesis that leads to cell division, proliferation and growth. In our study, we observed higher expression levels of p-rpS6 in the resistant cells compared to the parental cells (Figure 3a, Supplementary Figure S2). In addition, the levels of p-rpS6 expression remained unchanged in BT474R and SKBR3R cells following trastuzumab treatment. However, in the parental BT474 and SKBR3 cells, p-rpS6 expression was significantly suppressed by trastuzumab treatment (Figure 3a, b, and Supplementary Figure S2).
To further confirm our observation, we performed immunocytochemistry and incubated the parental and resistant cells with a p-rpS6 (s235/236) antibody. In concordance with the immunoblotting results detailed above, we observed more p-rpS6 positive cells with higher florescent intensity among the BT474R and SKBR3R resistant sublines compared to the parental lines (Figure 3b). ICC also confirmed treatment with trastuzumab did not decrease p-rpS6 expression in BT474R and SKBR3R cells, but did so in the parental BT474 and SKBR3 cells (Figure 3b).
Levels of p-rpS6 expression correlated with Ki67 status when these two markers were examined simultaneously
Uncontrolled cell proliferation is a hallmark of malignancy and usually measured by Ki67 antigen staining in both the research and clinic settings (Jalava et al., 2006). Therefore, we were interested in whether p-rpS6 expression levels correlated with Ki67 antigen levels in our models.
Figure 4 shows dual immunofluorescent staining of Ki67 and p-rpS6 in BT474R and SKBR3R and parental BT474 and SKBR3 cells pre- and post-trastuzumab treatment. The expression of p-rpS6 correlated with Ki67 expression in all cells and treatment conditions. In the resistant cells, both p-rpS6 and Ki67 levels remained high regardless of exposure to trastuzumab. Before trastuzumab exposure, the fluorescence intensity of p-rpS6 expression in BT474R was 27.0 and the percentage of Ki67 positive cell was 57.8%. After 24 h of trastuzumab treatment, the numbers did not significantly change: 23.0% for p-rpS6 and 57.0% for Ki67. The same trend was seen in SKBR3R, the numbers were 17.0 and 66.9% before treatment, 17.5 and 69.0% after treatment. However, in BT474 and SKBR3 cells, p-rpS6 and Ki67 expression decreased significantly after treatment (Figure 4).
Figure 4.
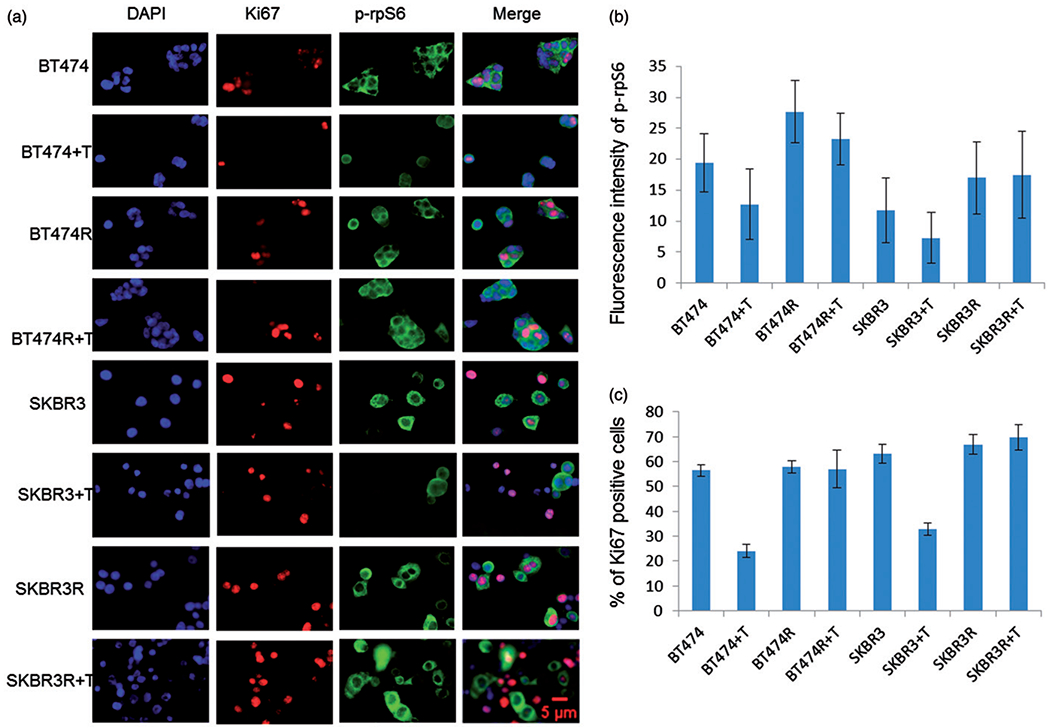
Higher intensity of p-rpS6 stain is correlated with a higher percentage of Ki67 positive cell numbers. (a) Dual staining of p-rpS6 (green in the cytoplasm) and Ki67 (red in the nuclei) of BT474R, SKBR3R treated or untreated with trastuzumab in comparison with their parental cells BT474 and SKBR3. (b) Quantified fluorescence intensity data from a fluorescence intensity of p-rpS6 was measured with imageJ, Ki67 positive cells were manually counted.
Clearly, trastuzumab treatment markedly suppressed p-rpS6 expression and simultaneously decreased the number of proliferating cells in trastuzumab-sensitive BT474 and SKBR3 cells; in trastuzumab-resistant BT474R and SKBR3R cells, the drug failed to suppress p-rpS6 expression as well as Ki67 expression.
Decreased p-rpS6 expression after drug treatments indicates drug response; constitutive p-rpS6 expression indicates drug resistance in BT474R and SKBR3R cells
We assessed the role of p-rpS6 as a predictive biomarker for sensitivity to HER2-targeted therapy using a panel of agents (Selleck Chemicals, Houston, TX) that target HER2 and downstream signalling pathway (Table 2). We first assessed the expression of p-rpS6 and other mediators in BT474R and SKBR3R treated with the drugs listed in Table 2. Immunoblotting showed differential expressions of these proteins associated with different drug treatments and responses (Figure 5a and c). We then tested the growth inhibition of these drugs after 5 days of treatment (Figure 5b and c).
Table 2.
HER2 signalling pathway inhibitor.
| Inhibitor (initial) | Rapamycin (R) | AZD2014 (A) | BEZ235 (B) | Erotinib (E) | Lapatinib (L) | MK2206 (M) | OSI-906 (O) |
|---|---|---|---|---|---|---|---|
| Target | mTOR | mTOR | PI3K/mTOR | EGFR | EGFR/HER2 | Akt1, Akt2, Akt3 | IGFR/IR |
| IC50a | 0.10nM | 335nM | 43.1nM | 551nM | 7.30nM | 250nM | 437nM |
| Concentration used | 10nM | 1 μM | 1μM | 1μM | 1μM | 1μM | 1μM |
IC50 values were obtained using inhibitors on SKBR3R cells.
Figure 5.
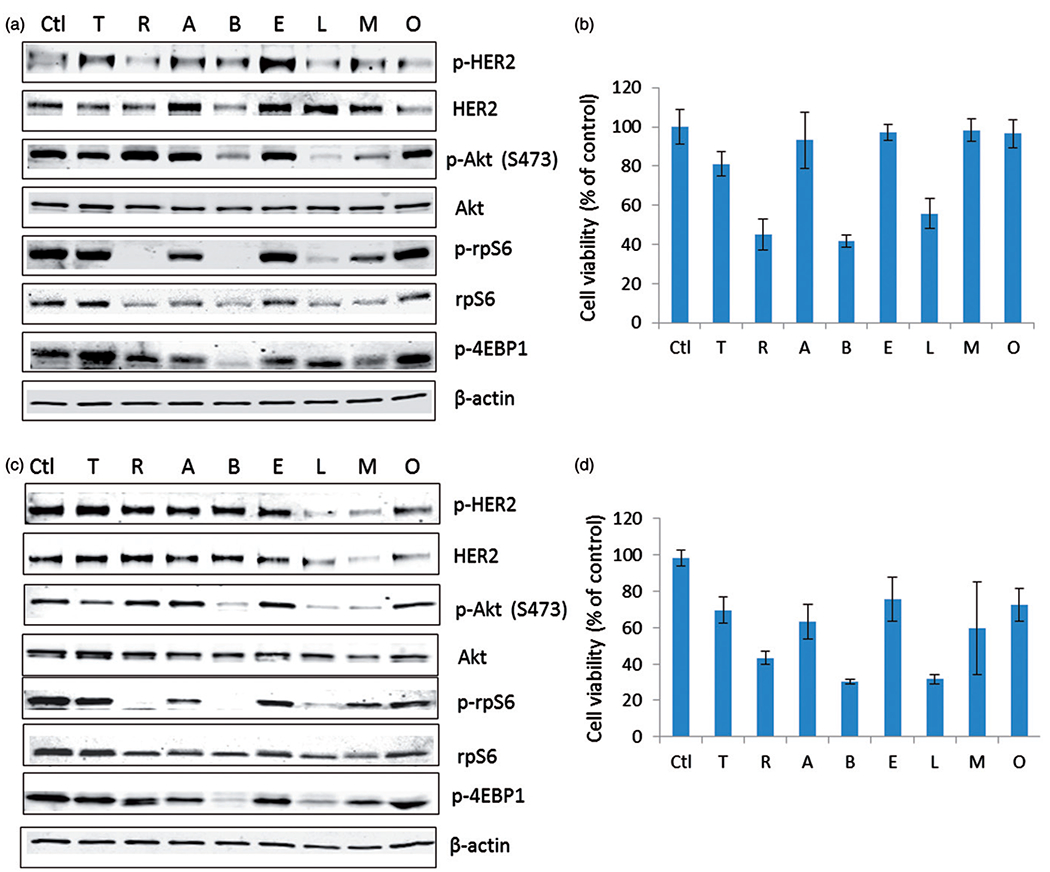
Decreased p-rpS6 expression correlates with the sensitivity of cells to HER2 signalling targeted drugs in BT474R and SKBR3R cells (r=0.8). (a) Immunoblotting of key proteins of HER2 signalling pathway in BT474R cells. (b) Growth inhibition of BT474R by HER2 signalling targeted drugs. Ctl: untreated; T: trastuzumab; R: rapamycin; A: AZD2014; B: BEZ235; E: erotinib; L: lapatinib; M: MK2206; O: OSI-906 (linsitinib). (c) Immunoblotting of key proteins of HER2 signalling pathway in SKBR3R cells. (d) Growth inhibition of SKBR3R by HER2 signalling targeted drugs.
The drugs that caused the greatest degree of growth inhibition in the trastuzumab-resistant cell sublines, such as lapatinib, rapamycin and BEZ235, also significantly suppressed p-rpS6 expressions in these cells with a tighter correlation than other pathway markers (Figure 5, Supplementary Figure S3). Those that did not inhibit BT474R and SKBR3R growth (trastuzumab, erotinib, MK2206 and OSI-906[linsitinib]) also failed to suppress p-rpS6 expression. The correlation between p-rpS6 expression and cell viability after drug treatment is statistically significant in both BT474R and SKBR3R cells (Table 3, r = 0.78, p = 0.013 for BT474R and r = 0.88, p = 0.002 for SKBR3R). These results point to p-rpS6 as the most predictive downstream biomarker of drug response in trastuzumab-resistant cells.
Table 3.
Correlation between marker expression and cell viability post-HER2 pathway-targeted drug treatment in trastuzumab-resistant cell models.
| Cell model | Marker | Correlation coefficienta | p Value |
|---|---|---|---|
| pERK1/2 | 0.13 | 0.73 | |
| pAkt | 0.17 | 0.67 | |
| BT474R | p-rpS6 | 0.78 | 0.013 |
| pS6K1 | 0.73 | 0.025 | |
| p-mTOR | 0.60 | 0.088 | |
| pHER2 | 0.067 | 0.86 | |
| p4EBP1 | 0.20 | 0.61 | |
| pERK1/2 | −0.05 | 0.9 | |
| pAkt | 0.35 | 0.36 | |
| SKBR3R | p-rpS6 | 0.88 | 0.002 |
| pS6K1 | 0.64 | 0.061 | |
| p-mTOR | 0.62 | 0.077 | |
| pHER2 | <0.01 | 0.99 | |
| p4EBP1 | 0.72 | 0.030 | |
Spearman correlation coefficient.
Synergistic effect of trastuzumab and lapatinib was observed with growth inhibition as well as p-rpS6 expression in trastuzumab-resistant cell models
Lapatinib (Tykerb, GlaxosSmithKline, Philadelphia, PA) is a dual EGFR/HER2 adenosine triphosphate competitive, small molecule tyrosine kinase inhibitor (Rusnak & Gilmer, 2011). It was hypothesized that dual blockade of HER2 with trastuzumab and lapatinib is more effective than either single therapy alone. Indeed, clinical data has shown combinatorial benefit from using trastuzumab and lapatinib in HER2-overexpressing breast cancers, exhibiting synergistic or additive effects (Baselga et al., 2012; Blackwell et al., 2012; Scaltriti et al., 2012).
In our preclinical models, we observed the combined growth inhibition of trastuzumab and lapatinib in the trastuzumab-resistant BT474R and SKBR3R cells (Figure 6). In BT474R cells, 7 days of trastuzumab treatment resulted in ~15% growth inhibition while lapatinib caused ~28% growth inhibition. However, when trastuzumab and lapatinib treatment were combined, the growth inhibition rate rose to ~51%. By using the formula developed by Colby (Colby, 1967), synergy was shown [(expected growth inhibition (E, 39%) < actual inhibition (A, 51%)]. The synergistic effect between trastuzumab and lapatinib was seen in SKBR3R cells as well (E, 49% < A, 61%). p-rpS6 expression levels in BT474R cells and SKBR3R cells remained unchanged following trastuzumab treatment, decreased slightly with lapatinib, and was significantly reduced with the combined treatment (Figure 6a and b); again, demonstrating an inverse correlation between the degree of growth inhibition when targeting HER2.
Figure 6.
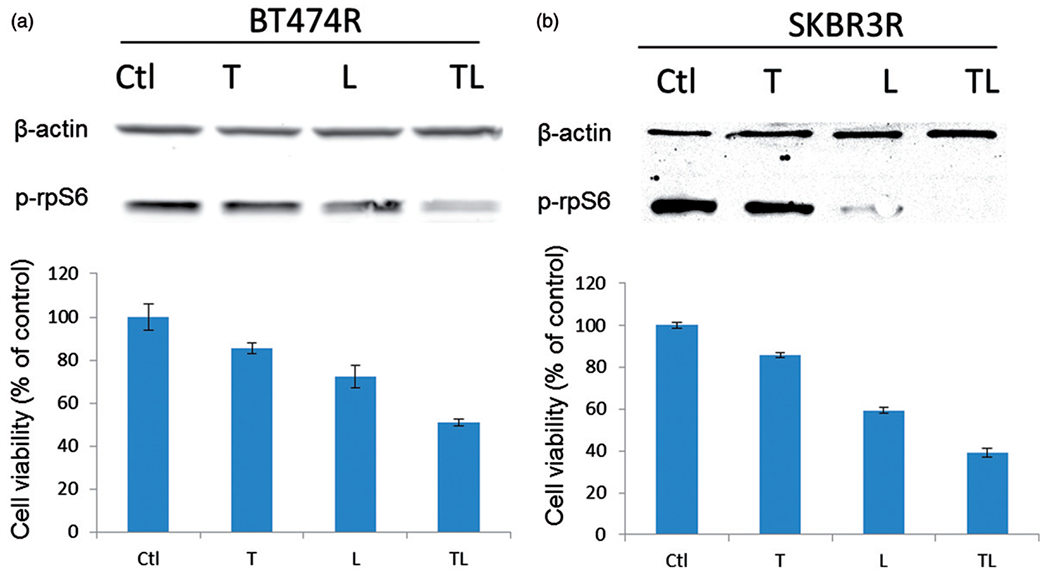
Synergistic effect of trastuzumab and lapatinib on growth inhibition (cell viability shown with bars) as well as p-rpS6 expression (shown with western blot bands on the top) of BT474R (a) and SKBR3R (b). Synergism was determined by Colby methods (Colby, 1967). T: trastuzumab; L: lapatinib; TL: trastuzumab +lapatinib.
Discussion
Predictive biomarkers are used to assess the probability that a patient will benefit from a particular treatment (Sawyers, 2008). The ones that best define of responsiveness to newer HER2-targeted agents and combinations need to be identified and validated to better select those likely to be successful for HER2+treatment-refractory breast cancers in the clinic. Mechanisms of resistance to growth factor pathway-targeted drugs are diverse and poorly understood but appear to involve bypass loops. Therefore, distal pathway readouts that correlate to cell proliferation and survival may represent the optimal pre- and post-treatment biomarkers to inform the development of novel targeted agents. In this report, we generated trastuzumab-resistant cell lines to better understand the mechanisms leading to resistance to targeted therapies in HER2+breast cancer. We further demonstrated that p-rpS6 most strongly corresponds with treatment response of trastuzumab and is a downstream effector of the HER2 signalling pathway. This is consistent with its key role as a master switch of protein synthesis regulation, a fundamental process that leads to cell division, proliferation and growth (Chaudhuri & Lieberman, 1968; Hagner et al., 2010; Holz 2012; Nielsen et al., 1982).
A deregulated HER2/Akt/mTOR pathway has been reported to be one of the major drivers of trastuzumab resistance (Chung et al., 2013). With our models, we were able to demonstrate altered protein expressions of key effectors in this pathway (Figure 3a, Supplementary Figure S2), especially the phosphorylated proteins, such as p-mTOR, p70S6K. p-rpS6, p-4EPB1, pHER2, the indicators of HER2 signalling activation. In fact, studies have shown activation of signalling pathways including HER2/PI3K/mTOR pathway often confers resistance to drugs. In breast cancer, PI3K/Akt/mTOR is the most frequently activated signalling pathway (Ciruelos Gio, 2014), supportive our observation that the increased expressions of phosphorylated proteins in this branch of HER2 signalling. Furthermore, using selected HER2 targeting drugs to treat the resistant cells, we found that drugs that remained potent to these cells are the ones targeting the mTOR and its effectors, such as rapamycin and BEZ235 (Figure 5). Considering the regulatory role of mTOR in cell growth, proliferation and survival, it is not hard to comprehend the benefit of targeting this branch of HER2 signalling. New clinical data has provided evidences (Ciruelos Gio, 2014; Jerusalem et al., 2014). As a direct effector of mTOR pathway, p-rpS6 holds the potential for being used clinically with further preclinical and clinical validation.
When HER2+breast cancer patients develop trastuzumab resistance, dual targeting thought to be a strategy to improve the effectiveness of trastuzumab or target cells that have developed resistance. Reported preclinical and clinical evidence demonstrated that dual blockdale has great advantage over trastuzumab alone (Lavaud & Andre, 2014; Kontzoglou et al., 2013; Kiimler et al., 2013). However, certain population of patients did not benefit from this regime. Therefore, there is an urgent need for better biomarkers to identify patients who will benefit from this therapy (Kümler et al., 2013). Our study showed that p-rpS6 expression was decreased as trastuzumab-resistant cells experiencing growth inhibition when they were treated with the combination of trastuzumab and lapatinib to target both intra and inter cellular domains of HER2 (Figure 6), exemplified the potential feasibility of p-rpS6 as an indicator for dual targeting therapy in trastuzumab resistant, HER2+breast cancers.
HER2-targeted therapies have made personalized medicine a reality in breast cancer management. However, the discorrelation of pathway alterations and outcome/treatment responses adds a layer of difficulty in consideration for patient and inhibitors selection (Ciruelos Gio, 2014). It is largely due to the disconnection of pathway alteration measures and response measures. Current common measures for pathway alteration are biomarkers at the molecular level, while outcome and treatment responses are majorly based on clinical data such as PCR, OS, PFS, etc. These measurements are taken over the course of months in which patients with de novo resistance may be exposed to unnecessary side effects and miss an earlier opportunity to change therapy—a paradigm that is currently being tested in the clinic.
A molecular marker, Ki67, is commonly used clinically to measure drug responses. Besides technical challenges, such as fixation and tissue storage, which make it difficult to accurately measure Ki67 expression in tissue (Kontzoglou et al., 2013), the nuclear antigen Ki67 is a proliferation marker. Its change can be a result of a variety of cellular processes. Here we suggest a more specific molecule, p-rpS6, a distal effector of HER2 signalling pathway, whose activity is directly related to cellular response of pathway inhibition. Furthermore, changes in p-rpS6 happen on a much shorter time scale, which makes it an ideal marker for tracking a patient’s response. In our preclinical cell line models, we were able to resolve expression changes within 24 h post-treatment (Supplementary Figure S4). p-rpS6 can be detected using immunoblotting techniques, which tend to have fewer issues with sample processing, analysis, standardization and interpretation. A recent report used IHC techniques to probe for p-rpS6 in mouse tissue to examine a connection between rpS6 phosphorylation, DNA damage, and tumour suppression during the development of pancreatic cancer (Khalaileh et al., 2013). This work supports the technical feasibility of measuring p-rpS6 as a predictive readout of cancer treatment response.
Conclusion
Our characterization of the trastuzumab-resistant BT474R and SKBR3R cell lines revealed (i) constitutive p-rpS6 expression in both pre- and post-trastuzumab treated resistant cells; (ii) an inverse correlation between p-rpS6 expression and cell viability after treatment with several HER2-targeted signalling drugs; (iii) a synergistic effect between trastuzumab and lapatinib combination therapy that resulted in stronger growth inhibition and suppression of p-rpS6 expression than was detected with either agent alone; and (iv) a correlation between p-rpS6 expression levels and the proliferation marker, Ki67. These results suggest that the expression level of p-rpS6 is an accurate indicator of response to HER2-targeted drugs whereby decreased p-rpS6 level indicates drug response and unchanged p-rpS6 predicts drug resistance.
Using our cell line models, we provide empirical evidence to better understand trastuzumab resistance mechanisms in HER2-overexpressing tumours. Furthermore, our models represent both ER/PR hormone receptor positive and negative breast cancers that will allow for p-rpS6 to be used as a marker in a broader cohort of patients. This work has identified a novel marker that could greatly improve clinical benefit from HER2-targeted treatment strategies, although it will require further validation using in vivo animal models and/or patient tissue samples before its application in the clinic.
Supplementary Material
Acknowledgments
This work is supported by Norris Cancer Centre, Women’s Cancer Program of the University of Southern California and was supported in part by award number P30CA015089 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Alvarez RH, Guarneri V, Icli F, et al. (2011). Bevacizumab treatment for advanced breast cancer. Oncologist 16:1684–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Bradbury I, Eidtmann H, et al. (2012). Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet 379: 633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell KL, Burstein HJ, Storniolo AM, et al. (2012). Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J Clin Oncol 30:2585–92. [DOI] [PubMed] [Google Scholar]
- Bottini A, Berruti A, Bersiga A, et al. (2000). p53 but not bcl-2 immunostaining is predictive of poor clinical complete response to primary chemotherapy in breast cancer patients. Clin Cancer Res 6:2751–8. [PubMed] [Google Scholar]
- Cameron D, Casey M, Oliva C, et al. (2010). Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: final survival analysis of a phase III randomized trial. Oncologist 15:924–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisuparat R, Rojanawatsirivej S, Yodsanga S. (2012). Ribosomal protein S6 phosphorylation is associated with epithelial dysplasia and squamous cell carcinoma of the oral cavity. Pathol Oncol Res 19: 189–93. [DOI] [PubMed] [Google Scholar]
- Chaudhuri S, Lieberman I. (1968). Control of ribosome syntesis in normal and regenerating liver. J Biol Chem 243:29–33. [PubMed] [Google Scholar]
- Chung A, Cui X, Audeh W, Giuliano A. (2013). Current status of anti-human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer 13:223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruelos Gio EM. (2014). Targeting PI3K/AKT/mTOR pathway in estrogen receptor positive breast cancer. Cancer Treat Rev 40: 862–71. [DOI] [PubMed] [Google Scholar]
- Cobleigh MA, Vogel CL, Tripathy D, et al. (1999). Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol 17:2639–48. [DOI] [PubMed] [Google Scholar]
- Colby SR. (1967). Calculating synergistic and antagonistic responses of herbicide combinations. Weeds 15:20–2. [Google Scholar]
- Dowsett M, Nielsen TO, Hern RA, et al. (2011). Assessment of Ki67 in Breast Cancer. Recommendations from the international Ki67 in breast cancer working group. J Natl Cancer Inst 103:1656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Tao Y, Luo J, et al. (2008). Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 100:1380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni L, Dafni U, Gelber RD, et al. (2011). Treatment with trastuzumab for 1 year after adjuvant chemotherapy in patients with HER2-positive early breast cancer: a 4-year follow-up of a randomized controlled trial. Lancet Oncol 12:236–44. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Blumenschein GR Jr. (2012). Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat Rev 39:313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Litton JK, Broglio KR, et al. (2009). High risk of recurrence for patients with breast cancer who have human epidermal growth factor receptor 2-positive, node-negative tumors 1 cm or smaller. J Clin Oncol 27:5700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Litton JK, Broglio KR, et al. (2009). High risk of recurrence for patients with breast cancer who have human epidermal growth factor receptor 2-positive, node-negative tumours 1 cm or smaller. J Clin Oncol 27:5700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagner PR, Schneider A, Gartenhaus RB. (2010). Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood 18;115:2127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Okumura Y, Osako T, et al. (2012). Time to first tumor progression as a predictor of efficacy of continued treatment with trastuzumab beyond progression in human epidermal growth factor receptor 2-positive metastatic breast cancer. Front Oncol 2:62. [DOI] [PubMed] [Google Scholar]
- Holz MK. (2012). The role of S6K1 in ER-positive breast cancer. Cell Cycle 11:3159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalava P, Kuopio T, Juntti-Patinen L, et al. (2006). Ki67 immunohistochemistry: a valuable marker in prognostication but with a risk of misclassification: proliferation subgroups formed based on Ki67 immunoreactivity and standardized mitotic index. Histopathology 48:674–82. [DOI] [PubMed] [Google Scholar]
- Jerusalem G, Rorive A, Gollignon J. ( 2014). Use of mTOR inhibitors in the treatment of breast cancer: and evaluation of factors that influence patient outcomes. Breast Cancer 6:43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümler I, Tuxen MK, Nielsen DL. (2013). A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev 40: 259–70. [DOI] [PubMed] [Google Scholar]
- Khalaileh A, Dreazen A, Khatib A, et al. (2013). Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res 73:1811–20. [DOI] [PubMed] [Google Scholar]
- Kontzoglou K, Palla V, Karaolanis G, et al. (2013). Correlation between Ki67 and breast cancer prognosis. Oncology 84:219–25. [DOI] [PubMed] [Google Scholar]
- La Thangue NB, Kerr DJ. (2011). Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat Rev Clin Oncol 23;8:587–96. [DOI] [PubMed] [Google Scholar]
- Lauring J, Park BH, Wolff AC. (2013). The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw 11:670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavaud P, Andre F. (2014). Strategies to overcome trastuzumab resistance in HER2-overexpressing breast cancers: focus on new data from clinical trials. BMC Med 12:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd Sharial MS, Crown J, Hennessy BT. (2012). Overcoming resistance and restoring sensitivity to HER2-targeted therapies in breast cancer. Ann Oncol 23:3007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinolo AA, Hewitt SM, Amornphimoltham P, et al. (2007). Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res 13:4964–73. [DOI] [PubMed] [Google Scholar]
- Nahta R (2012). Molecular mechanisms of trastuzumab-based treatment in HER2-overexpressing breast cancer. Oncol 2012:428062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahta R, O’Regan RM. (2010). Evolving strategies for overcoming resistance to HER2-directed therapy: targeting the PI3K/Akt/mTOR pathway. Clin Breast Cancer 10(Suppl. 3):S72–8. [DOI] [PubMed] [Google Scholar]
- Nielsen PJ, Manchester KL, Towbin H, et al. (1982). The phosphorylation of ribosomal protein S6 in rat tissues following cycloheximide injection, in diabetes, and after denervation of diaphragm. A simple immunological determination of the extent of S6 phosphorylation on protein blots. J Biol Chem 257:12316–21. [PubMed] [Google Scholar]
- Parker JL, Lushina N, Bal PS, et al. (2012). Impact of biomarkers on clinical trial risk in breast cancer. Breast Cancer Res Treat 136: 179–85. [DOI] [PubMed] [Google Scholar]
- Pernas S, Gil-Gil M, de Olza MO, et al. (2012). Efficacy and safety of concurrent trastuzumab plus weekly paclitaxel-FEC as primary therapy for HER2-positivebreast cancer in everyday clinical practice. Breast Cancer Res Treat 134:1161–8. [DOI] [PubMed] [Google Scholar]
- Rusnak D, Gilmer TM. (2011). The discovery of lapatinib (GW572016). Mol Cancer Ther 10:2019. doi: 10.1158/1535-7163.MCT-11-0697. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Sharon N, Lerer T, et al. (2005). Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 19:2199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaki M, Ito Y, Tada K, et al. (2004). Efficacy and safety of trastuzumab as a single agent in heavily pretreated patients with HER-2/neu-overexpressing metastatic breast cancer. Tumour 90: 40–3. [DOI] [PubMed] [Google Scholar]
- Sawyers CL. (2008). The cancer biomarker problem. Nature 452: 548–52. [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Chandarlapaty S, Prudkin L, et al. (2012). Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res 16:2688–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendur MA, Aksoy S, Altundag K. (2013). Cardiotoxicity of novel HER2-targeted therapies. Curr Med Res Opin 29:1015–24. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, et al. (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/ neu oncogene. Science 235:177–82. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, et al. (1989). Studies of the HER-2/ neu proto-oncogene in human breast and ovarian cancer. Science 244: 707–12. [DOI] [PubMed] [Google Scholar]
- Tan DS, Thomas GV, Garrett MD, et al. (2009). Biomarker-driven early clinical trials in oncology: a paradigm shift in drug development. Cancer J 15:406–20. [DOI] [PubMed] [Google Scholar]
- Tripathy D (2009). HER2 status and breast cancer therapy: recent advances. F1000 Med Rep 1:20. doi: 10.3410/M1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Diest PJ, van der Wall E, Baak JP. (2004). Prognostic value of proliferation in invasive breast cancer: a review. J Clin Pathol 57: 675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CL, Cobleigh MA, Tripathy D, et al. (2002). Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 20:719–26. [DOI] [PubMed] [Google Scholar]
- Yoshioka T, Hosoda M, Yamamoto M, et al. (2013). Prognostic significance of pathologic complete response and Ki67 expression after neoadjuvant chemotherapy in breast cancer. Breast Cancer 22: 185–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.