

Abstract
Improving the low-temperature activity (below 100 °C) and noble-metal efficiency of automotive exhaust catalysts has been a continuous effort to eliminate cold-start emissions, yet great challenges remain. Here we report a strategy to activate the low-temperature performance of Pt catalysts on Cu-modified CeO2 supports based on redox-coupled atomic layer deposition. The interfacial reducibility and structure of composite catalysts have been precisely tuned by oxide doping and accurate control of Pt size. Cu-modified CeO2-supported Pt sub-nanoclusters demonstrate a remarkable performance with an onset of CO oxidation reactivity below room temperature, which is one order of magnitude more active than atomically-dispersed Pt catalysts. The Cu-O-Ce site with activated lattice oxygen anchors deposited Pt sub-nanoclusters, leading to a moderate CO adsorption strength at the interface that facilitates the low-temperature CO oxidation performance.
Subject terms: Heterogeneous catalysis, Pollution remediation, Materials for energy and catalysis, Synthesis and processing
Improving low-temperature activity and noble-metal efficiency remains a challenge for next generation exhaust catalysts. Here, the authors achieve the activation of subnanometric Pt on Cu-modified CeO2 for low-temperature CO oxidation with an onset below room temperature.
Introduction
The fuel economy of vehicles has been greatly improved with the development of turbocharged engines, exhaust heat recovery systems and hybrid powertrains1,2. Due to the much lower exhaust temperature in these vehicles, aftertreatment catalysts must work efficiently at low temperatures for the recirculation usage of exhaust gas. The general consensus is that the next generation catalyst must be active in the sub 100 °C range to reduce or eliminate cold-start emissions3–5. To meet the requirement for >90% abatement of emissions (CO, HC, and NOx) at 150 °C6, the challenge is to develop highly efficient catalysts at low temperatures. Despite considerable progress made in low-temperature exhaust catalysts, such as Au nanoparticles and Co3O47,8, the harsh working environment of automotive catalysts would severely deteriorate the stability of such nanocatalysts. By far, Pt is still the most widely used catalyst component in the exhaust cleaning system due to its excellent reactivity and chemical stability9.
Since the discovery of the strong metal–support interaction, there has been an explosion of investigations on reducible oxide-supported Pt catalysts to enhance the low-temperature activity, such as Pt/CeO2, Pt/FeOx, and Pt/Co3O410–14. It has been demonstrated that the size of supported Pt catalysts is a key factor that affects the proportion of the interfacial active sites and interfacial interactions, directly correlating with the activity of Pt/oxide catalysts15. Pt nanoparticles with sizes around 1.6 nm on CeO2 supports have been reported to exhibit a turnover frequency (TOF) of 0.2 s−1 at 80 °C for CO oxidation, much greater than larger Pt nanoparticles with the same Pt mass loading16–18. To further increase the utilization rate of the Pt component, supported single atoms and sub-nanocluster catalysts have been extensively investigated due to their higher atomic efficiency19–21. While the atomically dispersed Pt atoms on oxide supports have been shown to exhibit higher activity than supported Pt nanoparticle catalysts in some reports22,23, it is still under heated debate as a number of studies pointed out that Pt single atoms and sub-nanoclusters are merely bystanders for low-temperature reactions in similar catalytic systems24–26. Despite researcher efforts to promote CeO2 supported Pt single atom catalysts using high temperature steam treatment, heteroatom doping and hydrogen thermal pretreatment, activities of such systems are nonetheless inferior to the activity of state-of-the-art Pt/CeO2 catalysts with Pt nanocluster sizes in the range of 1.2–1.6 nm5,13,16,27,28. In order to further improve the atomic efficiency, the highly efficient Pt subnanometric catalysts supported on CeO2 have been reported, which have shown enhanced low-temperature activity compared with atomically dispersed Pt catalysts29,30. It has also been reported that the catalytic activity of supported Pt single atoms and sub-nanoclusters is dependent on the dynamically evolving interfacial structures under reaction conditions31–33. At this juncture, a precise control of interfacial structures would have considerable potential in the development of highly efficient low-temperature catalysts. Recently, our group achieved such control of the interfacial structures of supported Pt nanoparticles and sub-nanoclusters by an atomic layer deposition (ALD) method34,35. The ALD method has also been successfully employed to prepare supported single atom catalysts by preventing adsorbed atoms on the supports from aggregating36,37. Nevertheless, since the deposition and diffusion of Pt atoms on oxide supports is sensitive to oxide surface structure and ALD protocols, a major challenge is the simultaneous control of the interfacial reducibility and structures for supported Pt catalysts, especially in the size range of sub-nanometer clusters to single atoms.
Here we report a strategy to precisely control interfacial structures of CeO2 supported Pt catalysts by coupling oxide doping and Pt size control via a redox-coupled ALD method. The remarkable performance with an onset of CO oxidation reactivity below room temperature and turnover frequency (TOF) of 0.26 s−1 at 80 °C is demonstrated for the Cu-modified CeO2 supported Pt sub-nanoclusters. Both experimental and theoretical results reveal the finely constructed interface composed of Cu–O–Ce site and with deposited Pt sub-nanoclusters contributes to the bifunctional active site, with enhanced lattice oxygen activity and moderate CO adsorption strength activating the low-temperature catalytic performance.
Results
Structural characterizations of catalysts
In our experiment we started from the CeO2 nanorod supports with exposed controllable facets of (220) and (200) (see Supplementary Fig. 1 in Supplementary Information), which were prepared by a hydrothermal method as reported in our previous study38. As illustrated in Fig. 1a, Cu dopants were introduced into CeO2 supports (denoted as Ce0.99Cu0.01O2) to tune the surface reducibility and to anchor Pt precursors during the ALD process. The atomically dispersed Pt catalysts were deposited on two supports via regular Pt ALD with the sequence of MeCpPtMe3-O2, which were denoted as Pt1/Ce and Pt1/CeCu. Notably, the Pt mass loading of Pt1/CeCu (1.51 wt%) is much larger than that of Pt1/Ce (0.63 wt%), due to the larger density of surface defects in Cu-doped samples for anchoring Pt precursors (Supplementary Fig. 2). The Cu K-edge X-ray absorption near edge structure (XANES) spectrum of Pt1/CeCu (Supplementary Fig. 3) shows that Cu dopants in Cu2+ state are atomically dispersed in ceria support, in good agreement with previous studies39. Meanwhile, the absence of signals in the Cu 2p spectra indicates that no segregated CuOx species are formed in CeO2 supports due to the low Cu concentration (0.20 wt% determined by ICP-OES). In order to fine tune the size of Pt, a redox-coupled ALD recipe of Pt with the sequence MeCpPtMe3–O2–H2 was performed on two supports (denoted as Ptn/Ce and Ptn/CeCu), utilizing the aggregation of Pt atoms under a reduced atmosphere40.
Fig. 1. Preparation processes and electron microscopy characterizations of catalysts.

a Schematic illustration of preparation processes for supported Pt catalysts, with Ce, O, Pt, and Cu atoms shown as Red, Cyan, Green, and Purple, respectively. HAADF-STEM images of (b) Pt1/Ce, (c) Ptn/Ce, (d) Pt1/CeCu, and (e) Ptn/CeCu. Inserts in (c) and (e) show HAADF-STEM images of the Ptn/Ce and Ptn/CeCu interfaces. Atomically dispersed Pt atoms are marked in (b) and (d) by white circles. f EDS analysis of Ptn/CeCu shows the locations of Ce, O, Cu, and Pt.
The morphology of prepared Pt catalysts is characterized by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The atomically dispersed Pt atoms (white circles) are shown for Pt1/Ce in Fig. 1b, which is consistent with the 2097 cm−1 peak detected in diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) spectrum of CO adsorption (Supplementary Fig. 4). Applying the same ALD process to Pt1/Ce, the HAADF-STEM image of Pt1/CeCu also shows atomically dispersed Pt atoms (Fig. 1d). However, the peak at 2097 cm−1 in the DRIFTS spectrum of CO adsorption on Pt1/CeCu is greater than that of Pt1/Ce (Supplementary Fig. 4), which can be attributed to the larger Pt mass loading of Pt1/CeCu induced by surface Cu dopants. The numbers of Pt atoms per unit area are also consistent with that calculated by the Pt mass loading of Pt1/Ce and Pt1/CeCu catalysts (Supplementary Fig. 5). Different from Pt1/Ce and Pt1/CeCu, subnanometric Pt clusters are observed in the HAADF-STEM images of Ptn/Ce and Ptn/CeCu catalysts with the redox-coupled ALD (Fig. 1c, e), with mean particle sizes of 0.63 ± 0.18 and 0.75 ± 0.11 nm, respectively (Supplementary Fig. 5). The average distances of Pt clusters in a ceria nanorod are 4.94 ± 1.21 and 4.60 ± 1.51 nm for Ptn/Ce and Ptn/CeCu, respectively (Supplementary Fig. 6). The inserted HAADF-STEM images also definitively show CeO2 (220) facet-supported Pt clusters composed of aggregated Pt atoms. In addition, the DRIFTS spectrum of CO adsorption on Ptn/Ce and Ptn/CeCu shows a mix of linear- and bridged-bonded CO on Pt clusters with peaks at 2083, 2064, 1883, and 1838 cm−1. No Pt diffraction signals were detected in the X-ray diffraction patterns (Supplementary Fig. 7), which is due to the high dispersion and low crystallinity of Pt clusters. The presence of Cu in Ptn/CeCu catalyst was further confirmed by energy dispersive spectroscopy (EDS) (Supplementary Fig. 8). STEM-EDS elemental maps (Fig. 1f) were produced to study the distribution of Cu in the catalyst. Clearly, the concentrations of Cu dopants near the Pt clusters are higher than other regions, which is beneficial to tune the reducibility of the support near the interfaces and affect the activity of composite catalyst.
CO oxidation performance evaluation
CO oxidation tests were then performed to investigate catalytic activities of supported Pt catalysts. Figure 2a shows the CO conversion rates of prepared catalysts as a function of reaction temperature. Interestingly, Ptn/CeCu exhibits an onset of CO oxidation reactivity below the room temperature, and the T50 (50% CO conversion temperature) is decreased to 34 °C, which is much lower than that of Ptn/Ce (91 °C), Pt1/CeCu (116 °C), and Pt1/Ce (166 °C). The slightly shifts of CO conversion curves in cycling tests of Ptn/CeCu (Supplementary Fig. 9) indicate the structural stability of the constructed Pt/oxide interface, which can be attributed to the gas-phase based ALD method that has minimal detrimental effect on the surface structure of oxide supports35. The concentration of Cu dopants is kept low in Ce0.99Cu0.01O2 to avoid the formation of CuOx species, and catalytic testing show that the contribution of our Ce0.99Cu0.01O2 support to reactivity below 100 °C is negligible (Supplementary Fig. 10), in agreement with the comparable activity of Cu-doped CeO2 catalyst in previous study41. It can be concluded that the enhanced activity of Ptn/CeCu originates from the highly dispersed Pt sub-nanoclusters and the synergy of introduced Cu dopants in the Ce0.99Cu0.01O2 supports. In order to eliminate the effects of Pt’s mass loading for fair comparison, Ptn/CeCu was diluted by an appropriate amount of Ce0.99Cu0.01O2, and CO oxidation activity was determined. The catalyst still exhibits better low-temperature CO oxidation activity with a T50 of 54 °C after dilution (Supplementary Fig. 11). In addition, the effect of Cu dopants concentration was also investigated (Supplementary Fig. 12). The CO oxidation activities go down when Ce/Cu molar ratio increases to 95:5, in part due to the separated CuOx species that adversely affect the interfacial structures of composite catalysts (Supplementary Fig. 13). Furthermore, the intrinsic activities of synthesized catalysts have been evaluated by kinetic tests as shown in Fig. 2b. The calculated apparent activation energy of Ptn/CeCu (39.49 kJ mol−1) is much lower than that of Ptn/Ce (97.62 kJ mol−1) and other two Pt single atoms catalysts. The reaction orders of CO and O2 over Ptn/CeCu are close to zero (Supplementary Fig. 14), which indicates the negligible competitive adsorption between CO and O2 during CO oxidation at the interfaces of Ptn/CeCu. The activation energy of Ce0.99Cu0.01O2 support is determined to be 77.60 kJ mol−1 (Supplementary Fig. 15), close to that in previous studies42–44. The TOF of Ptn/CeCu catalyst reaches 0.26 s−1 at 80 °C, which rivals other previously reported Pt/CeO2 catalysts including Pt single atoms, clusters and nanoparticles (Supplementary Table 1).
Fig. 2. Catalytic properties and surface activities characterizations of catalysts.

a CO conversion and (b) reaction rates of catalysts as a function of reaction temperature. The catalysts are Pt1/Ce (black filled square), Ptn/Ce (black square), Pt1/CeCu (red filled circle) and Ptn/CeCu (red circle). c Ce 3d XPS spectra of Pt1/Ce, Ptn/Ce, Pt1/CeCu and Ptn/CeCu. d In situ DRIFTS spectra of CO adsorption and oxidation of Ptn/CeCu. After CO exposure, He flow is continued and the spectra is recorded at 2, 4, 6, 8, 10 min. Subsequently, the flow is switched to 1% vol. O2 balanced by N2 and the spectrum is recorded at 2 min. Source data are provided as a Source Data file.
Interfacial structures and activities investigations
The interfacial oxygen activation capability and CO adsorption strength were then determined to clarify the root cause of the superior low-temperature CO oxidation activity of Ptn/CeCu. On one hand, the surface oxygen vacancies of catalysts were characterized by the concentrations of Ce3+ ([Ce3+]) calculated based on two sets of peaks in the Ce 3d X-ray photoelectron spectroscopy (XPS) spectra as shown in Fig. 2c. The calculated [Ce3+] of catalysts followed the order: Ptn/CeCu > Pt1/CeCu > Ptn/Ce > Pt1/Ce, implying that Cu dopants can considerably increase the concentrations of surface oxygen vacancy, which is consistent with the O 1s XPS results (Supplementary Fig. 16 and Supplementary Table 2). In addition, Raman spectrum analysis shows that Ptn/CeCu and Pt1/CeCu catalysts have a stronger defect-induced vibration mode than Ptn/Ce and Pt1/Ce (Supplementary Fig. 17). The temperature-programmed reduction by hydrogen (H2-TPR) has also been performed to investigate the surface reducibility of our prepared catalysts (Supplementary Fig. 18). The appearance of the reduction peak at 242 °C for Ce0.99Cu0.01O2 support can be assigned to the reduction of Cu–O–Ce species42,43. Cu dopants also cause the shift of reduction peaks of surface oxygen (250–550 °C) and bulk oxygen (about 710 °C) of CeO2 supports for Pt1/Ce and Ptn/Ce to low temperatures. The reducibility of surface oxygen follows the sequences of Ptn/CeCu > Pt1/CeCu > Ptn/Ce > Pt1/Ce, indicating that both Cu doping and change of Pt size can affect the surface reducibility of our prepared catalysts.
On the other hand, in situ DRIFTS spectra of Ptn/CeCu in Fig. 2d clearly show the loss of adsorbed CO molecules under He flow after CO exposure at the room temperature. The stretching signals of formed CO2 molecules under CO flow also decrease, which are related to the decreased CO adsorption signals (Supplementary Fig. 19). When the flow is switched from He to O2, the signal of adsorbed CO molecules is further weakened. The appearance of stretching bands of CO2 molecule at 2360 cm−1 and 2330 cm−1 suggests that the adsorbed CO molecules on Ptn/CeCu can be further oxidized by activated O2 molecules at the interface, which is typical in Pt/CeO2 catalysts45,46. In order to eliminate the effects of interfacial active oxygen, the in situ DRIFTS spectra of CO adsorption of Ptn/CeCu after pretreatment under CO flow have been collected, which still show the loss of adsorbed CO molecules indicating weakened CO bonding strength on Pt clusters due to Cu dopants (Supplementary Fig. 20). As a comparison, in situ DRIFTS spectra of Ptn/Ce (Supplementary Fig. 21) show that Pt sub-nanoclusters are poisoned by strongly adsorbed CO molecules. The temperature-programmed desorption of CO has also been performed to investigate the desorption behavior of CO on Ptn/CeCu and Ptn/Ce (Supplementary Fig. 22). The CO desorption found at −19 °C for Ptn/CeCu catalyst along with the formation of CO2 can be attributed to the CO oxidation reaction at interfaces. The desorption temperature of CO on Ptn/CeCu is much lower than that on Ptn/Ce, indicating the weakening of interfacial CO adsorption by Cu dopants. Thus, the interfacial oxygen site of Ptn/CeCu is activated and CO adsorption strength at the interface is weakened, contributing to the unique interfacial structure and reducibility in the Pt sub-nanoclusters and Cu-doped CeO2 support composites.
To further clarify the interfacial structures of supported Pt catalysts, X-ray absorption fine structure (XAFS) spectra tests were performed. The white line intensity of Pt LIII-edge XANES spectra in Fig. 3a indicates that oxidized Pt species exist in all composite catalysts, qualitatively indicating electron transfer between Pt and oxide supports. The white line intensity of the catalysts follows the order: Pt1/CeCu > Pt1/Ce > Ptn/Ce ≈ Ptn/CeCu, implying that Pt single atoms transfer larger numbers of electrons to oxide supports than Pt sub-nanoclusters. Moreover, the white line intensities of Pt1/CeCu and Pt1/Ce are close to that of PtO2, indicating that the supported Pt single atoms are mainly in oxidized states. The white line intensity of Ptn/Ce is similar to that of Ptn/CeCu, implying that the valence states of Pt catalysts are dependent on the morphologies, which is consistent with the Pt 4f XPS analysis (Supplementary Fig. 23 and Supplementary Table 2). The Fourier transform extended X-ray absorption fine structure (EXAFS) spectra of Pt1/CeCu and Pt1/Ce in Fig. 3b show one prominent peak at about 1.7 Å, which is assigned to the formed Pt–O bond between Pt single atoms and oxide supports. The EXAFS spectra of Ptn/CeCu and Ptn/Ce exhibit a decrease in signal from the Pt–O contribution and very weak signal at the range of Pt–Pt bonds compared to Pt foil. The small Pt–Pt contribution can be attributed to the low crystallinity and high disorder of Pt atoms for the deposited Pt clusters at relatively low temperature as presented in the HADDF-STEM images, which agrees with previously reports20,40.
Fig. 3. Interfacial structures characterizations of catalysts.

a Normalized Pt LIII-edge XANES and (b) k2-weighted Fourier-transformed EXAFS spectra of Pt1/CeCu, Pt1/Ce, Ptn/CeCu, Ptn/Ce, Pt foil and PtO2. c QPt, calculated Bader charges of interfacial Pt atoms; LPt–O, bond lengths between Pt atoms and interfacial oxygen; and CNPt–O, coordinate number of interfacial Pt atoms, of Pt1/CeCu, Pt1/Ce, Ptn/CeCu and Ptn/Ce. The black square points are the white line intensity from XANES spectra. The red and magenta square points representing LPt–O and CNPt–O are the fits to the EXAFS spectra. Source data are provided as a Source Data file.
By combining the results of HAADF-STEM images and EXAFS spectra, stable interface structures of Pt single atoms and Pt5 clusters supported on CeO2 (110) slabs (Pt1/Ce and Pt5/Ce) were constructed and optimized by density functional theory (DFT) calculations (Supplementary Figs. 24, 25). Pt1/CeCu and Pt5/CeCu models have an interfacial Cu dopant replacing a Ce atom (Supplementary Fig. 25). As shown in Fig. 3c, the calculated Bader charges47 of interfacial Pt atoms (QPt) show the same trend as the white line intensity that are related to electron transfer at the interface. Moreover, bond lengths between Pt atoms and interfacial oxygen (LPt–O), and coordinate number of interfacial Pt atoms (CNPt–O) also show the same trend as LPt–O, and CNPt–O from the fitting data of EXAFS results (Supplementary Fig. 26 and Supplementary Table 3), validating the constructed models. The formation of Pt sub-nanoclusters on Ptn/CeCu and Ptn/Ce leads to lengthened Pt–O bonds and decreased CNPt–O, which is associated with decreased electron transfer compared to Pt1/CeCu and Pt1/Ce. The relatively small CNPt–O values of Ptn/Ce and Ptn/CeCu can be related to the highly catalytic activity compared with Pt1/Ce and Pt1/CeCu, respectively29.
CO oxidation mechanism studies
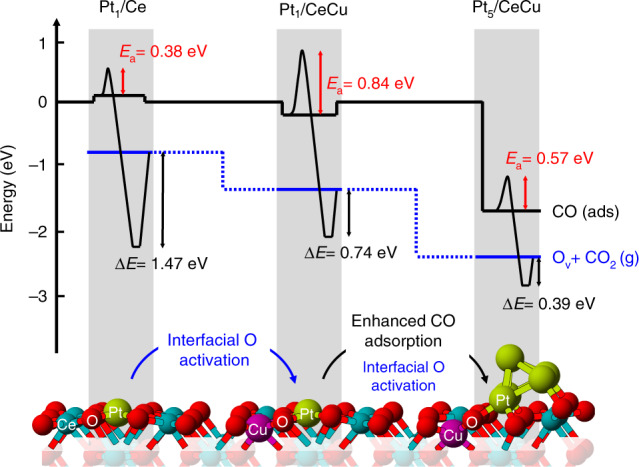
The adsorption energies of CO on the supported Pt single atom and cluster have been calculated as shown in Supplementary Fig. 27. The top Pt atoms for both Pt5/Ce and Pt5/CuCe will be poisoned by CO molecule with overbinding that is consistent with our DRIFTS results, i.e. some CO molecules remained after O2 flow was introduced (Fig. 2d). Therefore, we investigated the CO oxidation processes at the interface of catalysts based on the Mars-van Krevelen mechanism48, where adsorbed CO molecules first react with interfacial lattice oxygen along creating an oxygen vacancy (Ov). The formation energies of Ov at the interface of our constructed models have been presented in Supplementary Fig. 28. According to the CO adsorption energies and calculated oxygen vacancy formation energies, the energies of the initial states with an adsorbed CO molecule (black line) and the final states with oxygen vacancy after CO2 molecule desorption (blue line) are presented in Fig. 4. Although CO adsorbed on Pt1/Ce can easily react with the lattice oxygen by overcoming a low energy barrier (0.38 eV), the CO2 product at the interface (Supplementary Fig. 29) is very stable with a desorption energy of 1.47 eV, thus poisoning the active sites for subsequent reactions. The formation of interfacial Cu–O–Ce site in doped sample can decrease the desorption energy of formed CO2 to 0.74 eV for Pt1/CeCu, which is associated with the activated oxygen in Cu–O–Ce site. However, it needs to overcome an energy barrier of 0.84 eV to form the CO2 intermediate at the interface due to the weak adsorption of a CO molecule on Pt single atoms (–0.26 eV).
Fig. 4. Oxidation energetics of adsorbed CO molecules by interfacial oxygens of Pt1/Ce, Pt1/CeCu and Pt5/CeCu.
Source data are provided as a Source Data file.
After Pt single atoms are aggregated to a Pt5 cluster, the energy barrier is decreased to 0.57 eV due to the appropriately enhanced CO adsorption strength (−1.73 eV). Moreover, the more activated oxygen in the interfacial Cu–O–Ce site leads to a decrease in CO2 desorption energy (0.39 eV). However, in terms of Pt5/Ce (Supplementary Fig. 30), a high energy barrier of 1.10 eV is needed to overcome the strong CO poisoning effect on Pt clusters (−2.17 eV). The high energy barrier for Pt5/Ce also agrees well with the large activation barrier of Ptn/Ce in the kinetic test, which results from the strong CO poisoning effect on the subnanometric Pt clusters. The differential charge densities indicate that the bonding between interfacial Pt and oxygen is strengthened with an enhanced polarization due to the low-valence Cu dopant, which leads a weakened adsorption of CO molecule at interfacial Pt atoms (Supplementary Fig. 31). In order to eliminate potential variations due to the size of Pt clusters used in the calculation, the energetic routes of CO oxidation at the interfaces of oxide slabs supported a larger Pt14 cluster have also been calculated (Supplementary Fig. 32), which exhibit the similar results to supported Pt5 clusters. In addition, the oxidation processes of co-adsorbed CO and O2 molecules at interfaces containing an Ov show that Pt5/CeCu exhibits the highest activity with the lowest energy barrier of 0.53 eV (Supplementary Fig. 33). Therefore, both activated interfacial oxygen and moderate CO adsorption strength are keys to the excellent catalytic activity of Pt5/CeCu.
Discussion
Precisely controlled interfacial structures of Pt/CeO2 have been proposed by regulating oxide doping and Pt cluster size and employing the redox-coupled ALD method. Cu dopants have been introduced to modulate the surface reducibility of CeO2 support, which led to the formation of Cu2+ ions in the CeO2 lattice and helped create active Cu–O–Ce sites for tuning the interfacial structures and anchoring the Pt catalysts. The concentration of Cu dopants is kept low to avoid the segregation of CuOx species that are unfavorable for the activity enhancement of supported Pt catalysts. The size of deposited Pt catalysts in the range of sub-nanometer clusters to single atoms are well controlled by our proposed ALD recipe, which can directly affect the coordination environment of interfacial Pt atoms. The supported Pt single atoms with larger coordination number of Pt–O show lower activity than subnanometric Pt clusters with smaller coordination number of Pt–O29,30. Compared with subnanometric Pt clusters on CeO2 supports, Cu dopants at the interfaces can not only activate the interfacial oxygen, but also weaken the adsorption of CO molecule at interfacial Pt atoms, which are the keys to promoting CO oxidation with lattice oxygen at room temperature in Pt/CeO2 catalysts45,46. The experimental evidence agrees well with our proposed catalytic reaction path at the interfacial sites following the Mars-van Krevelen mechanism based on DFT calculations. The resulting Pt sub-nanoclusters on Cu-doped CeO2 exhibit excellent activity with an onset of CO oxidation reactivity below room temperature. Our study lays the foundation for designing and preparing highly efficient Pt catalysts for low-temperature exhaust abatement.
Methods
Catalysts preparation
The CeO2 nanorod supports were prepared by hydrothermal method. In details, we first dissolved Ce(NO3)3·6H2O and NaOH (1.96 and 16.88 g, Sinopharm Chemical Reagent Co., Ltd) into 40 ml and 30 ml of deionized water, respectively. After both solutions were cooled down to room temperature, the NaOH solution were slowly dropped into the Ce(NO3)3·6H2O solution with continuously magnetic stirring. Then, the mixture solution was poured in a 100 ml Teflon bottle, which was subsequently sealed in a stainless steel vessel autoclave. The hydrothermal reaction was performed at 100 °C for 24 h. After centrifugation collection, repetitive wash and vacuum dry (80 °C), the CeO2 precursors were calcined at 500 °C for 4 h to obtain the final CeO2 nanorod supports. In order to tune the reducibility of CeO2 supports, Cu dopants were introduced by substituting part of Ce(NO3)3·6H2O with a certain amount of Cu(NO3)2·3H2O (Sinopharm Chemical Reagent Co., Ltd) before being dissolved into deionized water. The molar ratio of Ce and Cu was kept as 99:1. The finally obtained Cu-doped CeO2 nanorod support was denoted as Ce0.99Cu0.01O2. As a comparison, the Cu-doped CeO2 nanorod support with the Ce/Cu molar ratio of 95:5 was also prepared, which was denoted as Ce0.95Cu0.05O2.
The Pt single atoms and sub-nanoclusters were loaded on CeO2 and Ce0.99Cu0.01O2 nanorod supports via ALD method. The ALD processes were performed in a custom-made fluidized-bed reactor (AngstromBlock Scale-F015 ALD system) as described in our previous studies34,35. For each ALD process, about 200 mg nanorod supports were loaded in a designed holder, which were firstly fluidized by 200 mL min−1 of N2 at 150 °C for 30 min. The Pt single atoms were loaded on CeO2 and Ce0.99Cu0.01O2 nanorod supports after one cycle of Pt ALD with trimethyl(methylcyclopentadienyl)platinum (MeCpPtMe3, 98%, Sigma-Aldrich) and ultrahigh purity O2 as precursors. The MeCpPtMe3 precursor, which was kept at 65 °C during Pt ALD, was introduced into the reactor at 150 °C with the pulse time and purge time of 100 s and 100 s. Then, the reactor was rapidly heated to 200 °C. 500 mL min−1 of O2 was subsequently introduced into the reactor with the pulse time and pure time of 600 and 200 s. For the preparation of Ptn/Ce and Ptn/CeCu, one cycle of redox-coupled Pt ALD was performed. The processes of Pt precursor and O2 were the same as that of Pt single atom preparation. After the purge step of O2, 500 mL min−1 of ultrahigh purity H2 was subsequently introduced into the reactor at 200 °C with the pulse time and purge time of 600 and 200 s.
Characterizations
The morphology of CeO2 nanorod supports was characterized by transmission electron microscopy (TEM, Tecnai G2 F30 electron microscope, FEI). The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and energy dispersive spectroscopy (EDS) spectra were obtained on a JEOL JEM-ARM200F TEM. The X-ray diffraction (XRD) patterns were recorded by the PANalytical X’Pert Pro with a Cu Kα1 radiation source. The mass loading of Pt on catalysts was analyzed by inductively coupled plasma atomic emission spectrometer (ICP-OES) on Optima 4300 DV spectrometer. The Raman spectra were recorded on a Renishaw inVia Reflex in a range from 200 to 800 cm−1 using an excitation laser line of 532 nm. The X-ray photoelectron spectroscopy (XPS) spectra were obtained with an AXIS-ULTRA DLD-600W XPS spectrometer in high vacuum environment (~10−7 Pa) after all samples are kept in the XPS chamber overnight, which were calibrated by the peak of C 1s at 284.8 eV.
The in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) spectra of CO on prepared catalysts were collected by wide band mercury cadmium telluride detector on the Nicolet iS50 FTIR spectrometer (ThermoFisher Scientific). The Praying MantisTM diffuse reflection accessory (Harrick Scientific Products Inc.) was used with ZnSe windows. The catalysts were first pretreated by 30 mL min−1 of ultrahigh purity N2 at room temperature for 30 min, and the background spectra were collected. Then, the gas was switched to 30 mL min−1 of 1% vol. CO balanced by N2. The CO DRIFTS spectra were collected after 10 min. In order to study the adsorption strength of CO on catalysts, the gas was subsequently switched to 30 mL min−1 of ultrahigh purity He. The DRIFTS spectra of CO adsorption on catalysts were collected every 2 min.
The temperature-programmed reduction by hydrogen (H2-TPR) was performed by a chemisorption analyzer (AMI-300 series, Altamira Instrument). Typically, 30 mg of the catalyst was supported by quartz wool in a U-type quartz tube reactor, which was pretreated using 30 mL min−1 of Ar at 100 °C for 30 min. The feed was switched to 30 mL min−1 of 10% vol. H2 balanced with Ar, when the catalyst was cooled down to room temperature. Then, the reactor was heated to 800 °C with a ramp rate of 5 °C min−1 and thermal conductivity detector was utilized to monitor the signal of H2 consumption.
The temperature-programed desorption of CO (CO-TPD) was performed by the VDSorb-91x chemisorption analyzer. 100 mg of the catalyst was supported by quartz wool in a U-type quartz tube reactor. After being pretreated using 50 mL min−1 of He at 200 °C for 30 min, the catalyst was cooled down to −100 °C by liquid nitrogen trap under He flow. The catalyst was exposed to 50 mL min−1 of 10 vol. % CO balanced by He for 30 min, then the feed was switched to He to purge the catalyst until the baseline was stable. CO-TPD curves were obtained under the He flow by using the AMETEK® quadrupole mass spectrometer to monitor the signal of CO (m/z = 28) and CO2 (m/z = 44), when the reactor was heated to 600 °C with a ramp rate of 5 °C min−1.
The X-ray absorption fine structure (XAFS) spectra were obtained at the 1W1B beamline of Beijing synchrotron radiation facility. The incident photon beam was selected by a double-crystal Si (111) monochromator after a collimating mirror and focused by a toroidal mirror. All XAS measurements were conducted in transmission mode using a 19-element high-purity germanium solid-state detector. The X-ray beam size on our prepared catalysts was about 0.9 × 0.3 mm2 at half-maximum (FWHM) with a photon flux of >4 × 1011 photons s−1 at 9 keV. Pt LIII-edge in the energy range of 11368–12463 eV was collected for catalysts. In order to avoid the influence of air, all catalysts were tested under N2 flow at room temperature in the cell as our previous study reported34. The Pt LIII-edge of Pt foil and PtO2 were also tested as references. By considering the parameters of coordinated number (N), bond length (R, Å), Debye-Waller factor (σ2, Å2) and shift in the edge energy (ΔE0, eV), the Pt k2-weighted Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectrum of catalysts was fitted by using the Pt foil and PtO2 modes in Demeter program49.
Activity evaluation
The CO oxidation activity evaluation of catalysts was performed by the VDSorb-91x chemisorption analyzer. 50 mg of catalysts without any pre-treatments were packed between quartz wool in a U-type quartz tube reactor. The flow rate of feed gas was 100 mL min−1, which was a mixture of CO (1% vol.), O2 (10% vol.) and N2. The temperature of reactor was linearly heated from −20 to 200 °C with a ramp rate of 2 °C min−1. The analysis of outlet gas mixture was performed by in situ HPR-20 mass spectrometer, which tested the partial pressure of CO2 . The CO conversion (XCO) was calculated by
| 1 |
where , , and are the partial pressure of CO2 in the outlet gas mixture before CO oxidation, after the total conversion of CO and at a reaction temperature (T), respectively. and were calibrated by the portable emission analyzer (MEXA-584L, Horiba). The kinetics tests were performed with CO conversion below 15% to eliminate the thermal and diffusion effects. The inert quartz was used to dilute the catalysts to make sure the appropriate CO conversion. The turnover frequency (TOF) of catalysts at a reaction temperature (T) was tested by decreasing the mass of catalysts, which was calculated by
| 2 |
where P, V, R and wCO were the pressure of feed gas (101325 Pa), the flow rate of feed gas (1.67 × 10−6 m3 s−1), universal gas constant (8.134 J mol−1 s−1) and vol. percentage of CO (1%), respectively. nPt was the mole mass of Pt in catalysts determined by ICP-OES method. According to the slope of the Arrhenius plots, the activation energies (Ea) of catalysts were calculated.
DFT calculations
Spin-polarized calculations were carried out based on DFT with the Vienna Ab initio Simulation Package (VASP) using the Perdew–Burke–Ernzerhof (PBE) functional50–53. The ionic cores were described by projector augmented wave method54. In order to correct the on-site Coulomb and exchange interactions, PBE+U method with U-J = 5.0 eV was applied to describe Ce 4f-orbital55,56. The energy cutoff of plane wave basis was set to 400 eV. The convergence criterion of atomic structural optimization was 0.05 eV Å−1. The CeO2 (110) slab was built with vacuum layer thicknesses larger than 15 Å to avoid the interaction between two periodic slabs. The Brillouin zone was sampled at the Γ point. The climbing image nudged elastic band (CI-NEB)57 method with six intermediate images was used to calculate the minimum energy paths of CO oxidation at the interfaces of Pt/CeO2 catalysts.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 51871103, 51835005, 51801067, and 51911540476) and Program for HUST Academic Frontier Youth Team (2019kfyXMBZ025 and 2018QYTD03). Xiao Liu gratefully acknowledges the support from the Postdoctoral Innovation Talents Support Program (BX20180104). Ming Yang acknowledges the support of General Motors Research and Development and the start-up funding provided by Clemson University. We would also like to acknowledge the technology support from the Analytic Testing Center, Flexible Electronics Research Center and SCTS/CGCL HPCC of HUST.
Source data
Author contributions
X.L. conducted the synthesis and characterizations of catalysts, and wrote the paper. S.F.J. and J.B.W. performed HAADF-STEM characterizations. X.L. and Y.T.T. performed catalytic activity evaluation. X.L. and Y.W.W. were responsible for the detailed DFT calculations. S.Q.C. performed XAFS characterizations. M.Y., B.S. and R.C. directed the project and mentored the paper writing. All authors discussed the results and commented on the paper.
Data availability
The source data underlying Figs. 2-4 and Supplementary Figs. 2–4, 7–23, 25, 26, 31 are provided as a Source Data file. Extra data are available from the corresponding authors upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Abhaya Datye and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Bin Shan, Email: bshan@mail.hust.edu.cn.
Rong Chen, Email: rongchen@mail.hust.edu.cn.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-18076-6.
References
- 1.Host R, Moilanen P, Fried M, Bogi B. Exhaust system thermal management: a process to optimize exhaust enthalpy for cold start emissions reduction. SAE Tech. Pap. 2017;2017:01–0141. [Google Scholar]
- 2.Getsoian A, Theis JR, Paxton WA, Lance MJ, Lambert CK. Remarkable improvement in low temperature performance of model three-way catalysts through solution atomic layer deposition. Nat. Catal. 2019;2:614–622. [Google Scholar]
- 3.Lambert CK. Current state of the art and future needs for automotive exhaust catalysis. Nat. Catal. 2019;2:554–557. [Google Scholar]
- 4.Lee J, Theis JR, Kyriakidou EA. Vehicle emissions trapping materials: successes, challenges, and the path forward. Appl. Catal. B: Environ. 2019;243:397–414. [Google Scholar]
- 5.Nie L, et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science. 2017;358:1419–1423. doi: 10.1126/science.aao2109. [DOI] [PubMed] [Google Scholar]
- 6.Zammit, M. et al. Future Automotive Aftertreatment Solutions: The 150 °C Challenge Workshop Report (US Department of Energy, 2013).
- 7.Min BK, Friend CM. Heterogeneous gold-based catalysis for green chemistry: low-temperature CO oxidation and propene oxidation. Chem. Rev. 2007;107:2709–2724. doi: 10.1021/cr050954d. [DOI] [PubMed] [Google Scholar]
- 8.Xie XW, Li Y, Liu ZQ, Haruta M, Shen WJ. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature. 2009;458:746–749. doi: 10.1038/nature07877. [DOI] [PubMed] [Google Scholar]
- 9.Wang AY, Olsson L. The impact of automotive catalysis on the United Nations sustainable development goals. Nat. Catal. 2019;2:566–570. [Google Scholar]
- 10.Tauster SJ, Fung SC, Garten RL. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978;100:170–175. [Google Scholar]
- 11.Farrauto RJ, Deeba M, Alerasool S. Gasoline automobile catalysis and its historical journey to cleaner air. Nat. Catal. 2019;2:603–613. [Google Scholar]
- 12.Vayssilov GN, et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles. Nat. Mater. 2011;10:310–315. doi: 10.1038/nmat2976. [DOI] [PubMed] [Google Scholar]
- 13.Kopelent R, et al. Catalytically active and spectator Ce3+ in ceria-supported metal catalysts. Angew. Chem. Int. Ed. 2015;54:8728–8731. doi: 10.1002/anie.201503022. [DOI] [PubMed] [Google Scholar]
- 14.Liu LQ, et al. Low-temperature CO oxidation over supported Pt, Pd catalysts: Particular role of FeOx support for oxygen supply during reactions. J. Catal. 2010;274:1–10. [Google Scholar]
- 15.Liu LC, Corma A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018;118:4981–5079. doi: 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cargnello M, et al. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science. 2013;341:771–773. doi: 10.1126/science.1240148. [DOI] [PubMed] [Google Scholar]
- 17.Gänzler AM, et al. Tuning the Pt/CeO2 interface by in situ variation of the Pt particle size. ACS Catal. 2018;8:4800–4811. [Google Scholar]
- 18.Lykhach Y, et al. Counting electrons on supported nanoparticles. Nat. Mater. 2016;15:284–289. doi: 10.1038/nmat4500. [DOI] [PubMed] [Google Scholar]
- 19.Beniya A, Higashi S. Towards dense single-atom catalysts for future automotive applications. Nat. Catal. 2019;2:590–602. [Google Scholar]
- 20.Qiao BT, et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011;3:634–641. doi: 10.1038/nchem.1095. [DOI] [PubMed] [Google Scholar]
- 21.Bruix A, et al. Maximum noble-metal efficiency in catalytic materials: atomically dispersed surface platinum. Angew. Chem. Int. Ed. 2014;53:10525–10530. doi: 10.1002/anie.201402342. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, et al. Identifying size effects of Pt as single atoms and nanoparticles supported on FeOx for the water-gas shift reaction. ACS Catal. 2018;8:859–868. [Google Scholar]
- 23.Qiao BT, et al. Ferric oxide-supported Pt subnano clusters for preferential oxidation of CO in H2-rich gas at room temperature. ACS Catal. 2014;4:2113–2117. [Google Scholar]
- 24.Stephens IEL, Elias JS, Shao-Horn Y. The importance of being together. Science. 2015;350:164–165. doi: 10.1126/science.aad3716. [DOI] [PubMed] [Google Scholar]
- 25.Ding KL, et al. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science. 2015;350:189–192. doi: 10.1126/science.aac6368. [DOI] [PubMed] [Google Scholar]
- 26.Jones J, et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science. 2016;353:150–154. doi: 10.1126/science.aaf8800. [DOI] [PubMed] [Google Scholar]
- 27.Feng YX, et al. Correlating DFT calculations with CO oxidation reactivity on Ga-doped Pt/CeO2 single-atom catalysts. J. Phys. Chem. C. 2018;122:22460–22468. [Google Scholar]
- 28.Li J, et al. In situ formation of isolated bimetallic PtCe sites of single-dispersed Pt on CeO2 for low-temperature CO oxidation. ACS Appl. Mater. Interfaces. 2018;10:38134–38140. doi: 10.1021/acsami.8b15585. [DOI] [PubMed] [Google Scholar]
- 29.Ke J, et al. Strong local coordination structure effects on subnanometer PtOx clusters over CeO2 nanowires probed by low-temperature CO oxidation. ACS Catal. 2015;5:5164–5173. [Google Scholar]
- 30.Wang H, et al. Surpassing the single-atom catalytic activity limit through paired Pt-O-Pt ensemble built from isolated Pt1 atoms. Nat. Commun. 2019;10:3808. doi: 10.1038/s41467-019-11856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeRita L, et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 2019;18:746–751. doi: 10.1038/s41563-019-0349-9. [DOI] [PubMed] [Google Scholar]
- 32.Therrien AJ, et al. An atomic-scale view of single-site Pt catalysis for low-temperature CO oxidation. Nat. Catal. 2018;1:192–198. [Google Scholar]
- 33.Liu LJ, et al. Determination of the evolution of heterogeneous single metal atoms and nanoclusters under reaction conditions: which are the working catalytic sites? ACS Catal. 2019;9:10626–10639. doi: 10.1021/acscatal.9b04214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, et al. Oxide-nanotrap-anchored platinum nanoparticles with high activity and sintering resistance by area-selective atomic layer deposition. Angew. Chem. Int. Ed. 2017;56:1648–1652. doi: 10.1002/anie.201611559. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, et al. Bifunctional CO oxidation over Mn-mullite anchored Pt sub-nanoclusters via atomic layer deposition. Chem. Sci. 2018;9:2469–2473. doi: 10.1039/c7sc05486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang CL, et al. Water-mediated Mars-van Krevelen mechanism for CO oxidation on ceria-supported single-atom Pt1 catalyst. ACS Catal. 2017;7:887–891. [Google Scholar]
- 37.Zhang L, Banis MN, Sun XL. Single-atom catalysts by the atomic layer deposition technique. Nat. Sci. Rev. 2018;5:628–630. [Google Scholar]
- 38.Liu X, et al. Effect of exposed facets and oxygen vacancies on the catalytic activity of PdxCe1-xO2-δ catalysts: a combined experimental and theoretical study. Catal. Sci. Technol. 2017;7:4462–4469. [Google Scholar]
- 39.Wang YF, et al. Single-atomic Cu with multiple oxygen vacancies on ceria for electrocatalytic CO2 reduction to CH4. ACS Catal. 2018;8:7113–7119. [Google Scholar]
- 40.Gänzler AM, et al. Tuning the structure of platinum particles on ceria in situ for enhancing the catalytic performance of exhaust gas catalysts. Angew. Chem. Int. Ed. 2017;56:13078–13082. doi: 10.1002/anie.201707842. [DOI] [PubMed] [Google Scholar]
- 41.Kappis K, et al. Tuning the catalytic properties of copper-promoted nanoceria via a hydrothermal method. Catalysts. 2019;9:138. [Google Scholar]
- 42.Yu WZ, et al. Construction of active site in a sintered copper-ceria nanorod catalyst. J. Am. Chem. Soc. 2019;141:17548–17557. doi: 10.1021/jacs.9b05419. [DOI] [PubMed] [Google Scholar]
- 43.Wang WW, et al. Crystal plane effect of ceria on supported copper oxide cluster catalyst for CO oxidation: importance of metal-support interaction. ACS Catal. 2017;7:1313–1329. [Google Scholar]
- 44.Safonova OV, et al. Elucidating the oxygen activation mechanism on ceria-supported copper-oxo species using time-resolved X-ray absorption spectroscopy. ACS Catal. 2020;10:4692–4701. [Google Scholar]
- 45.Pereira-Hernández XI, et al. Tuning Pt-CeO2 interactions by high-temperature vapor-phase synthesis for improved reducibility of lattice oxygen. Nat. Commun. 2019;10:1358. doi: 10.1038/s41467-019-09308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu YB, Thompson C, Kunwar D, Datye AK, Karim AM. Origin of the high CO oxidation activity on CeO2 supported Pt nanoparticles: weaker binding of CO or facile oxygen transfer from the support? ChemCatChem. 2020;12:1726–1733. [Google Scholar]
- 47.Henkelman G, Arnaldsson A, Jónsson H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006;36:354–360. [Google Scholar]
- 48.Mars P, van Krevelen DW. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954;3:41–59. [Google Scholar]
- 49.Ravel B, Newville MJ. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. Synchrotron Rad. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- 50.Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 1993;47:558–561. doi: 10.1103/physrevb.47.558. [DOI] [PubMed] [Google Scholar]
- 51.Kresse G, Hafner J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B. 1994;49:14251–14269. doi: 10.1103/physrevb.49.14251. [DOI] [PubMed] [Google Scholar]
- 52.Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996;6:15–50. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 53.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 54.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59:1758–1775. [Google Scholar]
- 55.Anisimov VI, Zaanen J, Andersen OK. Band theory and Mott insulators: Hubbard U instead of Stoner. Phys. Rev. B. 1991;44:943–954. doi: 10.1103/physrevb.44.943. [DOI] [PubMed] [Google Scholar]
- 56.Nolan M, Parker SC, Watson GW. The electronic structure of oxygen vacancy defects at the low index surfaces of ceria. Surf. Sci. 2005;595:223–232. [Google Scholar]
- 57.Henkelman G, Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000;113:9978–9985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source data underlying Figs. 2-4 and Supplementary Figs. 2–4, 7–23, 25, 26, 31 are provided as a Source Data file. Extra data are available from the corresponding authors upon request.