

Abstract
Na+/K+-ATPase is a transmembrane ion pump that is essential for the maintenance of ion gradients and regulation of multiple cellular functions. Na+/K+-ATPase has been associated with nuclear factor kappa B (NFκB) signalling, a signal associated with lipopolysaccharides (LPSs)-induced immune response in connection with activated Toll-like receptor 4 (TLR4) signalling. However, the contribution of Na+/K+-ATPase to regulating inflammatory responses remains elusive. We report that mice haploinsufficient for the astrocyte-enriched α2Na+/K+-ATPase isoform (α2+/G301R mice) have a reduced proinflammatory response to LPS, accompanied by a reduced hypothermic reaction compared to wild type litter mates. Following intraperitoneal injection of LPS, gene expressions of Tnf-α, Il-1β, and Il-6 was reduced in the hypothalamus and hippocampus from α2+/G301R mice compared to α2+/+ littermates. The α2+/G301R mice experienced increased expression of the gene encoding an antioxidant enzyme, NRF2, in hippocampal astrocytes. Our findings indicate that α2Na+/K+-ATPase haploinsufficiency negatively modulates LPS-induced immune responses, highlighting a rational pharmacological target for reducing LPS-induced inflammation.
Subject terms: Gene regulation in immune cells, Neuroimmunology, Neurological disorders
Introduction
The Na+/K+-ATPase is an integral membrane protein that spans the entirety plasma membrane of all animal cells. It exchanges three Na+ ions out of the cell for two K+ ions into the cell using energy generated from ATP hydrolysis1. The Na+/K+-ATPase activity is important for many cellular functions, such as maintenance of membrane potentials, cellular volume regulation and pH adjustments, and supporting secondary transport of transmitters2.
Studies have shown differences in the physiological functions of NKA isoforms. The α1-Na+/K+-ATPase isoform also appears to act as a receptor, a signal transducer, and a cell adhesion molecule3, involving various pathway components such as the membrane-associated non-receptor tyrosine kinase Src pathway4, Ras/Raf/ERK1/2 pathway activation5, the phosphate inositol 3-kinase (PI3K) pathway, the PI3K-dependent protein kinase B pathway, phospholipase C, [Ca2+]i oscillations6–8, and gene transcription9,10. Interestingly, Na+/K+-ATPase-mediated increases in Ca2+ concentration can affect gene transcription by promoting the translocation of nuclear factor-kappa B (NFκB) from the cytosol to the nucleus and by phosphorylating cAMP response element binding protein (CREB)11. However, elegant studies have shown that the a2 Na+/K+-ATPase isoform does not interfere with the signaling and activation of the Src, ERK and PI3K/Akt pathways12,13. In addition, it is important to highlight the isoforms of Na+/K+-ATPase are located in distinct regions in the plasma membrane of cells14, which confer different physiological functions in the regulation of intracellular Na+ and Ca2+. Within this context, studies with different cell types15, such as aortic smooth muscle and astrocytes, have demonstrated the importance of the a2Na+/K+-ATPase isoform in the maintenance of intracellular Na+, as well as the interaction of the α2 isoform with the Na+/Ca2+ exchanger for the reaction of intracellular Ca2+ levels and their signaling16,17. Furthermore, ifenprodil, an N-methyl-d-aspartate receptor antagonist, has been shown to restore GDNF‐evoked Ca2+ transients that are attenuated by LPS by inducing Na+/K+-ATPase expression18.
In mammals, four different Na+/K+-ATPase α isoforms (α1−4) are expressed, of which α1−3 are found in the central nervous system. While α1 is ubiquitously expressed and thought-about to maintain housekeeping cellular functions, the α2 isoform is functional primarily in astrocytes and developing neurons and the α3 isoform is restricted to neurons19,20.
In the adult brain, the Na+/K+-ATPase α2 isoform is enriched in astrocytes and important for extracellular K+ clearance and function to support glutamate uptake from the synaptic cleft21–23 and impair astrocytic K+ clearance24. Accordingly, mutations in the ATP1A2 gene, which encodes the α2 subunit isoform of the Na+/K+-ATPase, can cause familial hemiplegic migraine type 2 (FHM2)25, a subtype of migraine with aura26. Several Atp1a2 gene-modified mouse models have been made and extensively used to study the α2 isoform in vivo25. While KO and KI homozygous mice dies immediately after birth27–30 heterozygous α2 knock-out (KO) and knock-in (KI) mice are viable.
FHM2 patients with the G301R mutation resulting from a gene variant in the ATP1A2 gene have migraine comorbidity with epilepsy, coma, motor symptoms and psychiatric disorders such as depression and obsessive–compulsive disorder (OCD)31,32. Heterozygous KI mice containing the G301R disease mutation (α2+/G301R mice) displayed FHM2-related phenotypes, including mood depression and obsessive–compulsive disorder (OCD)-like symptoms33, besides showed a greater susceptibility to epilepsy and disseminated cortical depression (CSD)33,34. Moreover, astrocyte-neuron primary in vitro cultures from α2(G301R/G301R) mice revealed impaired glutamate uptake33. Surprisingly, when submitted to spinal cord injury, the α2+/G301R mice display improved functional recovery and decreased lesion volume compared to littermate controls (α2+/+)35. These phenotypes were associated with changes in pro- and anti-inflammatory cytokines, with the cytokines TNF, IL-6, and IL-10 upregulated in the spinal cord of α2+/G301R and α2+/+ mice with a spinal cord injury35. Interestingly, the functional recovery of the α2+/G301R mice was improved compared to α2+/+ mice and correlated with a significantly reduced lesion size35. In line with this, astrocytes deficient of the mutant superoxide dismutase 1 (SOD1) that also was depleted from the α2Na+/K+-ATPase, were able to protect motor neurons from degeneration in co-cultured primary motor neurons36. In these SOD-deficient astrocytes, mitochondrial respiration and inflammatory gene expressions appeared induced.
The genetic mechanisms of the Atp1a2 pathophysiology thus appear to reply on the ion balance, however, the impact of this on the immune response remains unknown.
As an initial attempt to understand this, we challenged heterozygous α2+/G301R mice by lipopolysaccharides (LPS) administration. We hypothesised that the mice would be compromised compared to wild type litter mates as ion channels-related diseases contribute to similar symptoms, and can be used as potential targets to modulate immune response and to treat inflammatory disorders and cancer.
LPSs, known as lipoglycans and endotoxins, acts as a prototypical endotoxin, binding the CD14/TLR4/MD2 receptor complex in many cell types, which promotes the production and secretion of proinflammatory cytokines, nitric oxide, and eicosanoids. LPS can induce neuroinflammation in the brain, including NF-κB activation in rodents, which can lead to impaired cognitive performance37,38.
Interestingly, short interference (si)RNA-mediated knockdown of the gene encoding the α2 isoform in primary astrocyte cultures prevented the LPS-mediated activation of ERK and NFκB39, but the precise mechanism through which the α2Na+/K+-ATPase pump regulates neuroinflammation remains elusive.
Here, we studied the α2Na+/K+-ATPase regulation of LPS-mediated neuroinflammation in the hypothalamus and hippocampus using a heterozygous mouse model (α2+/G301R) that exhibits α2 haploinsufficiency33. We found a reduction in the systemic production of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 following LPS administration, as well as a reduced hypothermic response in α2+/G301R mice.
Results
α2Na+/K+-ATPase haploinsufficiency reduce liposaccharide (LPS)-induced inflammation in vivo
To assess the effect of LPS in mice that are haploinsufficient for the α2 isoform, we used a knockin mouse model containing the FHM2-associated G301R disease mutation, which has been shown to harbour haploinsufficiency in heterozygotes α2+/G301R mice, associated with FHM2 traits33 . At such, α2+/G301R and α2+/+ mice were intraperitoneally injected with LPS (500 µg/kg). Intriguing, the LPS-injected α2+/G301R mice appeared much less affected by LPS and exhibited normal cage roaming in contrast to the α2+/+ littermates, which exhibited the expected severe sickness behaviour after LPS treatment (Supplementary Data, movie 1) (Fig. 1a).
Figure 1.

α2+/G301R mice display decreased sickness behaviour. α2+/G301R mice experience hypothermia, and systemic proinflammatory cytokine levels compared to α2+/+ mice following LPS administration (500 µg/kg, intraperitoneally (IP) injected). (a) Example of sickness behaviour 4 h after LPS administration. As illustrated, the α2+/+ mice displayed sickness behaviour, less locomotion, and, briefly, hypothermia, in contrast to the α2+/G301R mice that displayed less sickness behaviour after LPS administration. (b) The differences in body temperature 4 h after LPS administration compared to baseline (t = 0) in the α2+/+ and α2+/G301R animals treated with PBS or LPS, with a significant different after LPS treatment. (c–e) The α2+/G301R mice and their WT littermates (α2+/+) were injected with saline or LPS (500 µg/kg), and the levels of TNF-α (c) (n = 12 for the α2+/+ mice and n = 14 for α2+/G301R mice), IL-1β (d) [α2+/+ (n = 6) and α2+/G301R mice (n = 7)] and IL-6 (e) [α2+/+ (n = 6) and α2+/G301R (n = 7)] were measured in the serum 4 h after saline/LPS treatment. The data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test followed by Dunn’s post hoc test).
To test if the apparently unaffected LPS-treated α2+/G301R mice experienced changes in body temperature, a characteristic effect of LPS treatment, body temperatures were measured at baseline and 4 h after LPS injection. In line with the observed phenotypes, the α2+/+ mice exhibited a severe drop in body temperature (− 3.85 ± 3 °C) after LPS administration, whereas the α2+/G301R mice exhibited a significantly smaller drop in body temperature (− 1.40 ± 3.3 °C) (Fig. 1b). To evaluate whether cytokine levels were altered in the mice treated with LPS and if these alterations were different in the α2+/G301R mice, blood samples were collected 4 h after LPS administration and cytokine levels were measured using an enzyme-linked immunosorbent assay (ELISA). As expected, the LPS-treated α2+/+ mice exhibited significantly increased serum tumour necrosis factor-α (TNF-α), interleukin-1 beta (IL-1β) and IL-6 levels compared with those exhibited by the PBS-treated α2+/+ mice (Fig. 1c–e). The LPS-treated α2+/G301R mice had significantly reduced serum levels of TNF-α (Fig. 1c), and IL-6 (Fig. 1e) compared with those of the LPS-treated α2+/+ mice. The levels of IL-1β in the LPS-treated α2+/G301R mice were equal to those in the LPS-treated α2+/+ mice (Fig. 1d).
Combined, these results show a reduced sickness behavior and hypothermic response observed in the α2+/G301R mice compared to α2+/+ mice, and moreover, indicate that induction of TNF-α and IL-6 is compromised in response to LPS treatment in α2+/G301R mice. Our data show that LPS challenge promoted a serum increase of IL-1β that was not reversed in α2+/G301R mice, thus suggesting that α2 activity does not interfere with the release of IL-1β in its mature form.
α2Na+/K+-ATPase is not required for LPS-induced cytokine production in macrophages
Macrophages are the main producers of proinflammatory cytokines, including IL-6 and TNF-α40,41. Therefore, next we studied whether the decreased systemic levels of these cytokines in α2+/G301R mice were due to a reduced macrophage response and, hence, whether the macrophage response to LPS involves the Na+/K+-ATPase α2 isoform. To this end, we measured TNF-α production in response to LPS treatment in bone marrow-derived macrophages (BMDMs) isolated from α2+/G301R mice and wild-type (WT) littermates.
Surprisingly, the levels of TNF-α in the BMDMs from both α2+/+ and α2+/G301R did not differ significantly at any of the time points analysed (Fig. 2a–d). However, it is noteworthy that although the levels of TNF-α did not differ between α2+/+ and α2+/G301R, in both cases they are significantly increased after 1, 2, 4 and 6 h of LPS treatment. This suggested that the Na+/K+-ATPase α2 isoform may not be functionally required for LPS-induced TNF-α release in macrophages. Indeed, low levels of α2 protein were detected by Western blotting in BMDM cells and the levels diminished during BMDM differentiation in vitro (Fig. 2e and Supplementary Fig. 1). Furthermore, no significant differences in α2 isoform expression in the α2+/+ and α2+/G301R BMDM cells were observed (Fig. 2f), suggesting a low basal level of α2 protein not significantly affected by the α2 haploinsufficiency. Under these conditions, this implies that the α2Na+/K+-ATPase might regulate the proinflammatory response to LPS in other cell types.
Figure 2.

The α2 Na+/K+-ATPase isoform is not involved macrophage cytokine production in response to LPS. BMDMs derived from bone marrow cells from the femurs and tibias of α2+/+ and α2+/G301R mice were cultured in L929 cell supernatant for 6 days. After 6 days, the BMDMs were treated with LPS (100 ng/mL) or PBS (CTR). (a–d) The levels of TNF-α were measured in the supernatant 1 h (a), 2 h (b), 4 h (c), and 6 h (d) after LPS treatment. (e) α2 Isoform levels were measured by Western blotting in bone marrow derived macrophages from α2+/+ and α2+/G301R mice at the indicated times during differentiation. β-Actin was used as a loading control. A protein extract from the central nervous system (CNS) was loaded as a positive control. (f) Densitometric analysis from day 0 (arbitrary units, A.U.) showing that no obvious differences existed in the α2 protein levels in the BMDMs derived from the α2+/+ and α2+/G301R mice. The data are presented as the mean ± SEM from two individual experiments. The data are presented as the mean ± SEM. *p < 0.05 and **p < 0.01 (Kruskal–Wallis test followed by Dunn’s post hoc test).
LPS-induced neuroinflammation is suppressed in the hippocampus and hypothalamus of heterozygous α2+/G301R mice
Because steroid ouabain (OUA), a Na+/K+-ATPases inhibitor, has an anti-inflammatory effect in the rat hippocampus42–44, and because the hypothalamus endocrinologically regulates temperature control, we examined the α2 protein level in the hippocampus and hypothalamus of α2+/G301R mice.
No significant changes were observed in the levels of the α2 isoform in the hypothalamus between the α2+/G301R and α2+/+ mice (Fig. 3a, Supplementary Fig. 2). Like the observations in macrophages, this may imply that the α2 isoform may not be produced at levels that would permit a detectable difference. By contrast, the α2 isoform levels were significantly reduced in the hippocampus in the α2+/G301R mice compared with the α2+/+ mice (Fig. 3b, Supplementary Fig. 3). As a control, we also examined the levels of the housekeeping α1 isoform. No differences were detected in the α1 isoform levels in the hypothalamus and hippocampus (Fig. 3c,d, Supplementary Fig. 4, 5), confirming that the housekeeping α1 isoform is not affected by the G301R mutation in the α2 isoform. Moreover, LPS injection did not influence the levels of the α1 (Fig. 3c,d) or α2 (Fig. 3a,b) proteins in the hypothalamus or hippocampus in either the α2+/+ or α2+/G301R animals compared to those in the PBS-treated littermates.
Figure 3.

The levels of α2Na+/K+-ATPase isoform are reduced in the hippocampus of α2+/G301R mice. α1 and α2 isoform protein levels assessed by Western blotting in the hypothalamus (blue bars) and hippocampus (red bars). (a,c) Representative digital images of Western blots and densitometric analysis (arbitrary units) of hypothalamic lysates from the α2+/+ and α2+/G301R mice 4 h after saline or LPS injection showed no differences in α1Na+/K+-ATPase (a) (n = 8–10 mice/group) and α2Na+/K+-ATPase (c) (n = 6 mice/group). (b,d) Representative digital images of Western blots and densitometric analysis of hippocampal lysates from the α2+/+ and α2+/G301R mice 4 h after saline or LPS injection showed no differences in α2Na+/K+-ATPase (b) (n = 10–11 mice/group) but showed a reduction in α2Na+/K+-ATPase irrespective of LPS treatment (d) (n = 7–9 mice/group). The results are expressed relative to the control (PBS) as the mean ± SEM from three individual experiments. *p < 0.05 (Kruskal–Wallis test followed by Dunn’s post hoc test).
Since a significant reduction of α2 proteins levels was observed in hippocampus, but no difference in the hypothalamus, we next determined it this would influence the gene expression of TNF-α, IL-1β, and IL-6 genes.
Cytokine expression was measured specifically in the hypothalamus and hippocampus of the α2+/+ and α2+/G301R mice after PBS or LPS treatment by reverse transcription quantitative polymerase chain reaction (RT-qPCR). As expected, LPS treatment significantly upregulated the transcription of Tnf-α, Il-1β, and Il-6 genes in the hypothalamus (Fig. 4a–c) and hippocampus (Fig. 4d–f) of the α2+/+ mice compared with the PBS control group. By contrast, the transcription of these genes in the hypothalamus and hippocampus of the α2+/G301R mice was unaffected by LPS treatment compared with that in the PBS-treated littermates (Fig. 4a–f).
Figure 4.

The levels of cytokines are reduced in the hippocampus and hypothalamus of α2+/G301R mice. α2Na+/K+-ATPase haploinsufficiency decreases lipopolysaccharide (LPS)-induced hypothalamic (blue bars) and hippocampal (red bars) cytokine transcription. (a–f) TaqMan quantitative PCR analysis of TNF-α (a,d); IL-1β (b,e); and IL-6 (c,f) gene expression relative to the expression of β-actin, used as endogenous reference gene. LPS induced significant increases in the levels of TNF-α in the hypothalamus of both α2+/+ (n = 3), and α2+/G301R (n = 4), mice. LPS induced significant increases in the levels IL-1β and IL-6 in the hypothalamus (blue bars) of α2+/+ mice (n = 3), but not in α2+/G301R (n = 3) mice. In the hippocampus (red bars), LPS induced significant increases in the levels of TNF-α, IL-1β and IL-6 of α2+/+ mice (n = 3), but not in α2+/G301R (n = 4) mice. All data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (two-way ANOVA followed by Tukey’s post hoc test.
This indicates that part of the lack of a LPS immune response is associated with a significant reduced expression of the TNF-α, IL-1β, and IL-6 genes in both hippocampus and hypothalamus.
Tlr4 mRNA expression is reduced in astrocytes from α2+/G301R mice treated with LPS
It is well known that LPS acts as an agonist of TLR4 to induce neuroinflammation and the expression of proinflammatory cytokines. To determine whether the LPS-induced expression of proinflammatory cytokines is associated with a potential effect of α2 haploinsufficiency in the TLR4 induction in hypothalamus and hippocampus, we investigated the expression levels of Tlr4 in the hypothalamus and hippocampus by RT-qPCR. No difference in Tlr4 gene transcription between the α2+/+ and α2+/G301R mice in either the PBS or LPS treatment group was observed in the hypothalamus or hippocampus (Supplementary Fig. 6). However, as the α2 isoform is specifically expressed in astrocytes, we speculated that α2 haploinsufficiency would cause astrocytes from the α2+/G301R mice to have lower levels of α2-Na+/K+-ATPase, and therefore, an effect on Trl4 mRNA expression may be noted only in astrocytes and not in samples containing all cells from the hypothalamus or hippocampus. We therefore measured Tlr4 expression in astrocytes isolated by bead sedimentation using an antibody against astrocyte cell surface antigen-2 (ACSA-2)45. In agreement with the established LPS-induced upregulation of TLR4, Tlr4 expression was significantly increased in astrocytes from both the hypothalamus (blue in Fig. 5a) and hippocampus (red in Fig. 5b) in the α2+/+ mice after LPS treatment compared with PBS treatment. Tlr4 expression in the astrocytes from the α2+/G301R mice was comparable with that in the astrocytes from the α2+/+ mice treated with PBS; however, no increase in Tlr4 expression was observed in the hypothalamus (Fig. 5a) or hippocampus (Fig. 5b) of the α2+/G301R mice after LPS treatment, suggesting that the α2 isoform is required for LPS-mediated effect on Tlr4 expression in astrocytes during the acute LPS-induced neuroinflammation.
Figure 5.

The level of Tlr4 is reduced in the hippocampus and hypothalamus of α2+/G301R mice. The α2+/G301R mice exhibited reduced Tlr4 and Tnf-α mRNA expression in astrocytes isolated from the hypothalamus (blue bars) and hippocampus (red bars) compared with that in the α2+/+ mice after PBS treatment. (a,b) qPCR analysis of Tlr4 relative to β-actin expression in the hypothalamus (a) and hippocampus (b) showed a significant reduction of Trl4 in these brain areas, notably after LPS treatment, between the α2+/+ (n = 3) and α2+/G301R (n = 3) animals. The qPCR analysis of Tnf-α relative to β-actin expression in the hypothalamus (c) and hippocampus (d) showed that a significant decrease in Tnf-α mRNA expression was observed in astrocytes from α2+/G301R mice (n = 3) after LPS treatment compared with α2+/+ astrocytes (n = 3). All data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test followed by Dunn’s post hoc test).
Ligand-induced receptor dimerization is thought to initiate signal transduction through TLRs, which leads to the recruitment of signalling adaptor proteins and the regulation of TNF-α mRNA translation46,47. Therefore, we hypothesized that the reduced expression of Tlr4 in astrocytes from α2+/G301R mice could explain the reduced Tnf-α expression in hypothalamus and hippocampus of α2+/G301R mice. As the amount of astrocytes isolated form these brain areas are too low to measure TNF protein levels, we measured Tnf mRNA expression to investigate this, in astrocytes isolated from either the hypothalamus or the hippocampus of α2+/G301R mice compared to those isolated from α2+/+ (Fig. 5c,d), in line with the results of Tlr4 expression. In the hypothalamus there is a significant increase after LPS, though smaller than in α2+/+ astrocytes and in the hippocampus there is a significant reduction.
Our results demonstrated an increase in the proportion of Tnf/β-actin mRNA in α2+/+ mice astrocytes in the hypothalamus (Fig. 5c) and hippocampus (Fig. 5d) after treatment with LPS. Interestingly, we observed a reduction in Tnf transcription in hippocampal astrocytes from α2+/G301R mice after the challenge with LPS (Fig. 5d), supporting the results that astrocytes in these areas of the brain are differentially affected by α2 haploinsufficiency.
Together, these results show that the lack of the TNF-α response in the α2+/G301R mice is partly due to the lack of α2Na+/K+-ATPase expressing astrocytes that can sense LPS through induced Tlr4 expression.
Equal cytosolic levels of p65 in LPS-treated α2+/G301R hippocampus.
LPS binding to TLR4 initiates an intracellular signaling cascade that results in NFκB activation. The activation of the transcription factor NFκB is initiated upon a nuclear translocation. NFκB is an important transcriptional regulator of neuroinflammation that is activated by LPS and/or proinflammatory cytokines, such as TNF-α, which trigger intracellular localization of the NFκB complex. Therefore, next to further dissect the mechanisms of α2 isoform-mediated regulation of LPS response, we evaluated the nuclear translocation of NFκB in the hypothalamus and hippocampus of LPS-treated α2+/G301R mice. We examined nuclear protein levels via Western blotting using an antibody that recognize RelA (p65), a component of the NFκB complex. No significant changes were observed in cytoplasmic p65 levels in the hypothalamus (Fig. 6a, Supplementary Fig. 7), but there was a reduction in the hippocampus (Fig. 6b, Supplementary Fig. 8) of α2+/+ mice and the α2+/G301R mice treated with PBS or LPS. In the nucleus, active NFκB promotes the transcription of NFκB-responsive genes48,49, such as IL-1β and IL-6. Therefore, next we measured Il-6 and Il-1β mRNA levels in astrocytes isolated from both the α2+/+ and α2+/G301R mice subjected to PBS or LPS treatment. These results showed that Il-6 mRNA was upregulated in astrocytes isolated from the hypothalamus (Fig. 6c) and hippocampus (Fig. 6d) of α2+/+ and α2+/G301R mice after LPS treatment. Il-1β mRNA was significantly upregulated in astrocytes isolated from the hypothalamus (Fig. 6e) and the hippocampus of α2+/+ mice after LPS treatment (Fig. 6f). However, Il-1β mRNA was significantly reduced in hippocampal astrocytes of α2+/G301R mice after LPS treatment (Fig. 6f). Comparing the mRNA expression of Il-1b and Il-6 in whole hypothalamus (Fig. 4a–c) and Hippocampus (Fig. 4d–f), there is an overall agreement that the effect is very local to the astrocytes, as the specific cells that is subjected to α2 haploinsufficiency.
Figure 6.

LPS decreases the cytoplasmic fraction of of NFκB in the hippocampus. (a,b) Western blotting densitometric analysis (arbitrary units) and representative Western blots for p65 (NFκB) and β-actin in the hypothalamus (a) (blue bars) (n = 5 mice/group) and hippocampus (b) (red bars) of the α2+/+ (n = 5) and α2+/G301R (n = 5) animals 4 h after LPS treatment. All data are presented as the mean ± SEM. *p < 0.05 (Kruskal–Wallis test followed by Dunn’s post hoc test). RT-qPCR analysis of (c,d) Il-6 and (e,f) Il-1β relative to β-actin expression in astrocytes isolated from the hypothalamus (c,e) and hippocampus (d,f) showed significant increases after LPS treatment for all except Il-1β in hippocampus of α2+/G301R mice (n = 3 for both groups). All data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Kruskal–Wallis test followed by Dunn’s post hoc test).
This suggest that despite no significant nuclear translocation of NFκB was observed after LPS treatment in the α2+/G301R mice, there is however, a significant induction of Il-1b and Il-6 mRNA, suggesting either that the nuclear translocation assay needs to be performed in astrocyte populations, or supplementary mechanisms mediate the expression of the measured cytokines.
Hippocampal astrocytes from α2+/G301R mice express higher levels of Nrf2 than α2+/+ cells
The transcription factor NRF2 is an important regulator of the inflammatory response50,51. It regulates the expression of phase II detoxifying enzymes, including NADPH, NAD(P)H quinone oxidoreductase 1, glutathione peroxidase, ferritin hem oxygenase-1 (HO-1), and other genes that combat injury and inflammation52,53. Interestingly, p65 has been shown to activate NRF2 by sequestering the NRF2-regulator protein KEAP1 and thus leads to the transactivation of NRF2-dependent genes54–57. Therefore, next we investigated whether the NRF2 pathway is involved in the reduced LPS-induced neuroinflammation in the α2+/G301R mice by measuring Nrf2 expression in astrocytes of from hypothalamus and hippocampus. Interestingly, LPS induced Nrf2 upregulation in in hypothalamic (Fig. 7a) and hippocampal (Fig. 7b) astrocytes from the α2+/G301R and α2+/+ mice. Compared with the α2+/+ mice, the α2+/G301R mice constitutively expressed higher levels of Nrf2 in the hippocampus (Fig. 7b) suggesting that the astrocytes are affected by α2 haploinsufficiency even under naïve conditions.
Figure 7.

The α2 G301R mutation increases expression of the antioxidant enzyme NRF2-encoding transcript. (a–f) RT-qPCR mRNA expression analysis for Nrf2 (a,b); Ho-1 (c,d); Nqo-1 (e,f) in the hypothalamus (blue bars) and hippocampus (red bars). All data are the mean ± SEM of three individual experiments. *p < 0.05, *p < 0.01, and ***p < 0.001 (two-way ANOVA followed by Tukey’s post hoc test, n = 3–4 mice/group).
In conclusion, these results show that Nrf2 mRNA transcription is activated in the α2+/G301R mice and of relevance to LPS-induced response.
To study if the observed increase in Nrf2 mRNA expression in LPS-treated hypothalamus and hippocampus of the α2+/G301R and α2+/+ mice resulted in subsequent expression of NRF2-responsive genes, we measured the expression of the NRF2-responsive genes Ho-1 and Nqo158 in astrocytes from the hypothalamus and hippocampus of α2+/+ and α2+/G301R mice.
LPS treatment resulted in increased Ho-1 expression in astrocytes from the hypothalamus (Fig. 7c) and hippocampus (Fig. 7d) of α2+/+ mice compared to that in PBS-treated mice.
However, LPS did not lead to an increase in Ho-1 expression in the hypothalamus or in the hippocampus of α2+/G301R mice (Fig. 7c,d). No differences in Nqo1 expression between the hypothalamic astrocytes from the α2+/G301R and α2+/+ mice were observed after LPS treatment (Fig. 7e,f), while Nqo1 expression was increased in the hippocampal astrocytes from the of α2+/G301R mice 4 h after treatment with LPS (Fig. 7f).
Combined this indicate that Nfr2 expression increases after LPS treatment in both hypothalamus or in the hippocampus of α2+/+ mice and α2+/G301R mice. Of NRF2-responsive genes, Ho-1 mRNA appears reduced in astrocytes from hypothalamus of α2+/G301R mice compared to α2+/+ mice, however, only a difference was observed for Nqp-1 mRNA in astrocytes from hippocampus of α2+/G301R mice compared to α2+/+ mice.
α2 haploinsufficiency correlate directly with the increased expression of Cox2 transcripts in hippocampal astrocytes
LPS-mediated inflammation triggers the expression level of COX genes, major inflammatory factors59. To determine the role of the α2 isoform in LPS-induced Cox1 and Cox2 expression, we performed RT-qPCR analysis on astrocytes isolated from the hypothalamus and hippocampus of the PBS- and LPS-treated α2+/+ and α2+/G301R mice. We found that there was no difference in the expression of the Cox1 gene in the hypothalamic astrocytes of all groups evaluated (Fig. 8a). In α2+/G301R astrocytes from the hippocampus, we noted that Cox1 gene expression was significantly increased after LPS treatment, in contrast to PBS-treated α2+/G301R hippocampal astrocytes (Fig. 8b).
Figure 8.

α2 haploinsufficiency alters Cox1 and Cox2 gene expression in astrocytes from the hypothalamus and hippocampus of α2+/+ mice and α2+/G301R mice. (a–d). qPCR mRNA expression analysis for Cox1 (a,b); Cox2 (c,d). All data are the mean ± SEM of three individual experiments. *p < 0.05, *p < 0.01, and ***p < 0.001 (Kruskal–Wallis test followed by Dunn’s post hoc test, n = 3 mice/group).
The Cox2 mRNA expression in α2+/+ mice astrocytes was upregulated in hypothalamus after the challenge with LPS (Fig. 8c), but unchanged in hippocampus (Fig. 8d).
In astrocytes from the hypothalamus, Cox2 mRNA was significantly elevated in PBS-treated α2+/G301R mice, with no further induction was noted in astrocytes from the α2+/G301R mice upon LPS treatment (Fig. 8c). In astrocytes from the hippocampus, the Cox2 levels in the α2+/+mice were not altered (Fig. 8d). In contrast, Cox2 mRNA expression was significant upregulated in LPS-treated hippocampal astrocytes from the α2+/G301R mice (Fig. 8d).
Overall, this correlates very well with the tendency of increased nuclear levels of p65, which suggests increased NFκB activity in astrocytes from the hippocampus (Fig. 6b) and is responsible for high Cox2 expression59, as noted in the same cells (Fig. 8b).
LPS-induced memory impairment and anxiety-like behaviours are not significantly altered in α2+/G301R mice compared with WT littermates
Because long-lasting effects in memory and behavior have been observed following LPS administration (reviewed in60), we wished to examine if the α2+/G301R mice were susceptible for behavior changes upon LPS administration.
To assess whether the α2 isoform is involved in the cognitive and affective LPS-mediated effects, first, we examined the locomotor activity of the α2+/G301R mice versus the α2+/+ mice treated with saline or LPS using the open field test 4 h after PBS or LPS administration. LPS induced a reduction in locomotion and anxiety-like behaviours in both the α2+/+ and α2+/G301R mice (Fig. 9a,b). There was no difference in the total distance travelled (P = 0.5806) or the time spent in the middle zone of the arena (P = 0.7457) by LPS-treated α2+/+ and α2+/G301R mice. Similarly, no differences were observed between the two types of mice after PBS treatment.
Figure 9.

α2Na+/K+-ATPase isoform does not alter the memory impairment or anxiety that is induced by LPS. (a,b) Results of analysis of behaviour in the open-field test 4 h after LPS injection showing the total distance travelled (a) and time spent in the centre of the open field (b) (n = 10–12). (c) The passive avoidance test results (n = 10–12). The training and test were performed 24 and 72 h after LPS injection, respectively. All data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 (Kruskal–Wallis test followed by Dunn’s post hoc test).
Next, we investigated whether α2 isoform haploinsufficiency can influence LPS-induced memory impairment. To this end, we assessed fear memory using the passive avoidance test that measures the latency to enter a dark environment in which an aversive stimulus (foot shock) has been previously experienced using a light–dark box paradigm. All four groups (PBS- and LPS-treated α2+/+ and α2+/G301R mice) showed similar baseline latencies to enter the dark chamber in the training stage (Supplementary Fig. 9). We evaluated the latency for entry to the dark side of the box before the challenge with LPS and observed that the isoform α2-Na,K-ATPase mutation does not interfere with the time of entry to the chamber in the training stage (Supplementary Fig. 9a). Despite the increased latency time for α2+/G301R mice, in the test, after treatment with LPS, it showed that α2+/G301R mice are less cognitively affected than α2+/+ animals, after treatment with LPS (Fig. 9c), and we did not observe differences in latency between the mutated animals treated with PBS or LPS (Supplementary Fig. 9b).
The α2+/+ and α2+/G301R mice treated with PBS showed a significant increase in latencies in the probe stage, compared to LPS-treated α2+/+ mice (Fig. 9c). The increased time in latency was found significant for α2+/G301R mice, in the test, after LPS treatment, suggesting that the α2+/G301R mice are less cognitive affected than their littermates, after LPS treatment.
Collectively, our results suggest that the reduction of the α2 isoform does not interfere with the LPS-induced decrease in locomotor activity, increase in anxiety behaviours, or cognitive impairment. Activation of the immune system by LPS leads to production and release of proinflammatory cytokines such as TNF-α, IL-1β that act on the periphery and central nervous system leading to symptoms such as immobility and/or lethargy, piloerection, drowsiness, and ptosis, this symptoms are knowing as sickness behavior61, which may be accompanied by physiological changes such as hypothermia. The sickness behavior reduces with the process of resolving inflammation, but motivational deficits may last longer, such as anxiety. Given this, we can infer that the reduction in α2Na+/K+-ATPase activity interferes with sickness behavior, since we observed a less locomotion and hypothermia (Fig. 1a,b). However, there was no motivational change as observed by the open field test (Fig. 9a,b).
Discussion
Neuroinflammation is a critical factor in neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease. We used a gene modified mouse with a knock-in mutation to investigate the role of the vital membrane ion pump Na+/K+-ATPase in mediating neuroinflammation. In recent years, there has been increasing evidence that the pump, the Na+/K+-ATPase, serve not only to maintain the electrochemical gradient across the cell’s plasma membrane, but are highly involved in regulating intracellular signal cascades.
We report that mice that are haploinsufficient for the astrocyte-enriched α2Na+/K+-ATPase isoform (α2+/G301R mice) have a reduced proinflammatory response to LPS, accompanied by a reduced hypothermic reaction. Following administration of LPS, the gene expression of TNF-α, IL-1β, and IL-6 was reduced in the hypothalamus and hippocampus from α2+/G301R mice compared to WT littermates (Fig. 10).
Figure 10.
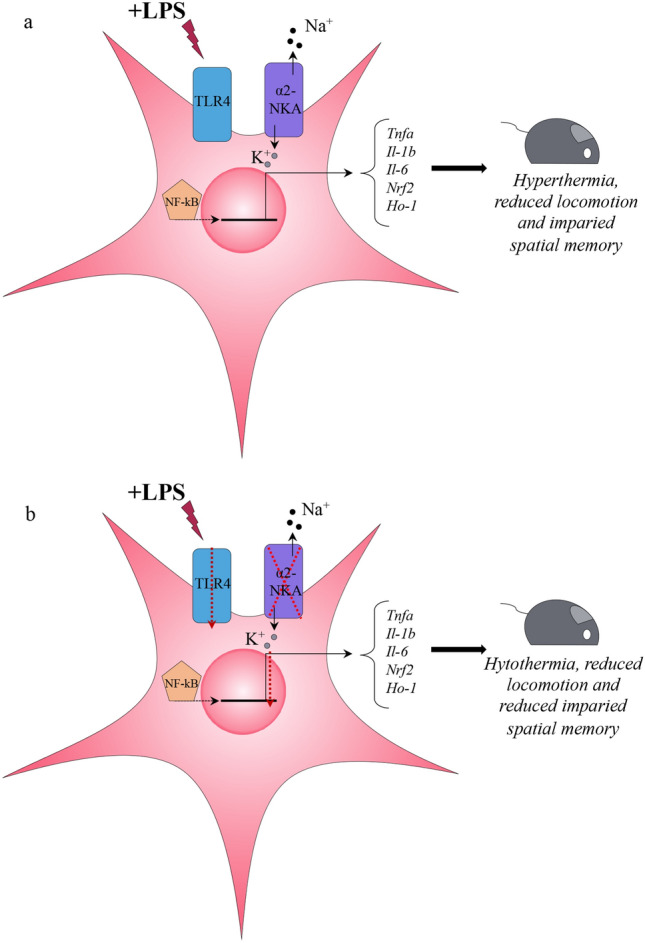
Schematic drawing of the proposed action upon LPS treatment in astrocytes expressing the α2Na+/K+-ATPase (noted α2NKA in the figure). (a) In astrocytes with normal α2Na+/K+-ATPase function, the intracellular responses to LPS will lead to an increasement of several inflammatory genes and cause LPS-related phenotypes in the mouse, including hypothermia, reduced locomotion and impaired spatial memory. (b) In astrocytes with α2Na+/K+-ATPase haploinsufficiency, the intracellular responses to LPS will lead to a significantly reduced expression of several inflammatory genes and compromise LPS-related phenotypes in the mouse, leading to less hypothermia, reduced locomotion and reduced impaired spatial memory.
The central nervous system regulates systemic inflammatory responses to endotoxin through humoral mechanisms via activation of the hypothalamic–pituitary–adrenal pathway, as well as through the activation of efferent vagal nerve pathways by reducing the production of pro-inflammatory cytokines produced by macrophages62,63. Therefore, we can assume that neuroinflammation reduction observed in the α2+/G301R animals could be due to a possible interference in the neuroimmune pathways, however studies must be carried out to understand this interference. Furthermore, activation of the immune system by LPS leads to production and release of proinflammatory cytokines such as TNF-α, IL-1β that act on the periphery and central nervous system leading to symptoms such as immobility and/or lethargy, piloerection, drowsiness, and ptosis, this symptoms are knowing as sickness behavior, which may be accompanied by physiological changes such as hypothermia. The sickness behavior reduces with the process of resolving inflammation, but motivational deficits may last longer, such as anxiety. Given this, we can infer that the reduction in α2Na+/K+-ATPase activity interferes with sickness behavior, since we observed a less locomotion and hypothermia (Fig. 1a,b). However, there was no motivational change as observed by the open field test (Fig. 9a,b). The α2+/G301R mice experienced increased expression of the gene encoding an antioxidant enzyme, Nrf2, in hippocampal astrocytes.
Here, we demonstrated by complementary in vitro and in vivo experimentation that the production of proinflammatory cytokines upon LPS stimuli is highly dependent on the astrocyte-specific α2 isoform of Na+/K+-ATPase. α2 Haploinsufficiency differentially altered the production of TNF-α, IL-6, and IL-1 in LPS-treated astrocytes. This appeared to be associated with a reduced Tlr4 expression.
The LPS-induced immune response in hypothalamus and hippocampus is different and may very well associated differential pathway activation promoted by TLR4, such as NFkB and MAPK, but may also involve other pathways64, yet to be elucidated. Interestingly, this caused a decrease in the hypothermic response in α2+/G301R mice compared with their α2+/+ littermates, which might be associated with a reduced expression of Nrf2, a mediator of inflammatory pathways50,51, in the hypothalamus in response to LPS. The LPS induced impairment of fear memory and anxiety-like behaviours observed in α2+/+ mice was not affected by α2 isoform haploinsufficiency.
Hypothermia is a thermoregulatory response to systemic inflammation that can be induced in rodents in response to systemic LPS challenge65,66. Although the molecular mechanisms of LPS-induced hypothermia are still poorly understood, the participation of cytokines has been observed in the development of hypothermia67. The reduced cytokine levels we found in α2+/G301R mice could explain the lower hypothermic response in these animals following LPS treatment.
Previous findings have shown that ouabain, which acts as a Na+/K+-ATPase inhibitor, can protect motor neurons from mutant SOD1–induced astrocyte degeneration and reduce the neuroinflammation resulting from LPS36,44. In addition, haploinsufficiency-causing Na+/K+-ATPase α2 isoform G301R mutation decreases lesion volume and improves functional outcomes after acute spinal cord injury in mice35. Furthermore, silencing of α2-Na+/K+-ATPase reduces LPS-induced inflammation in glial cells39.
We previously studied TNF, IL-6 and IL-10 production in the spinal cord of α2+/G301R and α2+/+ mice with spinal cord injury35. We found upregulation of these cytokines in mice with spinal cord injury compared to uninjured mice but we observed no significant differences between the two genotypes; only IL-1β appeared to display a tendency to be upregulated35, but it is important to consider that IL-1β is produced as precursor molecules, pro-IL-1β, which is cleaved in its active form by a caspase family cysteine protease, the IL-1β converting enzyme (ICE)68. Although there are different mechanisms proposed for post-translational processing of IL-1β, there is still no definite mechanism. Our data show that LPS challenge promoted a serum increase of IL-1β that was not reversed in a2 gene modified animals, thus suggesting that deficiency in a2 activity does not interfere with the release of IL-1β in its mature form. Combined, these previous and our new findings support a role for the α2 isoform of Na+/K+-ATPase in LPS-induced neuroinflammation, by means of mediating cytokine expression.
LPS induces an innate immune response and the production of various proinflammatory cytokines via TLR4 activation. In view of this, we hypothesized and confirmed that α2Na+/K+-ATPase haploinsufficiency prevents an increase in Tlr4 expression after LPS exposure in astrocytes of the hypothalamus and hippocampus.
Recently, a study showed that neuronal activity regulates the astrocytic signalling of the nuclear master transcription factor NRF2 through the secretion of glutamate and other soluble factors69. Thus, we hypothesized that α2+/G301R mice constitutively exhibit an increase in NRF2 activity that protects these animals from LPS-induced neuroinflammation, as NRF2 is capable of negatively regulating NFkB activity and the consequent reduced expression of pro-inflammatory cytokines. Confirming this hypothesis, we found that Nrf2 expression was upregulated in hippocampus, and Nrf2 expression was further enhanced upon LPS treatment, suggesting this as one mechanism that reduce NFkB activity in cells haploinsufficient for the α2 isoform.
Moreover, the NRF2-responsive antioxidant factor Nqo-1 mRNA was upregulated in the hippocampus in α2+/G301R mice in the absence of inflammatory stimulation, and only marginally increased upon LPS treatment, suggesting the involvement of NRF2 activation in the reduced neuroinflammatory response to LPS exposure in α2+/G301R mice. This is in line with the fact that NQO-1 exhibits anti-inflammatory activity by inhibiting the induction of TNF and IL-1β expression by LPS in human monocytes51,70.
Peripheral LPS administration promotes NFκB activation in various regions of the central nervous system, leading to the stimulation of proinflammatory cytokines71. Our results confirmed that LPS-induced activation of NFĸB occurred by measuring reduced levels of cytoplasmic p65, which increased proinflammatory genes within 4 h in the hippocampus of α2+/+ mice.
IL-1β has been associated with cognitive impairment during the inflammatory process, and the intra-hippocampal administration of IL-1β induces impaired memory consolidation and reconsolidation in rats72. Although our results showed a reduction in IL-1β expression in the hippocampus 4 h after LPS administration in α2+/G301R animals, we observed that that α2 haploinsufficiency does not affect the LPS-induced effect on memory impairment and anxiety.
While the present study addressed the regulation of cytokines in relation to the Na+/K+-ATPase in astrocytes from hypothalamus and hippocampus, future studies must delineate the mechanisms in other brain structures as well as the α2 isoform is expression in astrocytes throughout the brain. Of immediate interest in this context is the synergy between signaling pathways that mediate the α2-dependent LPS-induced neuroinflammation. Studies have demonstrated the existence of an important interaction between adipocytes and macrophages for the amplification of the inflammatory response induced by LPS73, and knowing that adipocytes express the α2Na+/K+-ATPase isoform74,75, it is likely that the a2 isoform in adipocytes contributes to the reduction of circulating levels of cytokines, however, this remains to be explored.
In summary, this study provides an undescribed link between the α2Na+/K+-ATPase and inflammation signaling in vivo. Overall, regulation of the astrocyte α2 isoform represents a significant regulator of inflammatory responses in the brain, with makes the α2 isoform a likely candidate for depressing neuroinflammation, and perhaps also neurodegenerative conditions.
Methods
Experimental animals
Mice were cared for in accordance with the protocols and guidelines that The Danish Animal Inspectorate approved under the Ministry of Food and Agriculture, Denmark (J. No. 2013-15-2934-00815 to KLH). We confirm that all experimental protocols were approved by the Animal Facility and the veterinary surgeon at the Department of Biomedicine, Aarhus University, Denmark.
Our mice were bred on a C57/BL6JRj (Janvier) background. All animals were housed under a reverse light/dark cycle to prevent daytime experiments from interfering with their normal sleep cycles.
All experiments were performed on 8- to 12-week-old α2+/G301R mice and α2+/+ mice, and all mice used are summarized in Table 1.
Table 1.
Distribution of genotype and sex of animals used.
| Sex | Genotype | Biochemical | Behavior | Macrophages culture | Body temperature | Total |
|---|---|---|---|---|---|---|
| Female | α2+/+ | 26 | 23 | 3 | 11 | 63 |
| Female | α2+/G301R | 12 | 22 | 3 | 5 | 42 |
| Male | α2+/+ | 16 | 10 | 3 | 9 | 38 |
| Male | α2+/G301R | 14 | 11 | 4 | 10 | 39 |
The animals were housed in ventilated cages with 1–3 cage mates under a 12-h light/dark cycle and controlled temperature and humidity and with free access to food and water. The animals were treated with 500 µg/kg LPS (#L2630, O11:B4) (Sigma-Aldrich, St. Louis, MO) or PBS. Four hours after LPS administration, the mice were anaesthetized by isoflurane inhalation and euthanized by decapitation, and the brains were immediately removed and immersed in cold PBS. The hippocampus and hypothalamus were rapidly dissected, quickly immersed in liquid nitrogen, and stored at − 80 °C for later use.
Genotyping
Heterozygous α2+/G301R mice and α2+/+ mice were genotyped33 via high resolution melt analysis (Roche LightCyclerR 96 Real-Time PCR System) using the following primers: F, 5′-ggatgagggacagaacgaag and R, 5′-catggagatcgagcatttca (Sigma-Aldrich).
Cell culture procedures
To isolate bone marrow–derived macrophages (BMDMs), bone marrow was isolated from the femurs and tibias of 9-week-old WT and α2+/G301R mice, and the cells cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% heat-inactivated foetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO), 100 U/mL penicillin/streptomycin, and 20% of L929 conditioned media (Sigma-Aldrich, St. Louis, MO) at 37 °C in 5% CO276,77. On day 2, 5 mL of supplemented RPMI 1640 medium and 40% L929 conditioned medium was added. On day 4, the non-adherent cells were removed from the flask, the media was replaced, and the remaining adherent cells were maintained in culture for 6 days in 20% L929 conditioned medium. On day 7, the cells were transferred to 24-well plates (4 × 105 cells per well) and cultured for 4 h before use. The cells were treated with PBS or LPS (100 ng/mL) for 1, 2, 4, and 6 h. Supernatant samples were collected for the analysis of TNF-α via ELISA.
Body temperature
The rectal body temperature was measured using a rectal probe (TFN 530 SMP Thermometer, Ebro) at the time of the first injection and 4 h after the injection of LPS or PBS.
Measurement of cytokine levels
Four hours after LPS or PBS injection, blood was collected in 15-mL conical tubes and centrifuged at 3,000 rpm for 10 min to obtain the serum. The concentrations of TNF-α (#88-7324), IL-1β (#MLB00C), IL-6 (#M6000B), and IFN-γ (#MIF00) were measured by mouse-specific sandwich ELISA according to the manufacturer’s instructions (eBioScience, Santa Clara, California, USA and R&D Systems, Minneapolis, USA)78. Briefly, samples were added to coated microwells with antibodies against TNF-α, IL-1β, IL-6, and IFN-γ along with a biotin-conjugated antibody (horseradish peroxidases; Mouse IL-1β Conjugate # 893830), polyclonal antibody specific for mouse IL-1β conjugated to horseradish peroxidase with preservatives (R&D Systems), Mouse IL-6 Conjugate # 892665, polyclonal antibody against mouse IL-6 conjugated to horseradish peroxidase with preservatives (R&D Systems), Mouse IFN-γ Conjugate (# 892666, polyclonal antibody specific for mouse IFN-γ conjugated to horseradish peroxidase with preservatives (R&D Systems). The Mouse TNF-α Conjugate (#88-7324), (eBioScience). The plate was incubated for 2 h at room temperature. The wells were washed, streptavidin-HRP was added to the entire plate, and the plate was incubated for 1 h at room temperature. Subsequently, the wells were washed and then incubated with TMB substrate (Thermo Fisher Scientific, Roskilde, Denmark) solution for 30 min while being protected from light. The reaction was stopped with a stop solution, and the absorbance was measured using a spectrophotometer at 450 nm. The concentrations of the cytokines were measured based on the standard curve.
Protein extraction and immunoblot analysis
The hippocampus and hypothalamus were isolated according to the protocol of the CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich, St. Louis, MO). In brief, the tissues were homogenized in lysis buffer containing 10 mM HEPES (pH 7.9) with 1.5 mM MgCl2, 10 mM KCl, 0.1 M dithiothreitol (DTT) solution and a protease inhibitor cocktail, and they were centrifuged at 10,000×g for 20 min. The supernatant representing the cytosolic fraction was transferred to a new tube. The pellet was resuspended in an extraction buffer containing 1.5 μL 0.1 M DTT and 1.5 μL protease inhibitor cocktail. The solution was allowed to stand on ice for 30 min with shaking at brief intervals followed by centrifugation at 20,000×g for 5 min. The supernatant, which contained the nuclear protein fraction, was transferred to a clean chilled tube. The proteins from the cytosolic fractions of the hippocampus and hypothalamus (20 μg) were separated by size by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The proteins were blotted onto a nitrocellulose membrane (Pharmacia-Amersham, Amersham, UK) and incubated with anti-α1 (1:500) (a6f.-c, Developmental Studies Hybridoma Bank, USA), anti-α2 (1:5,000) (07674, EMD Millipore, USA), anti-p65 (1:1,000) (CST-8242T, Cell Signalling), and actin (1:1,000) (A2066, Sigma-Aldrich,) primary antibodies overnight at 4 °C33. The secondary antibodies included horseradish peroxidase-conjugated pig anti-rabbit and pig anti-mouse (1:2,000) (Dako, Glostrup, Denmark) antibodies. The proteins recognized by the antibodies were revealed via an Amersham ECL Western Blotting Detection Kit, following the instructions of the manufacturer (GE Healthcare, Buckinghamshire, UK). To standardize and quantify the immunoblots, we used the photo documentation system of the LAS 3000 imager (Fujifilm, Tokyo, Japan) and ImageJ software (US National Institutes of Health, Bethesda, MD; https://rsb.info.nih.gov/ij), respectively. Several exposure times were analysed to ensure the linearity of the band intensities. β-Actin was used as an internal control for the experiments. The results are expressed in relation to the intensity of β-actin.
Astrocyte dissociation
The hippocampus and hypothalamus tissues were dissociated using the Neural Tissue Dissociation Kit—Postnatal Neurons (catalogue number 130094802), as described by the manufacturer. Briefly, the structures were weighed and transferred to a gentleMACS C tube containing 1960 μL of enzyme mix (50 μL of enzyme P + 1910 μL of buffer Z) and 45 μL of enzyme mix 2 (30 μL of buffer Y + 15 μL of enzyme A). Then, the tubes were connected to the gentleMACS Octo Dissociator with Heaters, and the gentleMACS 37C_NTDK_1 programme was used for tissue dissociation. Then, the samples were centrifuged briefly, and the cells were resuspended in D-PBS. The cell suspension was filtered with a 70-μm cell strainer (MACS SmartStrainer), which was washed with 10 mL of D-PBS supplemented with BSA (0.5%). Thereafter, the cell suspension was centrifuged at 300×g for 10 min at room temperature, and the cells were suspended in D-PBS supplemented with BSA (0.5%).
Myelin debris removal
Prior to the isolation of astrocytes, we performed a debris-removal step using protocols from Miltenyi Biotec’s Myelin Removal Kit (catalogue number 130096733). Following dissociation, the cells were incubated for 15 min at 4 °C with Myelin Removal Beads II, following the ratio of 500 mg of brain tissue in 1,800 μL of buffer + 200 μL of myelin removal beads)45. The cells were washed with 0.5% BSA in PBS and centrifuged at 300×g for 5 min to remove any unbound spheres from the pellet. After that, the pellet was resuspended in 500 μL of buffer, the suspension was added to a LS column prepared in the MACSMidi magnetic cell separator, and the flow-through was collected. The column was further washed three times with 3 mL of buffer to ensure the removal of the unlabelled cells. The cells retained on the column were eluted in 5 mL of buffer. The flow-through was used in subsequent steps for astrocyte isolation.
Isolation of astrocyte cells
The astrocytes were positively selected using the protocol of the Miltenyi Biotec Anti-ACSA-2 Kit (catalogue number 130097678). Up to 1 × 107 dissociated cells were suspended in 80 μL of buffer (0.5% BSA in PBS) and incubated with 10 μL of FcR blocking buffer for 10 min at 4 °C, followed by incubation with 10 μL of ACSA-2 MicroBeads for 10 min at 4 °C. Then, the cells were washed with 1 mL of buffer (0.5% BSA in PBS) and centrifuged at 300×g for 10 min to remove the excess beads from the solution. After the removal of the lavage solution, the pellet was resuspended in 500 μL of buffer, and the suspension was added to a prepared LS column installed in the MACSMidi magnetic cell separator. The column was washed with 3 mL of buffer three successive times to remove the unlabelled cells. After the column was removed from the magnetic separator, the astrocytes were eluted in 5 mL of buffer. The number of cells was then determined, and total RNA was extracted.
Reverse transcription quantitative PCR (RT-qPCR)
Total RNA was isolated from astrocytes isolated from astrocytes from the hippocampus and hypothalamus with the RNeasy Plus Mini Kit (Qiagen), according to the manufacturer’s instructions. Complementary deoxyribonucleic acid (cDNA) was generated from 500 ng total RNA using the PrimeScript RT Reagent Kit (Takara BIO INC). The Tnfα, Il-1β, Il-6, Tlr4, Nrf2, Ho-1, and Nqo-1β, Cox1/2, Ptgds and β-actin (our reference) gene expression levels were measured via quantitative PCR (qPCR) using the TaqMan gene expression assays (Thermo Fisher Scientific) noted below:
| Gene | Primer ID |
|---|---|
| Tnf-α | Mm00443258_m1 |
| Il-1β | Mm00434228_m1 |
| Il-6 | Mm00446190_m1 |
| Tlr4 | Mm00445273_m1 |
| Nrf2 | Mm00477784_m1 |
| Ho-1 | Mm00516005_m1 |
| Nqo-1β | Mm01253561_m1 |
| Cox1 | Mm04225243_g1 |
| Cox2 | Mm03294838_g1 |
| β-Actin | Mm02619580_g1 |
Real-time PCR analysis was performed in triplicate, with each reaction including 20 ng of cDNA, 5 μL of 2 × LightCycler 480 Probed Master Mix. (Roche, Basel, Switzerland), 0.5 μL of TaqMan Gene Expression Assay, and nuclease-free water up to a final volume of 10 μL. The PCRs were run in the Roche LightCycler 96-well system with the following protocol: 2 s at 50 °C for uracil-DNA glycosylase enzyme activation, 10 min at 95 °C for DNA Polymerase activation, 45 cycles of 15 s at 95 °C for denaturation, and 1 min at 60 °C for annealing and extension followed by a final denaturation step of 95 °C for 5 min79,80. The triplicate expression values of each gene were set relative to the expression of the reference gene via the delta-delta-Ct method81. As a negative control, RT-PCRs with no template were used.
Behavioural analysis
Open field test
The mice were placed in the centre of an open-field apparatus (50 × 50 cm) (Stoelting Europe; Dublin, Ireland) and monitored for 15 min using ANY-maze software V4.99 (Stoelting, USA)82. The system automatically recorded the total distance travelled (m) and the time (s) spent in the centre zone. Each mouse (n = 10–12 for each group) was tested once 4 h after PBS or LPS injection, and the open-field setup was cleaned with 70% ethanol and wiped with paper towels between each trial.
Passive avoidance test
The training was initiated on the acquisition day, 24 h after PBS or LPS injection. Each mouse was placed in a brightly lit compartment with an electronically controlled door leading into a dark compartment82. The latency (s) for the mouse to enter the dark compartment was recorded. Once in the dark compartment, the door closed, and the mouse received an electric shock (0.42 mA for 1 s). The test was performed 72 h after PBS or LPS injection, the mouse was reintroduced to the same brightly lit compartment, and the latency to enter the dark compartment was recorded as an indicator of memory for the shock.
Statistical analysis
The qPCR results were analysed via the delta-delta-Ct method, according to Schmittgen and Livak (2008) and calculated using REST 2009 Software (Qiagen, Duesseldorf, Ger). Normality was assessed through the D’Agostino and Pearson omnibus normality test, and, for parametric analyses. Parametric analyses were conducted through two-way ANOVA followed by Tukey’s post-test. Non-parametric analyses were conducted through Kruskal–Wallis test followed by Dunn’s post hoc test. Differences were considered to be significant at p < 0.05, and all results are expressed as the mean ± standard error of the mean (SEM) of the indicated number of experiments. All analyses were performed using the Prism 6 software package (GraphPad Software, San Diego, CA, USA).
Supplementary information
Acknowledgements
We are grateful to Dr. Rune Hartmann, Department of Molecular Biology and Genetics at Aarhus University for valuable suggestions regarding innate immunity and to Drs. Christian Kanstrup Holm and Martin Thomsen, Department of Biomedicine at Aarhus University. for their advice regarding BMDMs. The research was funded by Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP 2016/07427-8), Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq. J.A.L. is supported by a PhD fellowship from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2016/21343-1 and 2014/10171-0), and C.S. is a research fellow of CNPq. T.J.I. was 2/3 co-funded by The Danish research Centre of excellence, PUMPkin (DNRF85 to KLH) and 1/3 co-funded by the Graduate School of Health, Aarhus University. KLH received support through grants from the Lundbeck Foundation (J. Nr. 234/06), Th. Maigaards Eft. Fru Lily Benthine Lunds Fond, and Fonden til Lægevidenskabens Fremme.
Author contributions
J.A.L., C.S., and K.L.-H. conceived and designed the experiments. J.A.L., T.J.I., and A.H. performed the experiments. J.A.L, T.J.I., A.H., C.S., and K.L.-H. analyzed the data. J.A.L., C.S., and K.L.-H. composed the manuscript.
Data availability
We confirm that all relevant data from this study are available from the corresponding author upon request.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-71027-5.
References
- 1.Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta. 1957;23:394–401. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- 2.Dobretsov M, Stimers JR. Neuronal function and alpha3 isoform of the Na/K-ATPase. Front. Biosci. 2005;10:2373–2396. doi: 10.2741/1704. [DOI] [PubMed] [Google Scholar]
- 3.Cui X, Xie Z. Protein interaction and Na/K-ATPase-mediated signal transduction. Molecules. 2017 doi: 10.3390/molecules22060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian J, et al. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell. 2006;17:317–326. doi: 10.1091/mbc.e05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000;275:27832–27837. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 6.Aperia A. New roles for an old enzyme: Na, K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007;261:44–52. doi: 10.1111/j.1365-2796.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Xie ZJ. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta. 2010;1802:1237–1245. doi: 10.1016/j.bbadis.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 2007;293:C509–C536. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, Langhans SA. Transcriptional regulators of Na, K-ATPase subunits. Front. Cell Dev. Biol. 2015;3:66. doi: 10.3389/fcell.2015.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tupler R, Perini G, Green MR. Expressing the human genome. Nature. 2001;409:832–833. doi: 10.1038/35057011. [DOI] [PubMed] [Google Scholar]
- 11.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 12.Xie J, et al. Expression of rat Na-K-ATPase alpha2 enables ion pumping but not ouabain-induced signaling in alpha1-deficient porcine renal epithelial cells. Am. J. Physiol. Cell Physiol. 2015;309:C373–C382. doi: 10.1152/ajpcell.00103.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H, et al. Heterogeneity of signal transduction by Na-K-ATPase alpha-isoforms: role of Src interaction. Am. J. Physiol. Cell Physiol. 2018;314:C202–C210. doi: 10.1152/ajpcell.00124.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann. N. Y. Acad. Sci. 1997;834:524–536. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Songu-Mize E. Effect of Na+ on Na+, K+-ATPase alpha-subunit expression and Na+-pump activity in aortic smooth muscle cells. Eur. J. Pharmacol. 1998;351:113–119. doi: 10.1016/s0014-2999(98)00278-7. [DOI] [PubMed] [Google Scholar]
- 16.Golovina V, Song H, James P, Lingrel J, Blaustein M. Regulation of Ca2+ signaling by Na+ pump alpha-2 subunit expression. Ann. N. Y. Acad. Sci. 2003;986:509–513. doi: 10.1111/j.1749-6632.2003.tb07236.x. [DOI] [PubMed] [Google Scholar]
- 17.Correll RN, et al. Overexpression of the Na+/K+ ATPase alpha2 but not alpha1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 2014;114:249–256. doi: 10.1161/CIRCRESAHA.114.302293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundborg C, Westerlund A, Bjorklund U, Biber B, Hansson E. Ifenprodil restores GDNF-evoked Ca(2+) signalling and Na(+)/K(+)-ATPase expression in inflammation-pretreated astrocytes. J. Neurochem. 2011;119:686–696. doi: 10.1111/j.1471-4159.2011.07465.x. [DOI] [PubMed] [Google Scholar]
- 19.McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na, K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 1991;11:381–391. doi: 10.1523/JNEUROSCI.11-02-00381.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bottger P, et al. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 2011;519:376–404. doi: 10.1002/cne.22524. [DOI] [PubMed] [Google Scholar]
- 21.D'Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J. Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- 22.Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J. Physiol. 2000;522(Pt 3):427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorgensen PL, Hakansson KO, Karlish SJ. Structure and mechanism of Na, K-ATPase: functional sites and their interactions. Annu. Rev. Physiol. 2003;65:817–849. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- 24.Larsen BR, et al. Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia. 2014;62:608–622. doi: 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isaksen TJ, Lykke-Hartmann K. Insights into the pathology of the alpha2-Na(+)/K(+)-ATPase in neurological disorders; lessons from animal models. Front. Physiol. 2016;7:161. doi: 10.3389/fphys.2016.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietrobon D. Familial hemiplegic migraine. Neurotherapeutics. 2007;4:274–284. doi: 10.1016/j.nurt.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Leo L, et al. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. 2011;7:e1002129. doi: 10.1371/journal.pgen.1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda K, et al. Malfunction of respiratory-related neuronal activity in Na+, K+-ATPase alpha2 subunit-deficient mice is attributable to abnormal Cl- homeostasis in brainstem neurons. J. Neurosci. 2004;24:10693–10701. doi: 10.1523/JNEUROSCI.2909-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeda K, et al. Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump alpha2 subunit (Atp1a2)-deficient mice. J. Neurosci. 2003;23:4667–4676. doi: 10.1523/JNEUROSCI.23-11-04667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James PF, et al. Identification of a specific role for the Na, K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell. 1999;3:555–563. doi: 10.1016/S1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 31.Santoro L, et al. A new Italian FHM2 family: clinical aspects and functional analysis of the disease-associated mutation. Cephalalgia. 2011;31:808–819. doi: 10.1177/0333102411399351. [DOI] [PubMed] [Google Scholar]
- 32.Spadaro M, et al. A G301R Na+/K+ -ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics. 2004;5:177–185. doi: 10.1007/s10048-004-0183-2. [DOI] [PubMed] [Google Scholar]
- 33.Bottger P, et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016;6:22047. doi: 10.1038/srep22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kros L, Lykke-Hartmann K, Khodakhah K. Increased susceptibility to cortical spreading depression and epileptiform activity in a mouse model for FHM2. Sci. Rep. 2018;8:16959. doi: 10.1038/s41598-018-35285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellman DG, et al. The loss-of-function disease-mutation G301R in the Na(+)/K(+)-ATPase alpha2 isoform decreases lesion volume and improves functional outcome after acute spinal cord injury in mice. BMC Neurosci. 2017;18:66. doi: 10.1186/s12868-017-0385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallardo G, et al. An alpha2-Na/K ATPase/alpha-adducin complex in astrocytes triggers non-cell autonomous neurodegeneration. Nat. Neurosci. 2014;17:1710–1719. doi: 10.1038/nn.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawamoto EM, Scavone C, Mattson MP, Camandola S. Curcumin requires tumor necrosis factor alpha signaling to alleviate cognitive impairment elicited by lipopolysaccharide. Neurosignals. 2013;21:75–88. doi: 10.1159/000336074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasconcelos AR, et al. Intermittent fasting attenuates lipopolysaccharide-induced neuroinflammation and memory impairment. J. Neuroinflamm. 2014;11:85. doi: 10.1186/1742-2094-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinoshita PF, et al. Alpha 2 Na(+), K(+)-ATPase silencing induces loss of inflammatory response and ouabain protection in glial cells. Sci. Rep. 2017;7:4894. doi: 10.1038/s41598-017-05075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015;72:4111–4126. doi: 10.1007/s00018-015-1995-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lingrel JB. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na, K-ATPase. Annu. Rev. Physiol. 2010;72:395–412. doi: 10.1146/annurev-physiol-021909-135725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cavalcante-Silva LHA, et al. Much more than a cardiotonic steroid: modulation of inflammation by ouabain. Front. Physiol. 2017;8:895. doi: 10.3389/fphys.2017.00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinoshita PF, et al. Signaling function of Na, K-ATPase induced by ouabain against LPS as an inflammation model in hippocampus. J. Neuroinflamm. 2014;11:218. doi: 10.1186/s12974-014-0218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holt LM, Olsen ML. Novel applications of magnetic cell sorting to analyze cell-type specific gene and protein expression in the central nervous system. PLoS ONE. 2016;11:e0150290. doi: 10.1371/journal.pone.0150290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawai T, Akira S. TLR signaling. Semin. Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Gais P, et al. TRIF signaling stimulates translation of TNF-alpha mRNA via prolonged activation of MK2. J. Immunol. 2010;184:5842–5848. doi: 10.4049/jimmunol.0902456. [DOI] [PubMed] [Google Scholar]
- 48.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017 doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minogue AM, Barrett JP, Lynch MA. LPS-induced release of IL-6 from glia modulates production of IL-1beta in a JAK2-dependent manner. J. Neuroinflamm. 2012;9:126. doi: 10.1186/1742-2094-9-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thimmulappa RK, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thimmulappa RK, et al. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:585–597. doi: 10.1016/j.bbadis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Soares MP, Ribeiro AM. Nrf2 as a master regulator of tissue damage control and disease tolerance to infection. Biochem. Soc. Trans. 2015;43:663–668. doi: 10.1042/BST20150054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 55.Lau A, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jain A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Copple IM, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010;285:16782–16788. doi: 10.1074/jbc.M109.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rangasamy T, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wong JH, et al. Store-operated Ca(2+) entry facilitates the lipopolysaccharide-induced cyclooxygenase-2 expression in gastric cancer cells. Sci. Rep. 2017;7:12813. doi: 10.1038/s41598-017-12648-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anderson ST, Commins S, Moynagh PN, Coogan AN. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain Behav. Immun. 2015;43:98–109. doi: 10.1016/j.bbi.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Clark SM, et al. Dissociation between sickness behavior and emotionality during lipopolysaccharide challenge in lymphocyte deficient Rag2(−/−) mice. Behav. Brain Res. 2015;278:74–82. doi: 10.1016/j.bbr.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sternberg EM. Neural-immune interactions in health and disease. J. Clin. Investig. 1997;100:2641–2647. doi: 10.1172/JCI119807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 64.Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 65.Murray CL, Skelly DT, Cunningham C. Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1beta and IL-6. J. Neuroinflamm. 2011;8:50. doi: 10.1186/1742-2094-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Carvalho RVH, et al. Leishmanialipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep. 2019;26:429–437 e425. doi: 10.1016/j.celrep.2018.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang J, Ando T, Dunn AJ. Effect of homologous interleukin-1, interleukin-6 and tumor necrosis factor-alpha on the core body temperature of mice. NeuroImmunoModulation. 1997;4:230–236. doi: 10.1159/000097341. [DOI] [PubMed] [Google Scholar]
- 68.Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. 2011;22:189–195. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Habas A, Hahn J, Wang X, Margeta M. Neuronal activity regulates astrocytic Nrf2 signaling. Proc. Natl. Acad. Sci. U.S.A. 2013;110:18291–18296. doi: 10.1073/pnas.1208764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rushworth SA, MacEwan DJ, O'Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008;181:6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Glezer I, et al. MK-801 and 7-Ni attenuate the activation of brain NF-kappa B induced by LPS. Neuropharmacology. 2003;45:1120–1129. doi: 10.1016/S0028-3908(03)00279-X. [DOI] [PubMed] [Google Scholar]
- 72.Barrientos RM, et al. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav. Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakarai H, et al. Adipocyte-macrophage interaction may mediate LPS-induced low-grade inflammation: potential link with metabolic complications. Innate Immun. 2012;18:164–170. doi: 10.1177/1753425910393370. [DOI] [PubMed] [Google Scholar]
- 74.Young RM, Lingrel JB. Tissue distribution of mRNAs encoding the alpha isoforms and beta subunit of rat Na+, K+-ATPase. Biochem. Biophys. Res. Commun. 1987;145:52–58. doi: 10.1016/0006-291x(87)91286-1. [DOI] [PubMed] [Google Scholar]
- 75.Russo JJ, Manuli MA, Ismail-Beigi F, Sweadner KJ, Edelman IS. Na(+)-K(+)-ATPase in adipocyte differentiation in culture. Am. J. Physiol. 1990;259:C968–C977. doi: 10.1152/ajpcell.1990.259.6.C968. [DOI] [PubMed] [Google Scholar]
- 76.Pallai A, et al. Transmembrane TNF-alpha reverse signaling inhibits lipopolysaccharide-induced proinflammatory cytokine formation in macrophages by inducing TGF-beta: therapeutic implications. J. Immunol. 2016;196:1146–1157. doi: 10.4049/jimmunol.1501573. [DOI] [PubMed] [Google Scholar]
- 77.Marim FM, Silveira TN, Lima DS, Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE. 2010;5:e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miyazaki S, Ishikawa F, Fujikawa T, Nagata S, Yamaguchi K. Intraperitoneal injection of lipopolysaccharide induces dynamic migration of Gr-1high polymorphonuclear neutrophils in the murine abdominal cavity. Clin. Diagn. Lab. Immunol. 2004;11:452–457. doi: 10.1128/CDLI.11.3.452-457.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ernst EH, Franks S, Hardy K, Villesen P, Lykke-Hartmann K. Granulosa cells from human primordial and primary follicles show differential global gene expression profiles. Hum. Reprod. 2018;33:666–679. doi: 10.1093/humrep/dey011. [DOI] [PubMed] [Google Scholar]
- 80.Ernst EH, et al. Dormancy and activation of human oocytes from primordial and primary follicles: molecular clues to oocyte regulation. Hum. Reprod. 2017;32:1684–1700. doi: 10.1093/humrep/dex238. [DOI] [PubMed] [Google Scholar]
- 81.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 82.Isaksen TJ, Holm TH, Lykke-Hartmann K. Behavior test relevant to alpha2/alpha3Na(+)/K(+)-ATPase gene modified mouse models. Methods Mol. Biol. 2016;1377:341–351. doi: 10.1007/978-1-4939-3179-8_30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We confirm that all relevant data from this study are available from the corresponding author upon request.