

Abstract
The demyelinating disease progressive multifocal leukoencephalopathy (PML) is caused by the human polyomavirus, JCPyV, under conditions of prolonged immunosuppression. Initial infection is asymptomatic, and the virus establishes lifelong persistence in the host. Following the loss of immune surveillance, the virus can traffic to the central nervous system and infect oligodendrocytes to cause demyelination and PML. The mechanisms involved in glial cell infection are not completely understood. In a screen for N-glycosylated proteins that influence JCPyV pathology, we identified Adipocyte Plasma Membrane Associated Protein (APMAP), as a host cell modulator of JCPyV infection. The removal of APMAP by small interfering siRNA as well as by CRISPR-Cas9 gene editing resulted in a significant decrease in JCPyV infection. Exogenous expression of wild type APMAP in APMAP knockout cell lines rescued susceptibility to infection. These data suggest that virus infection of glial cells is dependent on APMAP.
Keywords: JC Virus, JCPyV, polyomavirus, PML, glial cell infection, APMAP, transmembrane protein, CRISPR
INTRODUCTION
The human polyomavirus JC virus (JCPyV) infects approximately 50 – 60% of the general population worldwide (Chang et al., 2002; Egli et al., 2009; Viscidi et al., 2011). Initial infection is asymptomatic, and the virus is thought to establish a life-long persistent infection in the urogenital tract and perhaps other peripheral organs of healthy individuals (Dorries et al., 1998; Egli et al., 2009; Imperiale and Jiang, 2016). Under conditions of immunosuppression, including patients with underlying immunosuppressive disorders (e.g.; AIDS, hematologic malignancies) and in patients treated with immunomodulatory drugs designed to treat autoimmune diseases, such as multiple sclerosis and Crohn’s disease, JCPyV can cause progressive multifocal leukoencephalopathy (PML), an often fatal demyelinating disease in the brain (Calabrese et al., 2015; Major, 2010; Monaco and Major, 2015; Tan and Koralnik, 2010). JCPyV invades the brain parenchyma following the loss of normal immune surveillance of the central nervous system (CNS) (Haley and Atwood, 2017). The subsequent infection and lytic destruction of glial cells causes demyelination and leads to the development of multifocal white matter lesions which are a hallmark of PML. Due to the absence of specific antiviral treatments for JCPyV, the prognosis of PML is often unfavorable. Currently, treatment involves removal of immunosuppressive therapies and immune reconstitution, which restores the host’s immune response against the virus (Dunham et al., 2020). However, this treatment may lead to immune reconstitution inflammatory syndrome (IRIS) and further neurological damage. Given the critical need for new therapeutic strategies, it is essential to understand the mechanism of JCPyV infection to prevent or treat PML.
JCPyV is a nonenveloped virus composed of an approximately 40 nm capsid that is comprised of one major protein (VP1) and two minor proteins (VP2, VP3) surrounding a double-stranded closed circular DNA genome of 5,130 bp (Ferenczy et al., 2012; Frisque et al., 1984). JCPyV infection is a multi-step process that requires both attachment and entry components (Assetta and Atwood, 2017). Infection is initiated by VP1 binding to the α2,6 sialic acid-linked glycan lactoseries tetrasaccharide c (LSTc) (Neu et al., 2010). While attachment to LSTc is sufficient for virus binding, JCPyV entry requires transient interaction with the 5-hydroxytryptamine (5-HT)2 family of serotonin receptors (Assetta et al., 2013; Assetta et al., 2019; Elphick et al., 2004; Maginnis et al., 2010). Infection of glial cells by JCPyV is inhibited in the presence of antibodies directed against these proteins, as well as in mutant cell lines where the receptors have been deleted (Assetta et al., 2019; Elphick et al., 2004). Following engagement of cell surface receptors the virus triggers clathrin-mediated endocytosis and the ERK signaling pathway (DuShane et al., 2018; Querbes et al., 2004; Querbes et al., 2006). The endocytosed virus is then trafficked to rab5+ endosomes and travels via retrograde transport to the endoplasmic reticulum (ER) where it is partially disassembled, and finally to the nucleus to undergo transcription, replication, and packaging of new virions (Maginnis et al., 2015; Nelson et al., 2012).
In this report, we aimed to identify additional host factor(s) that regulate JCPyV infection in glial cells. We used lectin affinity chromatography to isolate glycoproteins from SVG-A cell plasma membranes, followed by a virus interaction assay and individual protein identification by liquid chromatography with tandem mass spectrometry (LC-MS/MS) to determine potential proteins of interest. One of these proteins, Adipocyte Plasma Membrane Associated Protein (APMAP), was identified in this screen. APMAP is a 46 kDa, type I transmembrane protein with a short, N-terminal intracellular domain, a helical transmembrane domain, and an N-glycosylated, six-bladed β-propeller fold structure in the extracellular domain (Albrektsen et al., 2001; Bogner-Strauss et al., 2010). APMAP was first described as an upregulated protein in mouse and human models of adipogenesis and obesity (Albrektsen et al., 2001; Bogner-Strauss et al., 2010; Ilhan et al., 2008; Pessentheiner et al., 2017). APMAP has since been shown to have a wide tissue distribution, including the brain (Albrektsen et al., 2001; Ilhan et al., 2008; Mosser et al., 2015), where it has been described as suppressing the activity of gamma secretase and affecting the production of peptides associated with Alzheimer’s Disease (Gerber et al., 2019; Mosser et al., 2015). APMAP has been implicated in cancer progression, including colorectal cancer (Mekenkamp et al., 2013) and prostate cancer (Jiang et al., 2019). Notably, APMAP has been shown to modulate CMV infection in fibroblasts and epithelial cells (Ye et al., 2019). To test whether APMAP was involved in JCPyV infection we incubated cells with antibody directed against the protein and then challenged the cells with virus. We found that antibody to APMAP blocks infection. Inhibition of APMAP expression using siRNA or deleting APMAP using CRISPR-Cas9 gene editing led to reduced virus infection that could be rescued by overexpressing wild type APMAP.
MATERIALS AND METHODS
Cells and viruses
All cells were grown at 37°C with 5% CO2 in a humidified incubator. Cells were authenticated using the ATCC cell line authentication service (STR profile). SVG-A cells (Major et al., 1985) were grown in MEM media supplemented with 10% fetal bovine serum (FBS) and 1% each penicillin and streptomycin. Lenti-X HEK293T cells (Takara, #632180), used for lentivirus production, were cultured in DMEM supplemented with 10% fetal bovine serum and 1% each amphotericin B, penicillin and streptomycin. SVG-A cells were infected with the laboratory-adapted strain of JCPyV, Mad-1/SVE-delta (Vacante et al., 1989). CsCl purification of the virus was performed as described previously (Liu and Atwood, 2001; Nelson et al., 2013). Briefly, SVG-A cells were grown to 50% confluency in 1700cm2 roller bottles and infected with JCPyV at an MOI of approximately 0.1 FFU/cell. Cells were incubated for 14 days with a change of media after 7 days. Lysates were scraped in media and subjected to 3 freeze/thaw cycles. The lysates were then treated with neuraminidase (type II; Sigma) for 1 hr at 37°C to release any residual JCPyV bound to cells and then incubated in 0.25% deoxycholate (Fisher Scientific) for 30 min at 37°C and sonicated 3 times to further disrupt the cells. Lipid extraction was then performed with two rounds of Vertrel XF treatment (Fisher Scientific #NC9715008). Cell debris was removed by centrifugation and the viral supernatants were then pelleted through a 20% sucrose cushion in a Beckman SW40ti rotor at 150,000 × g at 4°C for 3 hr. Buffer A (10mM Tris-HCl, 50 mM NaCl, 0.1 mM CaCl2) was used to resuspend the viral pellet followed by sonication. This suspension was then loaded onto a CsCl step gradient (1.29–1.35 g/ml) and spun at 115,000 × g at 4°C for 18 hr in a Beckman SW55ti rotor. The band corresponding to DNA-containing virions was isolated and dialyzed extensively against buffer A. Direct labeling of JCPyV with Alexa Fluor 680 (AF680) was performed according to the manufacturer’s instructions and as described (Dugan et al., 2008). Briefly, 5.0 mg of CsCl-purified JCPyV was dialyzed against labeling buffer (0.1 M NaHCO3; pH 8.3) at 4°C overnight in 10,000 MWCO cartridges (Pierce). The virus was then incubated for 1 hr with constant rotation at room temperature with 0.5 μg of AF680-labeled succinimidyl ester (Thermo Fisher Scientific, #A20008) in 100 μl of DMSO, shielded from light. The AF680-labeled JCPyV was then extensively dialyzed in 10,000 MWCO cartridges against two changes of buffer A (10 mM Tris-HCl, 50 mM NaCl, 0.1 mM CaCl2) at 4°C for an additional 48 hr to remove excess dye.
Virus infection assay
Infection of cells was assessed by indirect immunofluorescence. Purified JCPyV (1.75 × 105 fluorescent forming units) was used to infect SVG-A cells (1.5 × 105) for 2 hr at 37°C in MEM supplemented with 2% serum. At four days post-infection, cells were washed twice in 1X PBS and fixed in ice-cold methanol for 30 min at −20°C. The cells were then washed, incubated with anti-VP1 antibody pab597 (kind gift of Ed Harlow; (Atwood et al., 1995; Dugan et al., 2007) for 2 hr at 37°C, washed again, incubated with anti-mouse secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific) for 1 hr at 37°C and then incubated with DAPI (Thermo Fisher Scientific). Slides were viewed using an epifluorescence microscope (Eclipse E800; Nikon) and scored by counting using Cell Profiler (Carpenter et al., 2006), with the number of positive cells reported as the percentage of infected cells per visual field. A minimum of ten fields were counted using the 10x objective for each experimental sample, in triplicate.
Lectin chromatography
The method to isolate N-glycosylated proteins from SVG-A cells was adapted from Lee et al. (Lee et al., 2008), employing a lectin from Polyporus squamosus (PSL), PSL specifically recognizes the trisaccharide motif Neu5Ac-α2,6-Gal-β1,4-Glc/GlcNAc (Mo et al., 2000; Toma et al., 2001). To immobilize PSL onto streptavidin-coupled magnetic beads, 50 μl of washed streptavidin coated beads (Thermo Fisher Scientific) and biotinylated PSL (0.625 mg/ml final concentration; E-Y Laboratories) in 1 ml TBS (150 mM NaCl and 50 mM Tris HCl, pH 7.4) were incubated together for 1 hr at 4°C with constant mixing and then washed 5 times, first with TBS, then with TBS with 1% Triton-X 100, then three times more in TBS and kept on ice. SVG-A plasma membranes were isolated from approximately 8×107 SVG-A cells were washed 3 times in TBS, detached from the flasks by scraping, and centrifuged for 4 min at 400×g. The pellet was then resuspended in 0.5 ml of cold TBS with protease inhibitors (Complete protease inhibitor cocktail (Roche) supplemented with 1.5 mM PMSF) and the cells disrupted by 30 strokes in a 2 ml Dounce homogenizer on ice. The SVG-A samples were then incubated with the PSL bound beads for 1 hr at 4°C on a rotator with constant mixing. The beads were then washed 5 times in TBS and eluted by incubating the beads on a rotator for 10 minutes with 0.5 ml of 250 mM sialic acid (Vector Laboratories) in TBS and 0.5% CHAPS. Duplicate sets of proteins were then separated on two separate 4–15% gradient SDS PAGE gels. The first gel was stained with Coomassie colloidal blue (Sigma). The second gel was blotted to PVDF and probed with AlexaFluor680 labeled JCPyV for 18 hr at 4°C shielded from light. Blots were then washed extensively in TBS-0.1% Tween (TBST) and visualized imaged using an infrared scanner (LICOR). Coomassie colloidal blue-stained bands were then excised corresponding to the fluorescent bands in the blot and subjected to LC-MS/MS analysis at The Mass Spectrometry (MS) & Proteomics Resource of the W.M. Keck Foundation Biotechnology Resource Laboratory. Proteins identified in our screen that were known to be N-glycosylated, extracellular transmembrane proteins were selected for further analysis for their potential involvement in JCPyV infection.
Infection blocking experiment
SVG-A cells were grown in 24 well dishes to 50% confluence. The cells were washed in cold 1X PBS and then incubated in either rabbit polyclonal anti-APMAP antibody (Sigma #HPA012863) or an equal concentration of isotype control rabbit IgG (Santa Cruz #sc-2027) diluted to 1.25 μg/ml in MEM supplemented with 2% FBS for 1 hr on ice. JCPyV is then added and incubated for 1 hr on ice. Then cells were then washed 3 times in cold MEM and fed with complete media. Virus infection was assayed 4 days post infection.
Transient knock-down of APMAP using small interfering RNAs
APMAP specific siRNA and control siRNA were obtained from Qiagen (APMAP siRNA Flexitube Gene Solution #GS57136; APMAP 1: CAAGGGACTATTTGAAGTAAA; APMAP 2: CTGGGTGGGCATGTCGACCAT; APMAP 3: CATGCTGGATTTCTTATCTGA; APMAP 4: TTCACCGATTCTAGCAGCAAA) and Flexitube All Stars Negative Control siRNA # SI03650318. In a 24 well dish, each siRNA was combined with 1.5 μl RNAimax (Thermo Fisher Scientific) in serum-free media for a final concentration of 10 nM. Subsequently, 1.6×105 SVG-A cells and 0.5 ml complete media were added per well and incubated for 72 hr prior to either infection with JCPyV or protein harvesting for immunoblot analysis.
APMAP Deglycosylation
To test the effect of tunicamycin on APMAP glycosylation in glial cells, SVG-A cells were grown to 50% confluence in 6 well dishes. Cells were treated with 0.25 μg/ul tunicamycin or vehicle control (methanol) for 24, 48, and 72 hr. After each time point, wells were washed twice in 1X PBS and removed from the plate using cell stripper (Mediatech). Cells were then centrifuged and resuspended in RIPA buffer (Pierce) containing both protease and phosphatase inhibitors (Complete protease inhibitor cocktail (Roche) supplemented with 1.5 mM PMSF and 1.0 mM sodium orthovanadate). For PNGaseF treatment, 10 μg of SVG-A membrane protein fraction was denatured at 100°C in 0.5% SDS, 40 mM DTT for 10 min then incubated with 500U PNGaseF (NEB; #P0704) in 50 mM sodium phosphate (pH 7.5) and 1% NP-40 for 2 hr at 37°C. For neuraminidase deglycosylation, 5 μg of SVG-A membrane protein fraction was incubated with 100U neuraminidase (derived from C. perfringens; NEB #P0720) in 50 mM sodium citrate (pH 6.0) for 12 hr at 37°C. Control experiments were performed in the absence of enzyme. All samples were then analyzed by immunoblotting.
Immunoblotting
Protein concentrations were determined using the BCA protein assay (Bio-Rad). For western blot analysis, 10 μg of protein per well was diluted in 4X Laemmli buffer with 15% BME (BioRad), heated to 95°C for 10 min, and separated on 4-to-15% Tris-HCl gradient gels (Bio-Rad). Samples were transferred to 0.2 μm nitrocellulose using a semidry transfer cell (Bio-Rad) and blocked with 1% BSA blocking buffer in TBST. Blots were incubated with anti-APMAP (1:500, LSBio #LS-C173702) and anti-actin (1:1000; Cell Signaling Technology #4970) diluted in blocking buffer overnight at 4°C. After washing, the membranes were incubated with IRDye 800CW goat anti-rabbit IgG (1:5,000; Li-Cor #926–32211) or IRDye 680 goat anti-mouse IgG (1:5,000; Li-Cor #926–68070) and imaged using a fluorescence imaging system (LICOR) and Odyssey software for densitometric analysis.
Cell fractionation assay
SVG-A cell membrane and cytoplasmic protein fractions were isolated using the Mem-PER Plus membrane protein extraction kit following the manufacturer’s protocol (Thermo Fisher Scientific #89842). Briefly, 4×106 cells were harvested by centrifugation at 300×g for 5 min and washed 3 times with cell wash solution. Cell pellets were then resuspended in 0.75 mL permeabilization buffer for 10 min at 4°C followed by centrifugation at 16,000×g for 15 min at 4°C. The resulting supernatant is enriched in cytoplasmic protein. The pellet is then resuspended in 0.5 mL solubilization buffer, and incubated for 30 min at 4°C. The membrane protein fraction is then recovered in the supernatant after centrifugation at 16,000×g for 15 min.
Flow cytometry
To assess expression of APMAP on the extracellular surface of glial cells, SVG-A cells were washed in 1X PBS and removed from wells using cell stripper (Mediatech) and incubated with primary antibody rabbit Anti-APMAP antibody (1:100; LSBio #LS-C173702) for 1 hr on ice, washed, then incubated in AF633-conjugated anti-rabbit secondary antibody diluted to 1:500 for 1 hr on ice. The cells were washed again, fixed with 1% paraformaldehyde, and analyzed by flow cytometry using a BD FACS-Canto II (BD Bioscience) and FlowJo software (Tree Star, Inc.).
CRISPR-Cas9-mediated knockout of APMAP in glial cells
APMAP knockout SVG-A cell lines were constructed essentially as previously described (Assetta et al., 2019; Sanjana et al., 2014). In brief, gRNA sequences targeting exon 1 of the human APMAP gene were designed using online software (http://crispr.mit.edu) and cloned into a lentiCRISPRv2 plasmid with puromycin resistance (Addgene #52961). APMAP sequence: CAGGTCGTCACAGACGATGA (Integrated DNA Technologies). Lentiviral particles were packaged in Lenti-X HEK293T cells (Clontech) using the pCMV-dR8.2 packaging plasmid (Addgene #8455) and the VSV-G envelope encoding plasmid (Addgene #8454) at a ratio of 3 (pCMV-dR8.91), 0.3 (VSV-G) and 3 (lentiCRISPRv2). Media was replaced after 18 hr with DMEM supplemented with 30% FBS and cells were incubated at 37°C for 24 hr. Supernatants containing lentivirus were harvested three times at 12 hr intervals. The supernatants were then filtered using a 0.45 μm filter, supplemented with 8 μg/ml polybrene and used to infect SVG-A cells. After lentivirus infection, SVG-A cells were selected for stable gRNA expression by incubation in media with 2 μg/ml puromycin for 48 hr, until an uninfected control plate had no live cells remaining. Following drug selection, single cells were isolated and expanded using the CellRaft System (Cell Microsystems). Clones of interest were identified by western blot analysis for the absence of APMAP expression and by DNA sequencing through the CRISPR deep sequencing service performed at the Center for Computational and Integrative Biology (CCIB) DNA core at Massachusetts General Hospital.
Transient transfection of APMAP for phenotypic rescue
APMAP knockout (KO), CRISPR modified SVG-A cells were cultured in 24-well plates in MEM containing 10% FBS without antibiotics. Cells at 80% confluence were transfected using Lipofectamine 3000 (Thermo Fisher Scientific) with 2 μg of DNA per well of human APMAP in pEZ-M14 (Genecopeia, #EX-V0537-M14). This vector expresses human APMAP with a 3X-flag tag fused to its C-terminal end. To permit robust expression of plasmid-derived APMAP in the KO cells, we altered the nucleotide sequence in the region of APMAP where we designed our guide RNA so it would be resistant to modification by the CRISPR-Cas9 system. Briefly, point mutations were introduced using the QuikChange II Site directed mutagenesis kit (Agilent, #200555) as per the manufacturer’s instructions: 5’-caggtcgtcacagacgat-3’ to 5’-caAgtTgtAacAgaTgaC-3’, where capital letters indicate the changed nucleotides. These mutations do not alter the amino acid sequence of APMAP in this region (QVVTDD), but will prevent editing by CRISPR-Cas9 and the exogenously expressed APMAP will be left intact in the KO cells. As a control, KO cells were also transfected with 2 μg of a corresponding empty expression vector. The cells were incubated at 37°C for 4 hr, then medium was replaced with MEM supplemented with 10% FBS and incubated at 37°C for 48 hr. The cells were then infected with JCPyV in MEM with 2% FBS for 1 hr at 37°C, washed and fed with complete media, and assayed for VP1 after 4 days.
Statistical analysis
GraphPad Prism 6 software and the Student’s t test was employed to analyze the data for two-group comparison. The results are expressed as means ± standard deviations (±SD) of three independent experiments. Statistical significance is marked by an asterisk (*). The statistical significance threshold was set at P < 0.05.
RESULTS
Identification of APMAP as a co-factor of JCPyV infection
To isolate additional glycoproteins that could potentially regulate JCPyV infection in glial cells, we incubated SVG-A membranes with magnetic beads coated with the lectin PSL, which is highly specific for the Neu5Ac-α2,6-Gal-β1,4-Glc/GlcNAc trisaccharide sequence of asparagine-linked oligosaccharides (Kadirvelraj et al., 2011; Zhang et al., 2001). Eluted proteins from the pulldown assay were separated via SDS-PAGE, transferred to nitrocellulose membranes, and probed with labeled JCPyV. Areas of the gel that corresponded to virus binding were then excised and subjected to LC-MS/MS analysis. We identified APMAP as a protein of interest on this list, as it is known to be an N-glycosylated, transmembrane protein. To test the hypothesis that APMAP facilitates JCPyV infection, we used anti-APMAP antibodies to block access of the virus to extracellular APMAP by pre-incubating SVG-A cells with anti-APMAP antibodies and then infecting the cells with JCPyV. The cells were washed, challenged with JCPyV and the infection allowed to proceed for 4 days. As a control for specificity, we also preincubated glial cells with an equivalent amount of control isotype antibody and in buffer alone. Cells were stained for late protein VP1 and positive nuclei counted to indicate a productive infection. Incubation with anti-APMAP antibodies blocked JCPyV infection of SVG-A cells (Fig. 1A) compared to controls as measured by quantification of VP1(+) nuclei (Fig. 1B). Other candidates on the list, such as CD44, were analyzed but we did not observe inhibition of JCPyV infection in antibody blocking assays (data not shown).
Figure 1. Anti-APMAP antisera reduces JCPyV infection of glial cells.
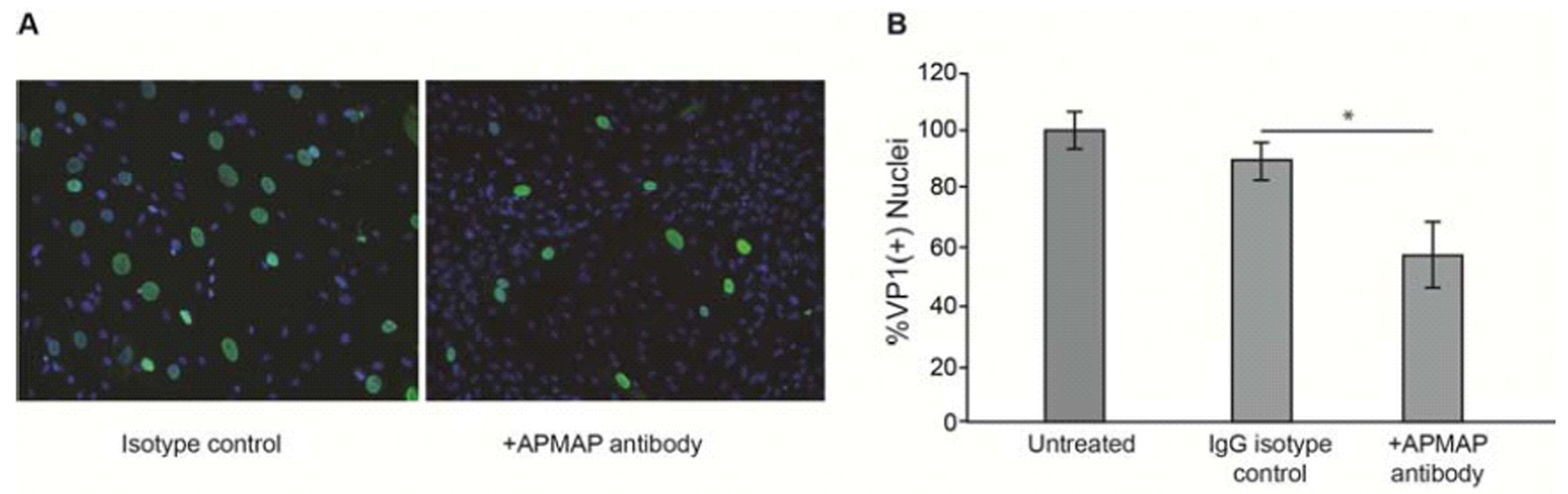
(A) SVG-A cells preincubated with anti-APMAP antisera and then infected with JCPyV exhibit reduced infection compared to cells preincubated with isotype control antibody. Post infection, cells were fixed and stained with anti-VP1 antibody (green) and nuclei were counterstained with DAPI (blue). (B) Quantification of VP1(+) nuclei. Experiments were performed three times in triplicate. Error bars represent SD. *p<0.05
The reduction of APMAP protein results in a reduced number of infected cells, suggesting that APMAP is important for JCPyV infection
We next asked if reducing the expression of APMAP in glial cells could affect the extent of JCPyV infection. APMAP protein was knocked down by transient transfection of SVG-A cells with four different small-interfering RNA (siRNA) targeted to APMAP. Following transfection, cells were infected with JCPyV for 1 hour and stained after 4 days for the presence of late protein VP1 and positive nuclei counted to indicate a productive infection. Specificity controls included transfection with a non-specific siRNA, and mock infection using transfection reagent only. Each APMAP-specific siRNA treatment led to a decrease of JCPyV infection compared to untreated cells and cells treated with either transfection reagent alone or non-targeting siRNA controls (Fig. 2A). The decrease in APMAP protein expression in SVG-A cells was confirmed by western blot and densitometric analysis (Fig. 2B & C).
Figure 2. Knockdown of APMAP protein by siRNA reduces JCPyV infection of glial cells.
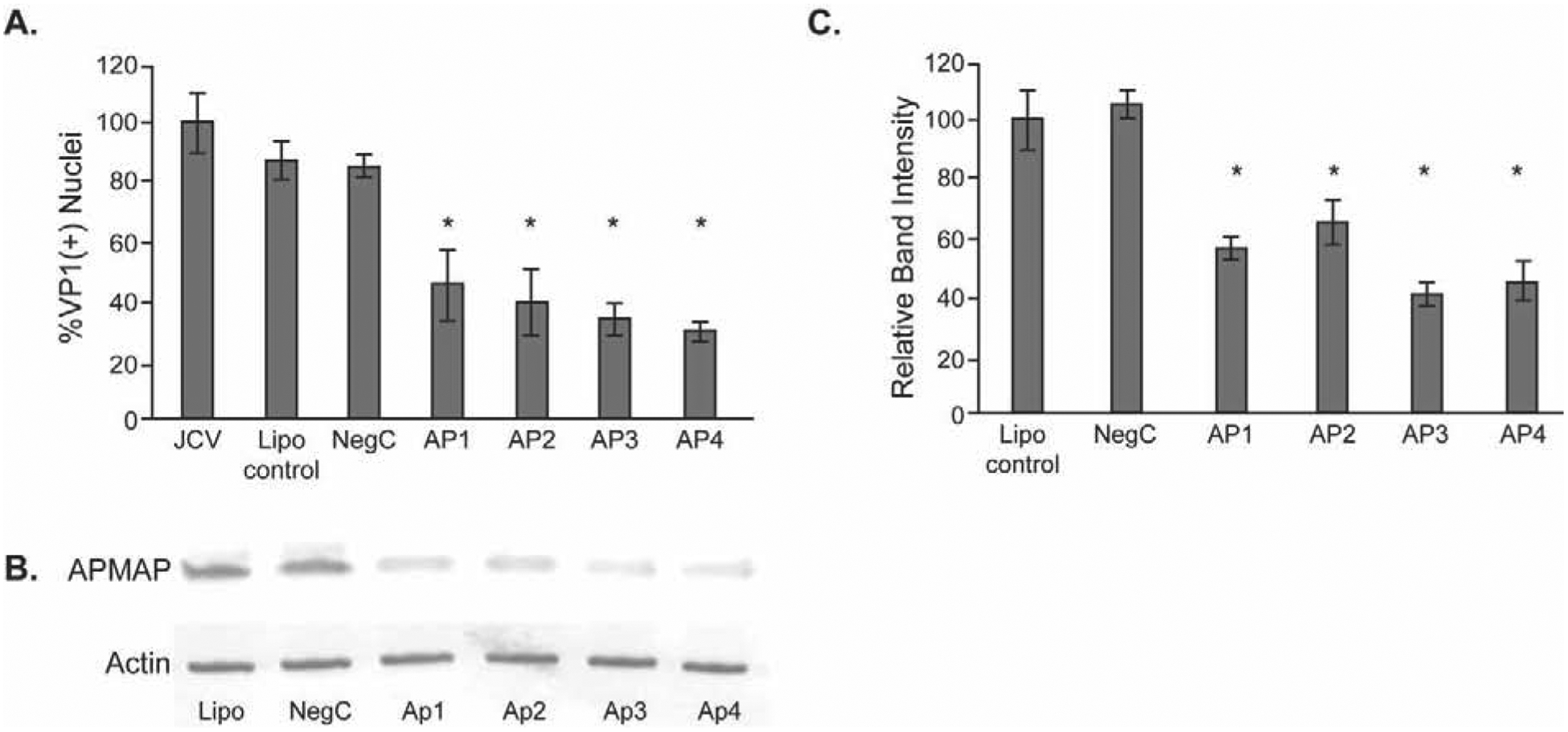
(A) SVG-A cells were untreated or treated with control nontargeting siRNA (NegC) or one of four different APMAP-specific siRNAs for 72 hours and then infected with JCPyV. Post-infection, cells were fixed and stained for VP1 and counterstained with DAPI, and VP1(+) nuclei quantified. (B) siRNA knockdown of APMAP decreases APMAP protein in SVG-A cells. (C) Quantification of western blot band intensity using Odyssey software. The results represent the average of two independent experiments performed in triplicate. Error bars represent SD. *p<0.05
APMAP is N-glycosylated and predominantly on the plasma membrane of glial cells
In other mouse and human cell types, APMAP has been described as an N-glycosylated, extracellular protein (Albrektsen et al., 2001; Bogner-Strauss et al., 2010; Ilhan et al., 2008). Human APMAP is predicted to be N-glycosylated at two asparagine residues, N160 and N196. To examine the nature of APMAP in human glial cells, we treated live SVG-A cells with tunicamycin. We have previously shown treatment with tunicamycin, an inhibitor of N-linked glycosylation, blocks JCPyV infection (Liu et al., 1998; Maginnis et al., 2010). Here, tunicamycin treatment of live SVG-A cells showed progressive deglycosylation of APMAP (Fig. 3A). In addition, immunoprecipitated APMAP treated with either PNGase or neuraminidase, enzymes that remove N-linked glycans from glycoproteins, also demonstrated deglycosylation (Fig. 3B). Previous work has shown treatment of SVG-A cells with neuraminidase will inhibit JCPyV activity (Dugan et al., 2008; Liu et al., 1998). To determine if glycosylated APMAP is expressed extracellularly, fixed, intact SVG-A cells were labeled with anti-APMAP antibody, washed, labeled with fluorescent secondary antibody, and subjected to flow cytometric analysis. This analysis demonstrated that APMAP is present on the plasma membrane of intact cells (Fig. 3C). Cell fractionation analysis suggests that the majority of APMAP in glial cells is found in membrane fractions and not the cytoplasm (Fig. 3D).
Figure 3. In human glial cells, APMAP is an N-glycosylated, plasma membrane protein.
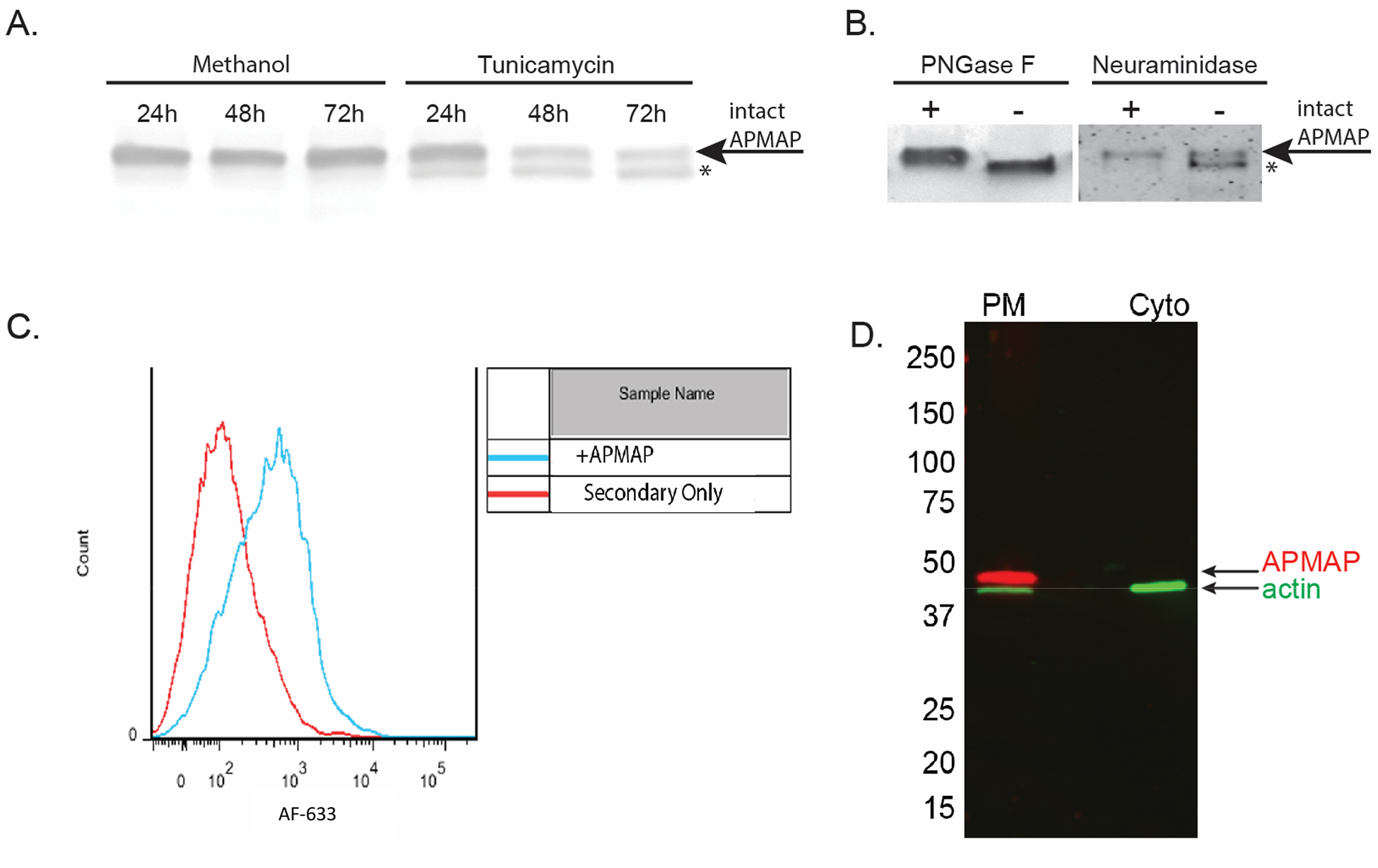
(A) SVG-A cells were treated with either methanol control or an inhibitor of N-glycosylation, tunicamycin. Cells were assayed by western blot for deglycosylation after 24, 48 and 72 hours. (B) Immunoprecipitated APMAP treated with either PNGaseF or neuraminidase from C. perfringens also demonstrated deglycosylation. * = deglycosylated APMAP. (C) APMAP is found primarily on the cell surface of SVG-A cells. Cells were labeled with APMAP antisera, washed and incubated with fluorescently labeled secondary antibody, then analyzed by flow cytometry. (D) Cellular fractionation further supports APMAP is localized to the plasma membrane.
CRISPR-Cas9-mediated deletion of APMAP correlates with reduced susceptibility to JCPyV Infection in glial cells
To further verify the participation of APMAP in JCPyV infection, we used CRISPR-Cas9-mediated genome editing to create stable glial cell lines lacking APMAP. A guide RNA (gRNA) targeting exon 1 of the APMAP gene was synthesized using sequence predicted from online software as described (Cong and Zhang, 2015; Sanjana et al., 2014). Exon 1 corresponds to the small, intracellular domain of APMAP, which precedes the transmembrane and extracellular domains. The gRNA sequence was cloned into the lentiCRISPRv2 plasmid. After lentiviral infection and drug selection, single cells were isolated on rafts which were then expanded to generate clonal populations. Cells lacking APMAP were identified by western blotting (Fig. 4A), and two distinct CRISPR-modified clones (KO1 and KO2) displaying successful ablation of APMAP were selected for further analysis. Figure 4B shows that without APMAP expression, JCPyV infection is decreased significantly in the APMAP KO cells compared to WT SVG-A cells. However, susceptibility to infection can be rescued by the reintroduction of wild type APMAP. APMAP KO cells were transiently transfected with a plasmid expressing APMAP or an empty control vector (Fig. 4C), and after 24 hours the cells were challenged with JCPyV. Infection was assayed 4 days after infection. SVG-A APMAP KO cells transfected with the control construct remained refractory to JCPyV infection (Fig. 4D). In contrast, transient transfection of the mutant cell line with a plasmid expressing APMAP partially rescued infection (Fig. 4D). These data suggested that APMAP plays a critical role in facilitating virus infection.
Figure 4. CRISPR-Cas9 targeted APMAP gene disruption decreases JCPyV infection.
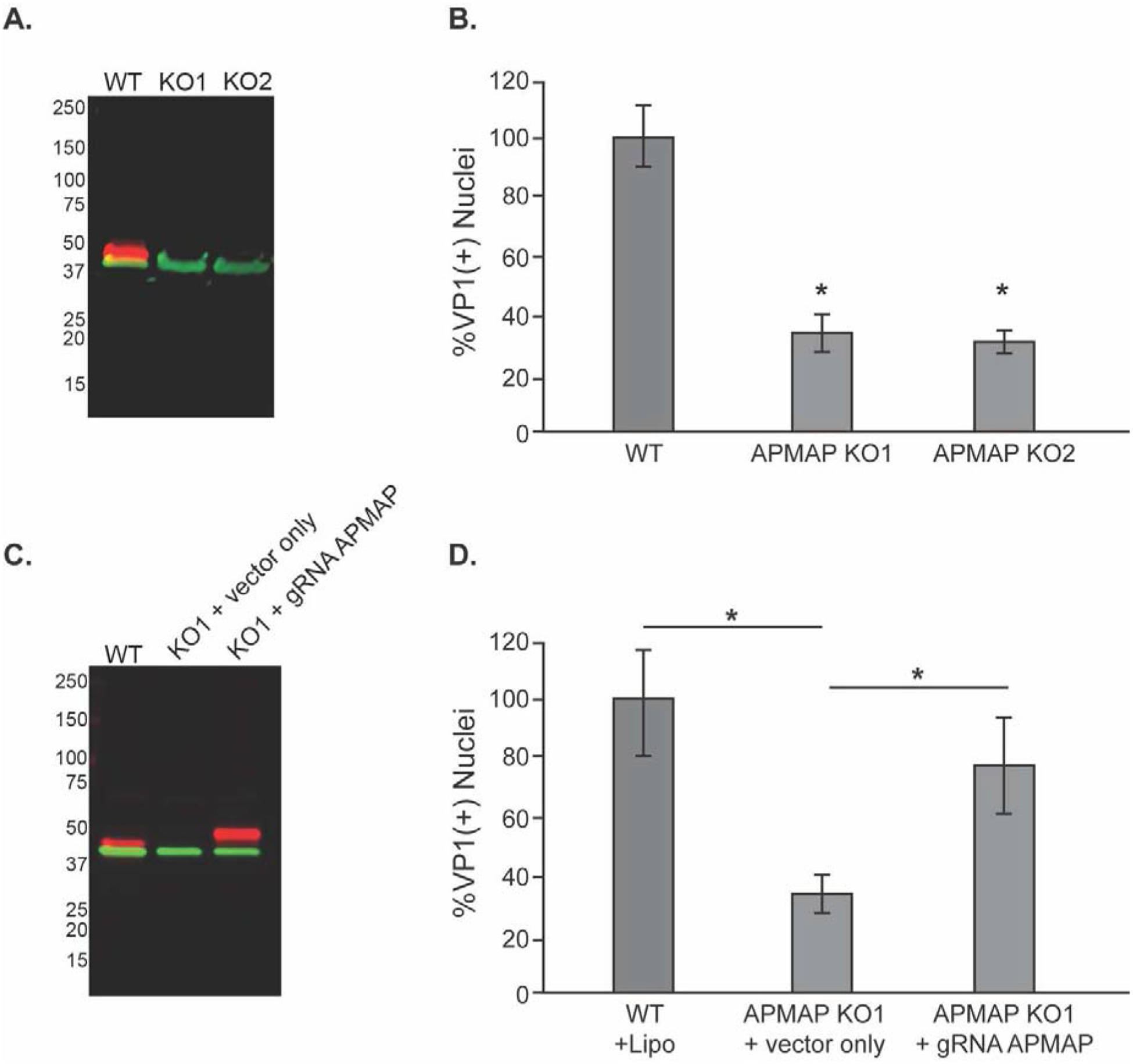
(A) Western blot analysis of wild type SVG-A (WT) cells or two APMAP null clones (KO1 & KO2) using antibodies to APMAP (labeled in red) and actin (green) demonstrates the loss of APMAP expression in the null clones. (B) SVG-A (WT) cells and APMAP null clones KO1 and KO2 were challenged with JCPyV and infection scored by indirect immunofluorescence for the viral protein VP1. (C) Western blot demonstrating APMAP expression in KO1 cells is restored by transient transfection with a plasmid expressing human APMAP with a mutated guide sequence region (gRNA APMAP). APMAP is labeled in red, actin in green as in (A). The vector-derived APMAP is slightly larger than wildtype APMAP due to a 3X flag-tag sequence from the vector that adds an additional 2.8 KDa to the C-terminal end of endogenous APMAP. (D) Susceptibility to JCPyV infection of APMAP KO1 cells was rescued by transfecting the cells with the APMAP expression plasmid expressing APMAP compared to empty control Flag expression vector without the APMAP insert. The results represent the average of three independent experiments performed in triplicate. Error bars represent SD. *p<0.05
DISCUSSION
In this study, we identified an N-glycosylated protein on the cell surface of human glial cells that modulates JCPyV infection. We surmise extracellular APMAP is acting at an early stage in infection, as anti-APMAP bound to the surface of intact glial cells inhibits subsequent infection by JCPyV. APMAP is an N-glycosylated transmembrane protein expressed on the surface of glial cells. Reduction of APMAP either transiently by siRNA depletion or in CRISPR-mediated stable knockout cell lines causes a significant decrease in susceptibility to JCPyV infection. This reduction appears to be specifically dependent on the presence of APMAP, as rescue of infection is achieved with the reintroduction of wild type APMAP. Therefore, we conclude that APMAP is an important regulator of JCPyV infection in glial cells.
Other groups have shown that APMAP is implicated in a variety of important pathologies, including obesity, Alzheimer’s disease, cancer metastasis, and virus infection, in a variety of human and mouse cell types. In an obesity model in APMAP deficient mice, APMAP promotes a profibrotic phenotype in adipose tissue by its interaction with extracellular matrix (ECM) proteins (Pessentheiner et al., 2017). APMAP interacts with extracellular oxidases that drive ECM collagen crosslinking, suggesting APMAP can regulate ECM remodeling. In addition, APMAP appears to have additional roles in the brain. The depletion of APMAP in a mouse model of Alzheimer’s disease (Gerber et al., 2019) has increased memory deficits as well as increased Aβ deposition in senile plaques associated with brain pathology. APMAP has been shown to interact with proteins in the gamma secretase complex that regulate Aβ production.
Although other groups have shown evidence of alternative splicing/multiple isoforms of APMAP in both mouse and human tissues, we have not observed multiple isoforms by western blot in our glial cell knockouts (Gerber et al., 2019; Pessentheiner et al., 2017). However, while infection in our APMAP depleted cells is significantly decreased, infection is not completely eliminated. One explanation of residual infectivity could be the presence of minor splice variants of APMAP that remain following CRISPR treatment that are below our level of detection. Another possibility is that there are alternative and unknown mechanisms of JCPyV infection independent of APMAP. Previous studies have implicated APMAP in cancer progression (Jiang et al., 2019; Mekenkamp et al., 2013). In prostate cancer cells, high cholesterol drives APMAP interaction with eps15R at the cell surface, which leads to increased ERK1/2 signaling and a metastatic phenotype (Jiang et al., 2019). Interestingly, JCPyV infection requires eps15, a distinct, but related, protein as well as ERK pathway activation (DuShane et al., 2018; Querbes et al., 2004; Querbes et al., 2006).
While our blocking antibody results suggest APMAP is affecting a step at the cell surface, it is also possible APMAP is modulating a step in the cytoplasm, later in the virus life cycle. APMAP has been shown to be involved in infection of cells by the β-herpesvirus, human cytomegalovirus (CMV) (Ye et al., 2019). At low pH, APMAP interacts with the gH/gL glycoprotein complexes of the CMV envelope, and enhances immediate early gene transcription and pp65 tegument protein translocation to the nucleus, implicating an intracellular role for APMAP in infection. Therefore, APMAP may be a central modulator (throughout the cell) for infection by a variety of viruses. Future studies are aimed at defining the stages in the viral life cycle that are facilitated by APMAP. PML is a devastating disease with few treatment options so it is therefore critical to identify all the mechanisms that drive infection and subsequent pathology.
Highlights.
Adipocyte Plasma Membrane Associated Protein (APMAP) was identified as a modulator of JCPyV infection of human glial cells.
Antibody to APMAP reduced infection of glial cells by JCPyV
siRNA and CRISPR-CAS9 mediated knockdown and knockout strategies respectively reduced JCPyV infection.
Infection could be rescued in the APMAP knockouts by overexpressing APMAP with a mutation rendering it resistant to CRISPR-CAS9.
APMAP is expressed at the plasma membrane of glial cells and is glycosylated.
ACKNOWLEGMENTS
We thank all members of the laboratory for helpful discussions. This work was supported by the National Institute of Neurological Disease and Stroke grant P01NS065719.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- Albrektsen T, Richter HE, Clausen JT, Fleckner J, 2001. Identification of a novel integral plasma membrane protein induced during adipocyte differentiation. Biochem J 359, 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assetta B, Atwood WJ, 2017. The biology of JC polyomavirus. Biol Chem 398, 839–855. [DOI] [PubMed] [Google Scholar]
- Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O’Hara BA, Allen Ramdial SA, Atwood WJ, 2013. 5-HT2 receptors facilitate JC polyomavirus entry. J Virol 87, 13490–13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assetta B, Morris-Love J, Gee GV, Atkinson AL, O’Hara BA, Maginnis MS, Haley SA, Atwood WJ, 2019. Genetic and Functional Dissection of the Role of Individual 5-HT2 Receptors as Entry Receptors for JC Polyomavirus. Cell Rep 27, 1960–1966 e1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood WJ, Wang L, Durham LC, Amemiya K, Traub RG, Major EO, 1995. Evaluation of the role of cytokine activation in the multiplication of JC virus (JCV) in human fetal glial cells. J Neurovirol 1, 40–49. [DOI] [PubMed] [Google Scholar]
- Bogner-Strauss JG, Prokesch A, Sanchez-Cabo F, Rieder D, Hackl H, Duszka K, Krogsdam A, Di Camillo B, Walenta E, Klatzer A, Lass A, Pinent M, Wong WC, Eisenhaber F, Trajanoski Z, 2010. Reconstruction of gene association network reveals a transmembrane protein required for adipogenesis and targeted by PPARgamma. Cell Mol Life Sci 67, 4049–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese LH, Molloy E, Berger J, 2015. Sorting out the risks in progressive multifocal leukoencephalopathy. Nat Rev Rheumatol 11, 119–123. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM, 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Wang M, Tsai RT, Lin HS, Huan JS, Wang WC, Chang D, 2002. High incidence of JC viruria in JC-seropositive older individuals. J Neurovirol 8, 447–451. [DOI] [PubMed] [Google Scholar]
- Cong L, Zhang F, 2015. Genome engineering using CRISPR-Cas9 system. Methods Mol Biol 1239, 197–217. [DOI] [PubMed] [Google Scholar]
- Dorries K, Arendt G, Eggers C, Roggendorf W, Dorries R, 1998. Nucleic acid detection as a diagnostic tool in polyomavirus JC induced progressive multifocal leukoencephalopathy. J Med Virol 54, 196–203. [PubMed] [Google Scholar]
- Dugan AS, Gasparovic ML, Atwood WJ, 2008. Direct correlation between sialic acid binding and infection of cells by two human polyomaviruses (JC virus and BK virus). J Virol 82, 2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan AS, Gasparovic ML, Tsomaia N, Mierke DF, O’Hara BA, Manley K, Atwood WJ, 2007. Identification of amino acid residues in BK virus VP1 that are critical for viability and growth. J Virol 81, 11798–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham SR, Schmidt R, Clifford DB, 2020. Treatment of Progressive Multifocal Leukoencephalopathy Using Immune Restoration. Neurotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuShane JK, Wilczek MP, Mayberry CL, Maginnis MS, 2018. ERK Is a Critical Regulator of JC Polyomavirus Infection. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH, 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis 199, 837–846. [DOI] [PubMed] [Google Scholar]
- Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ, 2004. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306, 1380–1383. [DOI] [PubMed] [Google Scholar]
- Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO, 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 25, 471–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ, Bream GL, Cannella MT, 1984. Human polyomavirus JC virus genome. J Virol 51, 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H, Mosser S, Boury-Jamot B, Stumpe M, Piersigilli A, Goepfert C, Dengjel J, Albrecht U, Magara F, Fraering PC, 2019. The APMAP interactome reveals new modulators of APP processing and beta-amyloid production that are altered in Alzheimer’s disease. Acta Neuropathol Commun 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley SA, Atwood WJ, 2017. Progressive Multifocal Leukoencephalopathy: Endemic Viruses and Lethal Brain Disease. Annu Rev Virol 4, 349–367. [DOI] [PubMed] [Google Scholar]
- Ilhan A, Gartner W, Nabokikh A, Daneva T, Majdic O, Cohen G, Bohmig GA, Base W, Horl WH, Wagner L, 2008. Localization and characterization of the novel protein encoded by C20orf3. Biochem J 414, 485–495. [DOI] [PubMed] [Google Scholar]
- Imperiale MJ, Jiang M, 2016. Polyomavirus Persistence. Annu Rev Virol 3, 517–532. [DOI] [PubMed] [Google Scholar]
- Jiang S, Wang X, Song D, Liu X, Gu Y, Xu Z, Wang X, Zhang X, Ye Q, Tong Z, Yan B, Yu J, Chen Y, Sun M, Wang Y, Gao S, 2019. Cholesterol Induces Epithelial-to-Mesenchymal Transition of Prostate Cancer Cells by Suppressing Degradation of EGFR through APMAP. Cancer Res 79, 3063–3075. [DOI] [PubMed] [Google Scholar]
- Kadirvelraj R, Grant OC, Goldstein IJ, Winter HC, Tateno H, Fadda E, Woods RJ, 2011. Structure and binding analysis of Polyporus squamosus lectin in complex with the Neu5Ac{alpha}2–6Gal{beta}1–4GlcNAc human-type influenza receptor. Glycobiology 21, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YC, Block G, Chen H, Folch-Puy E, Foronjy R, Jalili R, Jendresen CB, Kimura M, Kraft E, Lindemose S, Lu J, McLain T, Nutt L, Ramon-Garcia S, Smith J, Spivak A, Wang ML, Zanic M, Lin SH, 2008. One-step isolation of plasma membrane proteins using magnetic beads with immobilized concanavalin A. Protein Expr Purif 62, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CK, Atwood WJ, 2001. Propagation and assay of the JC virus. Methods Mol Biol 165, 9–17. [DOI] [PubMed] [Google Scholar]
- Liu CK, Wei G, Atwood WJ, 1998. Infection of glial cells by the human polyomavirus JC is mediated by an N-linked glycoprotein containing terminal alpha 2–6 linked sialic acids. J. Virol 72, 4643–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Haley SA, Gee GV, Atwood WJ, 2010. Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J Virol 84, 9677–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Nelson CD, Atwood WJ, 2015. JC polyomavirus attachment, entry, and trafficking: unlocking the keys to a fatal infection. J Neurovirol 21, 601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO, 2010. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med 61, 35–47. [DOI] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J, 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A 82, 1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekenkamp LJ, Haan JC, Koopman M, Vink-Borger ME, Israeli D, Teerenstra S, Ylstra B, Meijer GA, Punt CJ, Nagtegaal ID, 2013. Chromosome 20p11 gains are associated with liver-specific metastasis in patients with colorectal cancer. Gut 62, 94–101. [DOI] [PubMed] [Google Scholar]
- Mo H, Winter HC, Goldstein IJ, 2000. Purification and characterization of a Neu5Acalpha2–6Galbeta1–4Glc/GlcNAc-specific lectin from the fruiting body of the polypore mushroom Polyporus squamosus. J Biol Chem 275, 10623–10629. [DOI] [PubMed] [Google Scholar]
- Monaco MC, Major EO, 2015. Immune System Involvement in the Pathogenesis of JC Virus Induced PML: What is Learned from Studies of Patients with Underlying Diseases and Therapies as Risk Factors. Front Immunol 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser S, Alattia JR, Dimitrov M, Matz A, Pascual J, Schneider BL, Fraering PC, 2015. The adipocyte differentiation protein APMAP is an endogenous suppressor of Abeta production in the brain. Hum Mol Genet 24, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CD, Carney DW, Derdowski A, Lipovsky A, Gee GV, O’Hara B, Williard P, DiMaio D, Sello JK, Atwood WJ, 2013. A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 4, e00729–00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CD, Derdowski A, Maginnis MS, O’Hara BA, Atwood WJ, 2012. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 428, 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T, 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessentheiner AR, Huber K, Pelzmann HJ, Prokesch A, Radner FPW, Wolinski H, Lindroos-Christensen J, Hoefler G, Rulicke T, Birner-Gruenberger R, Bilban M, Bogner-Strauss JG, 2017. APMAP interacts with lysyl oxidase-like proteins, and disruption of Apmap leads to beneficial visceral adipose tissue expansion. FASEB J 31, 4088–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, Benmerah A, Tosoni D, Di Fiore PP, Atwood WJ, 2004. A JC virus induced signal is required for infection of glial cells by a clathrin and eps15 dependent pathway. J. Virol 78, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, O’Hara BA, Williams G, Atwood WJ, 2006. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J Virol 80, 9402–9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F, 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CS, Koralnik IJ, 2010. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 9, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma V, Zuber C, Winter HC, Goldstein IJ, Roth J, 2001. Application of a lectin from the mushroom Polysporus squamosus for the histochemical detection of the NeuAcalpha2,6Galbeta1,4Glc/GlcNAc sequence of N-linked oligosaccharides: a comparison with the Sambucus nigra lectin. Histochem Cell Biol 116, 183–193. [DOI] [PubMed] [Google Scholar]
- Vacante DA, Traub R, Major EO, 1989. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 170, 353–361. [DOI] [PubMed] [Google Scholar]
- Viscidi RP, Rollison DE, Sondak VK, Silver B, Messina JL, Giuliano AR, Fulp W, Ajidahun A, Rivanera D, 2011. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin Vaccine Immunol 18, 1737–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Gui X, Freed DC, Ku Z, Li L, Chen Y, Xiong W, Fan X, Su H, He X, Rustandi RR, Loughney JW, Ma N, Espeseth AS, Liu J, Zhu H, Wang D, Zhang N, Fu TM, An Z, 2019. Identification of adipocyte plasma membrane associated protein as a novel modulator of human cytomegalovirus infection. PLoS Pathog 15, e1007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Palcic MM, Mo H, Goldstein IJ, Hindsgaul O, 2001. Rapid determination of the binding affinity and specificity of the mushroom Polyporus squamosus lectin using frontal affinity chromatography coupled to electrospray mass spectrometry. Glycobiology 11, 141–147. [DOI] [PubMed] [Google Scholar]