

Summary
Translational control targeting the initiation phase is central to the regulation of gene expression. Understanding all of its aspects requires substantial technological advancements. Here we modified yeast translation complex profile sequencing (TCP-seq), related to ribosome profiling, and adapted it for mammalian cells. Human TCP-seq, capable of capturing footprints of 40S subunits (40Ss) in addition to 80S ribosomes (80Ss), revealed that mammalian and yeast 40Ss distribute similarly across 5′TRs, indicating considerable evolutionary conservation. We further developed yeast and human selective TCP-seq (Sel-TCP-seq), enabling selection of 40Ss and 80Ss associated with immuno-targeted factors. Sel-TCP-seq demonstrated that eIF2 and eIF3 travel along 5′ UTRs with scanning 40Ss to successively dissociate upon AUG recognition; notably, a proportion of eIF3 lingers on during the initial elongation cycles. Highlighting Sel-TCP-seq versatility, we also identified four initiating 48S conformational intermediates, provided novel insights into ATF4 and GCN4 mRNA translational control, and demonstrated co-translational assembly of initiation factor complexes.
Keywords: gene expression, translational control, mRNA, UTR, ribosome profiling, Ribo-seq, TCP-seq, ribosome, eIF3, eIF2, ATF4, GCN4, co-translational assembly
Graphical Abstract
Highlights
-
•
Selective TCP-seq dissects translation mechanisms in mammals and yeast
-
•
40S-bound eIF2 and eIF3 traverse 5′ UTRs and sequentially dissociate at AUG codons
-
•
AUG recognition involves four conformational intermediates of the 48S PIC
-
•
Initiation factors can assemble co-translationally into higher-order complexes
Changing environmental conditions require reprogramming of gene expression for cells to adapt and survive. Translational control is central to this complex response. Wagner et al. developed assays capturing footprints of mRNA-bound ribosomal complexes associated with selected translation-promoting factors to uncover mechanisms underlying translation in the context of its regulatory potential.
Introduction
Changing environmental conditions require rapid reprogramming of gene expression to enable cells to adapt and survive. Regulation of mRNA translation is central to this complex response (Sonenberg and Hinnebusch, 2009), with the initiation phase the main target of regulatory inputs that promote or attenuate some of its steps. Therefore, it is of fundamental importance to fully understand the molecular mechanism of all initiation steps in the context of their regulatory potential.
In eukaryotes, the Met-tRNAiMet is delivered to the small ribosomal subunit (40S) by translation initiation factor 2 (eIF2) bound to guanosine triphosphate (GTP) in the form of a ternary complex (eIF2-TC) (Hinnebusch, 2014, Shirokikh and Preiss, 2018, Valásek, 2012). This step is further promoted by eIF1, eIF1A, eIF3, and eIF5, producing the 43S pre-initiation complex (PIC). In fact, the eIF2-TC and eIF1, eIF3, and eIF5 can assemble into a multifactor complex (MFC), ensuring more efficient delivery of Met-tRNAiMet to the 40S (Asano et al., 2001a, Dennis et al., 2009, Sokabe et al., 2012). The eIF4F cap-binding complex, in cooperation with eIF3, then loads mRNA to the 43S PIC to form the 48S PIC. In a scanning-conducive open conformation, with the anticodon of Met-tRNAiMet in the ribosomal P site in a POUT conformation, the 48S PIC scans the mRNA 5′ UTR for the proper initiation site (Hinnebusch, 2017). Upon start codon recognition accompanied by GTP hydrolysis on eIF2, eIF1, eIF1A, eIF2, eIF3, and eIF5 promote a series of rearrangements leading to a scanning-arrested conformation of the 48S PIC, before it is joined by the large ribosomal subunit (60S). This entails complete accommodation of Met-tRNAiMet in the ribosomal P site (PIN conformation), closure of the 40S mRNA binding channel, and ejection of the majority of eIFs. Although the functions of most eIFs are well understood biochemically, information is sparse about when and where along the mRNA 5′ UTR interactions among eIFs, mRNA, and ribosomal subunits are established and broken during the steps of initiation.
The eIF3 complex, composed of five subunits in yeast (a/Tif32, b/Prt1, c/Nip1, g/Tif35, and i/Tif34) and 12 in mammals (a, b, c, d, e, f, g, h, i, k, l, and m) (Figures S1A and S1B), is known to be critical for efficient progression of most of the initiation steps, but its complete structure has not been determined from any organism (Cate, 2017, Valášek et al., 2017). The assembly pathway for human and N. crassa (similar in composition to human) eIF3 was recently described (Smith et al., 2016, Wagner et al., 2014, Wagner et al., 2016), but it remains to be examined for the most extensively studied S. cerevisiae eIF3 complex (Zeman et al., 2019). Recent structural studies of various PICs revealed several well-resolved but otherwise discontinuous densities attributed to various eIF3 modules that together nearly embrace the entire 40S (Llácer et al., 2018, Llácer et al., 2015, Simonetti et al., 2016). Intriguingly, besides initiation, eIF3 has also been shown to (1) control translation termination and ribosomal recycling (Beznosková et al., 2013, Beznosková et al., 2015, Pisarev et al., 2007) and (2) stimulate reinitiation (REI) on downstream cistrons after translation of short upstream open reading frames (uORFs) due to its ability to remain bound to elongating ribosomes immediately following termination (Mohammad et al., 2017, Park et al., 2001, Szamecz et al., 2008). Owing to the manifold functions of eIF3, deregulated eIF3 expression is associated with numerous pathologies (reviewed in Gomes-Duarte et al., 2018, Robichaud and Sonenberg, 2017, Valášek et al., 2017).
Translation REI can occur on mRNAs carrying more than one open reading frame (ORF), involving post-termination 40Ss or 80Ss, and is highly regulated in response to various stresses and other intra- or extracellular signals (Gunišová et al., 2018). The mammalian master transcriptional activator ATF4 and its yeast functional ortholog GCN4 are among the most studied examples of genes whose expression is regulated by REI in response to stress. The GCN4 mRNA 5′ UTR contains four very short AUG-initiated uORFs preceded by a longer uORF that can be initiated by either of two consecutive non-AUG codons in the same frame (creating nAuORF1 and nAuORF2) (Gunišová et al., 2018; Figure S1C). nAuORF1 and nAuORF2 have been reported to be ribosome occupied (Ingolia et al., 2009) but with unknown functions (Zhang and Hinnebusch, 2011). GCN4 expression is regulated by delayed REI, enacted by AUG-initiated uORFs 1–4, which is exquisitely sensitive to eIF2-TC availability (Gunišová and Valášek, 2014, Hinnebusch, 2005). The 5′-proximal uORF1 and uORF2 are positive REI-promoting sequences whose ability to allow efficient REI is determined by five cis-acting REI-promoting elements (RPEs i–v) mapping upstream of these uORFs and making contacts with eIF3 (Figure S1C). In contrast, the 5′-distal uORF3 and uORF4 are negative REI-non-permissive sequences lacking any RPEs (Gunišová et al., 2016, Munzarová et al., 2011).
ATF4 expression is also governed by delayed REI, here relying on two short uORFs (Vattem and Wek, 2004) that are preceded by an additional AUG-initiated uORF upstream of uORF1, in humans referred to as uORF0 (Lu et al., 2004), whose role has not been examined. In analogy to GCN4’s uORFs, uORF1 is a positive stimulatory sequence allowing efficient REI after its translation, whereas translation of uORF2 inhibits ATF4 expression. Translation of uORF1 combined with low levels of the eIF2-TC is then required to overcome the uORF2 inhibitory effect. Similar to GCN4’s uORF1 and uORF2, ATF4’s uORF1 is surrounded by cis-acting, REI-promoting sequences. The upstream sequences most probably form a specific secondary structure contacting eIF3 (Hronová et al., 2017; Figure S1D). Extensive genetic and biochemical efforts by numerous labs have been devoted to fully grasp the molecular details of these canonical examples of gene-specific translational control; however, further progress has been hindered by the limitations of standard ex vivo approaches.
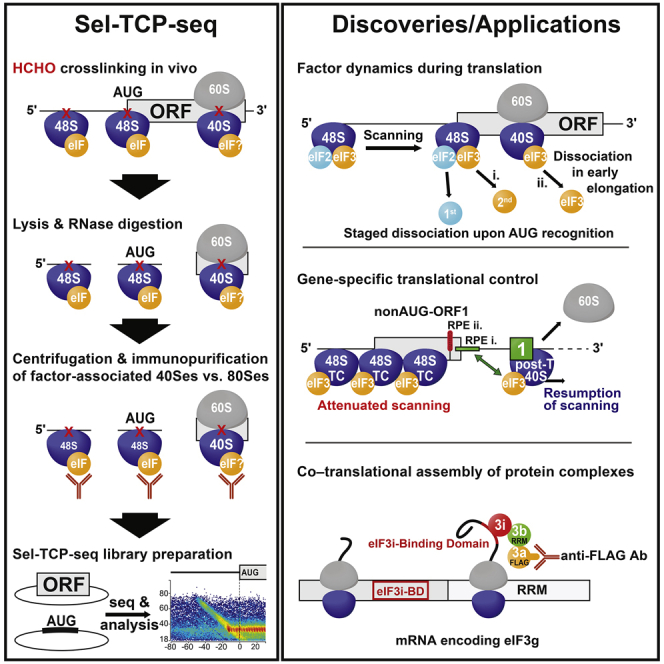
A breakthrough in this direction has been the development of ribosome profiling (Ribo-seq) to study protein synthesis in living cells. Cell lysates are typically prepared in the presence of the elongation inhibitor cycloheximide to preserve polysomes and treated with RNase I, and then regions of protected mRNA (i.e., the ribosome footprints [FPs] are isolated bound to ribosomes and identified by high-throughput sequencing (Ingolia et al., 2009, McGlincy and Ingolia, 2017). Ribo-seq gives access to many aspects of translation and even some coupled events, such as protein folding and localization (reviewed in Ingolia et al., 2019). Translation complex profile sequencing (TCP-seq) extends the Ribo-seq approach in yet another direction. Here, rapid formaldehyde fixation of live yeast cells is used to cross-link any type of ribosomal complex bound to mRNA at native positions. The separate purification and sequencing of FPs from 40Ss and 80Ss adds the ability to survey all mRNA-associated steps of translation, including, uniquely, the initiation phase (Archer et al., 2016).
In this study, we developed TCP-seq for human cells and further devised selective TCP-seq (Sel-TCP-seq) for yeast and human cells by adding a prior step of immunopurifying ribosomes associated with an initiation factor of interest (FOI). Using these novel techniques we studied the association of eIF3 and eIF2 with 40Ss globally as well as on 5′ UTRs of GCN4 and ATF4, and, finally, we also described co-translational assembly of factors within the yeast MFC.
Results and Discussion
TCP-Seq for Yeast and Human Cells
The methodology development for this study commenced independent of the original yeast TCP-seq work but yielded largely convergent approaches for the yeast protocol (Shirokikh et al., 2017); the published version included an initial separation of mono- and poly-ribosomal complexes from mRNPs and ribosomal subunits not associated with mRNAs, which was omitted here (Figure 1A). For human TCP-seq, HEK293T cell cultures were incubated with formaldehyde at 0.3% (w/v) for 5 min, followed by detergent lysis, digestion with RNase I for 30 min to release 40Ss and 80Ss from polysomes, and sucrose density gradient ultracentrifugation to separate them. These conditions were chosen to effectively cross-link polysomes (Figure S2A) and achieve subsequent conversion of most polysomes into well-separated 40S and 80S fractions (Figure S2B). Separate 40S and 80S FP cDNA libraries were then generated, allowing a wide range of insert sizes, and sequenced in parallel with standard RNA sequencing (RNA-seq) analysis of total RNA (see Tables S1 and Table S3 for an overview of generated libraries). As shown in Figure S2C, sequencing and mapping of the resultant libraries consistently gave 40S and 80S FPs with the expected characteristics (see below) and with FP coverage per mRNA showing good correlation between biological replicates.
Figure 1.

TCP-Seq in Human Cells: Full Ribosome FPs
(A) Outline of translation complex profile sequencing (TCP-seq) and selective (Sel)-TCP-seq. 40S, small ribosomal subunit; 80S, ribosome; FOI, factor of interest.
(B) Human 80S footprint (FP) length versus 5′ end position relative to the first nucleotide (position 0) of start (left, 10,778 sites) or stop codons (right, 16,803 sites). The color scale represents FP count as indicated on the right.
(C) Length distribution of human 80S FPs within 9,241 coding sequences (CDS; excluding start and stop codon-associated FPs).
(D) Schematic to facilitate interpretation of (B). Bars illustrate FP lengths as opposed to 5′ end position only.
Shown are representative data from biological replicate 3 (see Table S1 for a full listing).
Metagene plots show that human 80S FPs primarily map within the coding sequence (CDS) of mRNAs and exhibit “translating” 3-nt periodicity in position (Figure 1B). The predominant human 80S FP type detected by TCP-seq is slightly longer (between ∼32–36 nt; Figures 1C and S2G) compared with the ∼28/29-nt FP length typically seen with conventional Ribo-seq (McGlincy and Ingolia, 2017). This is also somewhat larger than the 80S FP size measured by TCP-seq in yeast (∼29/30 nt measured here; Figures S2H and S2I; ∼30/31 nt in Archer et al., 2016). The small differences in FP size between studies are likely due to variable efficiency of formaldehyde cross-linking and its effect on RNase I accessibility. TCP-seq further detects a less abundant, smaller 80S FP type in humans and more distinctly in yeast (∼21–25 nt and 21–23 nt, respectively; Figures 1B, 1C, and S2F–S2I). This smaller FP is thought to originate from 80Ss with an empty A site and was also observed in Ribo-seq studies; for example, when omitting the cycloheximide pre-treatment (Lareau et al., 2014, Wu et al., 2019). Of note, a proportion of 80Ss residing at the start codon produces substantially 5′-extended FPs, protecting a stretch of up to ∼35 nt emerging from the exit channel (Figures 1B and 1D). This is also observable to a lesser degree in yeast TCP-seq (Figure S2F). It can be explained by residual binding of eIFs at the exit channel or by “queuing” of a trailing scanning 40S, which impedes RNase I cleavage because of steric accessibility restraints (Figures S3A and S3B). Collectively, these data validate the TCP-seq protocols used here by presenting observations with 80S FPs that are largely equivalent to conventional Ribo-seq.
Human 40S FPs mapped throughout the 5′ region of mRNA, including the initiation codon (Figure 2A), whereas some are also found in the CDS (Figure S2D). As shown in Figures S4A and S4B, the latter display a higher proportion of the smaller FP type than what is seen in the 80S FP libraries, suggesting that they mostly derive from a small proportion of inefficiently cross-linked 80Ss that released their 60S subunits during purification and predominantly contain an empty A site. The 40S FPs also accumulate at stop codons (Figures 2A, right panel, and S2D), likely representing authentic products of the first stage of 80S recycling, where the 60S subunit (60S) has been released (Hellen, 2018). Finally, the presence of some 40S FPs in the 3′ UTR (Figure S2D) is consistent with recently observed evidence of scanning 40S subunits engaged in REI in 3′ UTRs in yeast (Young et al., 2018); it could also indicate non-canonical ribosomal recycling (Guydosh and Green, 2014, Skabkin et al., 2013). Of note, 5′ UTR 40S coverage, which should represent primarily scanning PICs, extends up to the mRNA 5′ ends (Figures 2B and S4E). The broad size range of these 5′ UTR 40S FPs, protecting mRNA fragments from a size corresponding approximately to the span of the 40S alone (∼24 nt) up to ∼70 nt (Figures 2C and S4H), is likely due to residual eIFs binding at the exit channel or by 40S queuing, as noted above for 80S FPs (Figures S3A and S3B). Together, these 40S FP distributions are consistent with human 40Ses binding to the mRNA’s 5′ cap structure and then scanning the 5′ UTR as a larger assembly with the assistance of eIFs. All these observed patterns are similar to those seen in yeast (Figures S2E, S2F, S4C, S4D, S4F, S4G, S4I, and S4J; Archer et al., 2016).
Figure 2.

TCP-Seq in Human Cells: Small Ribosomal Subunit FPs
(A) Human 40S FP length versus FP 5′ end positions relative to start (left, 10,778 sites) or stop codons (right, 16,803 sites) and count (color scale).
(B) Human 40S FP coverage and mRNA coverage (from “regular” RNA-seq) versus distance from transcription start sites (cap; 1,046 mRNAs, excluding start codon-assigned FPs). RPM, reads per million.
(C) Length distribution of human 40S FPs assigned to the 5′ UTR of 9,241 mRNAs (excluding start codon-assigned FPs).
(D) Yeast start codon-aligned 40S FP 5′ and 3′ end positions (5,994 sites). FPs with 5′ ends between the −14 to −12 positions (black rectangle in left plot) were chosen to display their 3′ end distribution (right; shading around the line indicates the 95th percentiles of random gene-wise resampling). Gray vertical bars on the right indicate discernible 3′ end peaks; see illustrative black bars on the left.
(E) As (D) but for human 40S FPs (11,174 start sites). Gray vertical bars in the right plot show yeast 3′ end positions. Human 3′ end distribution peaks at +20 and +24 nt, cf. illustrative black FP bars on the left.
Shown are representative data from human biological replicate 3 and yeast replicate 1-1.
The original TCP-seq protocol yielded three main yeast 40S FPs at start codons. Their 5′ ends predominantly mapped to position −12 (the A of the AUG is set to 0), with 3′ ends mostly found at the +6, +16, and +24 positions, likely representing sequential intermediates of AUG recognition (Archer et al., 2016). We broadly replicate this 3′ extended pattern of predominant 5′ end FPs here, with only minor changes in the preferred yeast 40S FP 5′ and 3′ end positions (at −13, +7, +15, and +23, respectively) and in the relative proportions between FPs of different sizes, with +15 representing the major 3′ end intermediate (Figures 2D, S3C, S3D, and S4L). Another characteristic of start codon-associated FPs is that, besides these 3′ extensions, some exhibited extended 5′ ends beyond the predominant −13 position (Figures S3A, S3B, S3D, and S3E). Because their 3′ ends are unchanged, we assume that these 5′-extended FPs also have the start codon set in the 40S P site. As mentioned above, the 5′ extensions likely represent eIF interactions at the 40S exit channel, expected for a scanning PIC, that are still partly in place (for FPs with a 3′ end reaching the +5 to +8 positions) or a second queuing 40S extending protection upstream (for FPs with a 3′ end reaching +15 to +18 and even more pronounced for those with a 3′ end reaching +23 to +25) (Figures S3A and S3B).
Human initiating 40Ss show a slightly different pattern. A major FP 5′ end position at −12/−14 is also seen; however, we observed only two 3′ ends at comparable frequency, around +20 and +24 (Figures 2E and S4K). In addition, although in yeast all three 3′ end intermediates feature 5′ extensions of comparable intensity, as mentioned above (Figure S3E), notably, in humans, only the +24 3′ end intermediate displays a prominent 5′ extension (Figures 2A and S3F). We speculate that this species-specific distinction might point to biological differences in AUG recognition kinetics, such as faster conversion of human 40Ss from early to late AUG recognition states and/or slower 60S joining to convert initiating 40Ss into full 80Ss.
Taken together, our yeast findings obtained with the revised TCP-seq largely mirror those found in the original TCP-seq study (Archer et al., 2016), illustrating high reproducibility of this approach. Despite the considerable evolutionary conservation of the initiation mechanism among eukaryotes, mammalian TCP-seq points to numerous differences between yeast and humans that probably reflect variations in the number of involved proteins, composition of initiation factor complexes, and the means of their interactions with 40Ss as well as with each other (Hinnebusch, 2017, Shirokikh and Preiss, 2018, Valásek, 2012).
Sel-TCP-Seq Detects Staged Dissociation of eIFs from Translation Initiation Complexes and Resolves Individual Start Codon Recognition Steps
Next we sought to deploy TCP-seq to better understand the role of individual eIFs during initiation. Because different steps of the process involve particular 40S positions along the mRNA, and the factor composition of PICs at these positions may vary, our rationale was to compare the FPs of 40Ss associated with an FOI (FOI::40S) with those of unselected 40Ss. Thus, we subjected RNase I-digested 40Ss, prepared as described above, to an immunoprecipitation step using anti-FOI antibodies, creating Sel-TCP-seq (Figure 1A).
Our first FOI was eIF3 because it is implicated in promoting numerous steps throughout the translation cycle and because most of its subunits resides on the 40S solvent-exposed side (Valášek et al., 2017). For human cells we used our efficient eIF3b co-immunoprecipitation protocol (Wagner et al., 2014, Wagner et al., 2016; Figures S5F and S5G); for yeast, we used our well-established affinity tag pull-downs (Nielsen et al., 2006, Valásek et al., 2002),where genes encoding the Tif32 and Nip1 subunits of eIF3 with a C-terminal FLAG tag were expressed from plasmids in a strain deleted for the corresponding genomic allele (Figures S5H–S5J). Analogously, we generated a strain expressing a FLAG-tagged β subunit of the trimeric eIF2 complex chosen as our second FOI because it resides at the 40S interface side (Llácer et al., 2015). We sequenced FOI::40S FPs and compared them with FPs of the unselected pool of 40Ss isolated from the same experiment. Reassuringly, we observe a high correlation between replicates and between the FOI::40S and the total 40S FP count per gene for each targeted factor and organism (Figures 3A–3C and S5A). Further, eIF2::40S and eIF3a/c::40S FP coverage in yeast and eIF3b::40S coverage in humans begins immediately at the 5′ end of mRNAs (Figures 3D–3F and S6A). This provides direct evidence for the long-standing expectation that most 40Ss have both factors bound upon mRNA recruitment.
Figure 3.

Sel-TCP-Seq Detects Staged Dissociation of eIFs from Translation Initiation Complexes
(A) Human unselected 40S FP versus eIF3b::40S FP counts per mRNA. r, Pearson correlation coefficient.
(B) As (A) but for yeast unselected 40S versus eIF3a::40S.
(C) As (A) but for yeast unselected 40S versus eIF2β::40S.
(D) Human eIF3b::40S FP and unselected 40S FP coverage aligned to annotated transcription start sites (cap; 1,046 mRNAs, excluding start codon-associated FPs). Averages of replicates 1 and 3 are shown.
(E) As (D) but for yeast eIF3a::40S and unselected 40S (198 mRNAs).
(F) As (E) but for yeast eIF2β::40S and unselected 40S (198 mRNAs).
(E and F) Averages of replicates 1 and 2 are shown.
(G) Human eIF3b::40S versus unselected 40S FP count ratios within indicated mRNA regions (n = 1,389; dashed line, median; dotted lines, first and third quartiles; ∗∗∗p < 0.001, two-sample t test). Data from human replicate 3 are shown.
(H) As (G) but for yeast eIF3a::40S versus unselected 40S FP count ratios (n = 2,096, ∗∗∗∗p < < 0.0001). Data from yeast replicate 1-1 are shown.
(I) As (H) but for yeast eIF2β::40S versus unselected 40S FP count ratios (n = 1,734). Data from yeast replicate 1-3 are shown.
(J) Enrichment of tRNA read counts in human eIF3b::40S versus an unselected 40S library. Averages of human replicates 1 and 3 are shown.
(K) As (J) but for yeast eIF3a::40S and eIF2β::40S versus the corresponding unselected 40S libraries. Averages of replicates 1 and 2 are shown.
(L) Differences in FOI (indicated below the x axis) persistence upon initiation complex arrival at start codons. The ratio of FOI::40S FP counts in the 5′ UTR versus start codon regions of 1,218 mRNAs was divided by the equivalent ratio of unselected 40S FP counts. Both FOI::40S complexes redistribute to 5′ UTRs relative to the unselected 40S (∗∗∗∗p < < 0.0001, one-sample t test), but this is more pronounced for eIF2β than eIF3a (∗∗p < 0.005, two-sample t test). Data from yeast replicate 1 are shown.
(M) Human unselected 80S FP and eIF3b::80S FP coverage frequency at the beginning of the CDS of 10,778 mRNAs. Averages of replicates 1 and 3 are plotted.
Error bars in (D)–(F), (J), (K), and (M) represent ± standard deviation.
As detailed above, unselected 40S libraries likely contain a proportion of elongating 80S-derived 40S FPs that should be devoid of either of the examined eIFs. They also feature fragments of elongator tRNAs and Met-tRNAiMet, as seen before (Archer et al., 2016). Consistently, compared with unselected 40S FPs, eIF2::40S and each of the eIF3::40S FPs are significantly underrepresented in the CDS and 3′ UTRs but enriched in 5′ UTRs (Figures 3G–3I, S5B–S5D, S6B, and S6C), and their FP libraries are dramatically enriched for Met-tRNAiMet fragments (Figures 3J, 3K, and S6D), demonstrating the selectivity of our approach. Importantly, we found that, compared with unselected 40S, eIF2::40S and each of the eIF3::40S are less enriched in the start codon region relative to the 5′ UTR (Figures 3G–3I, S5B–S5D, S6B, and S6C), supporting the assumption that both eIFs dissociate from PICs during late stages of AUG recognition. The fact that this enrichment is significantly more pronounced for eIF2::40Ss than eIF3::40Ss (Figures 3L, S5E, S6E, and S6F) indicates that some PICs at start codons still contain eIF3 but not eIF2, which is consistent with the model that eIF2 leaves the PIC upon AUG recognition (Hinnebusch, 2017, Shirokikh and Preiss, 2018, Valášek et al., 2017), whereas eIF3 lingers during the initial elongation cycles (Mohammad et al., 2017; see also the related papers in this issue of Molecular Cell, Bohlen et al., 2020 and Lin et al., 2020). In support, we observed markedly increased coverage of yeast and human eIF3::80 FPs over unselected 80S FPs within the first ∼20–25 codons past the AUG (Figures 3M and S6J).
Comparing the FP 3′ end frequency of unselected start codon 40Ss with eIF2::40Ses or eIF3::40Ses reveals an enrichment of the longest 3′ end position (+23–25) at the expense of the shorter 3′ ends (mostly +15–18) for the FOI::40Ss (Figures 4A and S6G). Furthermore, the indicated enrichment is stronger for eIF3 versus eIF2, which is again consistent with (1) the expectation that eIF2 leaves the initiating PIC before eIF3 and (2) our earlier interpretation that FPs with the longest 3′ end represent a late PIC intermediate (Archer et al., 2016). The technical explanation for why we see stronger enrichment for eIF3 versus eIF2 is illustrated in Figure 4B. All 3′ FP intermediates show extended 5′ ends, some of which exceed the length of the typical 40S FP (∼30 nt; Figures 4D, 4E, S6H, S7A, and S7B). We assume that the latter FPs originated from two queuing 40S species at the start codon region; i.e., the phenomenon that was demonstrated recently (Shirokikh et al., 2019; Figure S3A). With Sel-TCP-seq, an equal pull-down efficiency can be expected for early AUG recognition intermediates with a queuing 40S because both 40S species should still contain eIF2 and eIF3 (Figure 4B). In contrast, late intermediates might have already lost eIF2, specifically increasing pull-down efficiency with eIF3.
Figure 4.

Sel-TCP-Seq Resolves Individual Start Codon Recognition Steps
(A) Yeast start codon-aligned, unselected, and FOI::40S FP 3′ end position frequencies (5,994 sites; shading around lines indicates ± standard deviation). Gray vertical bars are shown as in Figure 2D.
(B) Suggested influence of queuing 40Ses on co-immunoprecipitation (coIP) efficiency.
(C) 5′ end position frequencies of unselected 40S FPs ending at specific 3′ end positions, as indicated to the right of each row. Selection matches the gray bars in (A) but respects partition of the +15 to +18 area into two peaks. For orientation, bars representing the −30 and −13 positions are highlighted in black. Error bars indicate ± standard deviation.
(D) As (C) but for eIF2β::40Ss.
(E) As (C) but for eIF3a::40Ss.
Shown are averages of yeast replicates 1 and 2.
Another interesting observation is the apparent split of the FP population with 3′ ends around +15–18 into two sub-populations, which are differently enriched with eIF2::40S (+15 and +18) or eIF3::40S (primarily +18), suggesting that Sel-TCP-seq dissects the yeast AUG recognition process into four stages (Figures 4A and S6G). To better interpret these distinct FP types, we grouped them by their 3′ end position and examined their extended 5′ ends (Figures 4C–4E; S6H,S7A, and S7B). Unselected 40S FPs sorted by increasing 3′ end positions show a transition from a smooth trail up to around position −60 (representing scanning-type interactions) to an increasingly pronounced “bump” at around −30 (representing a second, queuing 40S) (Figure 4C). Therefore, we propose that the FPs with their 3′ end at +15 represent the stage that follows the “+5–8 stage” but precedes the “+18 stage.” Implying that the +15–18 set of FPs exemplifies PICs in the closed conformation, we further propose that the +15 peak embodies 40Ss that have undergone the conformational switch from the open to closed state with the eIF2-TC still in the POUT arrangement, whereas the +18 stage reflects the closed state with the TC already stabilized in the PIN arrangement (Hinnebusch, 2017).
Focusing on FPs with 3′ ends at +5–8, a marked enrichment of the longest 5′ end extensions in the eIF2::40S and eIF3::40S versus unselected 40S libraries can be seen (Figures 4C–4E, S6H, S7A, and S7B, top panels). Because eIF2 and eIF3 are firmly established as scanning-promoting factors (Hinnebusch, 2017), this finding supports our earlier proposal that the +5–8 stage represents PICs that are still partially in the scanning configuration (Archer et al., 2016). Interestingly, compared with unselected libraries, a distinct peak (at −32/33) is revealed for FPs with 3′ ends at +15 and mainly at +18 positions (Figures 4C–4E; S6H,S7A and S7B, center panels). Considering that these two 3′ end positions likely represent the open-to-closed transition, which is known to be accompanied by substantial conformational rearrangements (Llácer et al., 2015, Simonetti et al., 2016, Valášek et al., 2017), we speculate that this peak could represent one of the specific AUG recognition intermediates involving both eIFs under study; for example, a proposed temporal eIF3 relocation step around the entry channel toward the ribosomal A site, where both complexes transiently contact each other (Llácer et al., 2018).
These findings demonstrate that deploying a selective approach to translation complex isolation (Sel-TCP-seq) enables fine dissection of the individual initiation phases as well as of the compositional/conformational states of PICs. Importantly, Sel-TCP-seq can help answer questions concerning functions of particular eIFs directly in the cell. It will be intriguing to employ Sel-TCP-seq in deciphering physiological roles of, for example, the mRNA-recruiting eIF4F complex and the subunit joining-promoting eIF5B factor.
Sel-TCP-Seq Reveals Another Layer of Regulation of GCN4/ATF4 mRNAs
As described above, the ATF4 and GCN4 genes encode master transcriptional activators and represent classic examples of genes under REI-mediated translational control in response to stress (Gunišová et al., 2018). Because the key tenets of the GCN4 regulatory mechanism were established mostly based on extensive yeast genetic analysis (Figure S1C), we examined whether they would find additional support in our TCP-seq data of non-stressed cells (Figure 5). Although unselected 40S and 80S coverage frequency is high across the 5′ UTR of GCN4 mRNA, broadly tracking the presence of uORFs but with distinct local differences in the extent of their accumulation, it is absent over the main ORF, as expected for translational repression by uORFs in non-stressed cells (Figure 5A). Within the nAuORF1/2 region, 40Ss do not noticeably accumulate at the two potential non-AUG start codons; instead, there is a broad and pronounced 40S peak in the region between them. Because there are only modest levels of 80Ss throughout nAuORF1/2, these findings indicate that initiation events are rare at both nAuORF start sites, consistent with the observation that they are dispensable for GCN4 translational control (Zhang and Hinnebusch, 2011). Instead, most 40Ss appear to slow or stall in their scanning some distance into nAuORF1/2, just upstream of one of the RPEs linked to uORF1, RPE ii, positioned 69 nt downstream of the nAuORF1 start site, which forms a 9-bp-long hairpin (Figure 5B; Gunišová et al., 2016, Munzarová et al., 2011). This suggests that one of the main functions of the nAuORF region might be exerted by this RPE, forming a barrier scanning 40Ss have to overcome to reach the regulatory AUG-initiated uORFs. We propose that these stalled 40Ses constitute a reservoir of initiation-competent (eIF2-TC-containing) PICs, built up under normal growth conditions, that can be utilized under stress conditions. Consistent with this, eIF3::40S and, to a lesser extent, eIF2::40S are enriched relative to unselected 40S in the nAuORF region, whereas this ratio inverts over several of the uORFs, uORFs 1–3 in case of eIF3::40S, and still notable over uORFs 1 and 3 for eIF2::40S (Figures 5C, 5D, S6I, and S7G). This indicates that, although some 40S species in the uORF regions have already lost one or both of these factors (the proportion of those unselected is higher than of those selected), this is not the case for the PICs built up in the RPE region. Combined with a high abundance of GCN4 mRNA (Hinnebusch, 2005), this reservoir might assist with fast and sustained Gcn4 upregulation because it will be unaffected by the limiting amounts of the eIF2-TC responsible for the general translational shutdown during stress (Figure S1C). In support, deletion of RPE ii reduces the inducibility of GCN4 under stress by ∼30% (Munzarová et al., 2011).
Figure 5.

Sel-TCP-Seq Dissects REI-Mediated Control of GCN4 mRNA Translation
(A) Yeast unselected 40S and 80S FP coverage frequency along the 5′ UTR of GCN4 mRNA (schematic with regulatory elements shown on top). uORF locations are emphasized by vertical gray shading; positions of REI-promoting elements (RPEs) i, ii, and iv are further indicated by colored bars on the x axis. Averages of replicates 1-1, 1-2, and 1-3 are shown.
(B) Schematic of the yeast GCN4 translational control mechanism.
(C) As (A) but showing eIF3a::40S FP coverage frequency (the unselected 40S track is indicated as a dashed line). Averages of replicates 1 and 2 are shown.
(D) As (C), but eIF2β::40S and unselected 40S tracks are shown.
Error bars in (A), (C), and (D) indicate ± standard deviation.
Interestingly, coverage in general is less for the second uORF in each pair (1 versus 2 and 3 versus 4), supporting the recently proposed “fail-safe” model of GCN4 translational control postulating that uORF2 serves as the REI-promoting backup for uORF1, whereas uORF4 serves as an inhibitory backup for uORF3 (Gunišová and Valášek, 2014). Further, we noticed substantial differences in the 40S/80S ratios between the uORF1,2 and uORF3,4 pairs (Figure 5A). The relatively low 40S/80S ratios over uORF3 and uORF4 can be explained by efficient conversion of 48S PICs into elongation-competent 80Ss followed by rapid termination/recycling for these uORFs. In contrast, the relatively high 40S/80S ratios over uORF1 and uORF2 might stem from slow conversion of the post-termination 40Ss into those that resume 5′ UTR traversal/scanning downstream; the level of leaky scanning over the latter uORFs is negligible (Grant et al., 1994, Szamecz et al., 2008). The selected eIF3::40S and eIF2::40S FPs over uORFs did not reveal any specific differences.
Comparing the 40S and 80S coverage along the 5′ UTR of ATF4 (for its current regulatory model, see Figure S1D) reveals some similarities to GCN4. Unselected 40S and 80S coverage broadly tracks with the presence of uORFs along the 5′ UTR, showing distinct local differences. The 40S coverage frequency in a region overlapping with uORF0 is significantly higher than that of 80S (Figure 6A), which resembles the high occupancy of 40Ss in the nAuORF1/2 region of GCN4 and could suggest a similar build-up of a reservoir of initiation-competent 40Ss upstream of the ATF4 uORF1. However, the uORF0 proximity to the mRNA cap, allowing only one initiation-competent 40S to constitute this reservoir, makes this option less likely. Interestingly, uORF0 has a special character because its “CDS” consists of just the AUG start codon immediately followed by the stop codon. Therefore, the 40S and 80S coverage frequency over this peculiar uORF might rather reflect the particular kinetics of non-canonical translation events; i.e., the balance between the 48S PIC conversion into the 80S initiating complex, its direct transition into the 80S termination complex, and emergence of the 40S post-termination recycling intermediate. As for the other ATF4 uORFs, the higher 40S/80S ratio for uORF1 compared with uORF2 is broadly similar to that seen for uORF1,2 versus uORF3,4 on GCN4 mRNA (Figure 6A versus Figure 5A). We also detected slightly increased occupancy of heIF3::40Ses relative to unselected 40Ses at uORF1 compared with uORF0 and uORF2; however, this observation lacks statistical significance (Figure 6B). Our observations thus suggest that the uORF0 region might form an active road block preventing uORF1 translation, which is permissive for REI, to keep the basal level of ATF4 expression down (Figure 6C). Taken together, we conclude that the translation of these two functional orthologs is controlled by similar molecular mechanisms despite the differences in the arrangement of their 5′ UTRs.
Figure 6.

Sel-TCP-Seq Dissects REI-Mediated Control of ATF4 mRNA Translation
(A) Human unselected 40S and 80S FP coverage frequency along the ATF4 5′ UTR (schematic with regulatory elements shown on top). uORF locations are emphasized by vertical gray shading. Averages of replicates 1, 2, and 3 are shown.
(B) As (A) but showing eIF3b::40S FP coverage frequency (the unselected 40S track is indicated as a dashed line). Averages of replicates 1 and 3 are shown.
(C) Schematic of ATF4 translational control.
Error bars in (A) and (B) indicate ± standard deviation.
Sel-TCP-Seq Enables Monitoring of the Co-Translational Assembly of Higher-Order Protein Complexes in the Cell
Sel-TCP-seq also affords a comparison of the FPs of 80Ss associated with a FOI with those of unselected 80Ss (Figure 1A). Translatome-wide analysis of yeast eIF3::80S FPs revealed a dramatically uneven distribution in the CDS of several mRNAs encoding non-targeted eIF3 subunits or other eIF3-associated eIFs. These patterns can be accounted for by co-translational assembly of the targeted FOI with its interacting partner(s) under synthesis. In particular, we saw a sudden increase in 80S FP coverage on the mRNA encoding the interacting partner at positions consistent with emergence of the nascent binding domain for the FOI from the ribosome exit tunnel (Figure 7A).
Figure 7.

Sel-TCP-Seq Detects Co-translational Assembly within the MFC in Yeast
(A) Co-translational interactions between a tagged “bait” FOI (e.g., FLAG-tagged eIF3a) and its nascent assembly “target” (e.g., yeast eIF3b via its RNA recognition motif [RRM]) lead to a characteristic increase of selected 80S FP coverage within the assembly partner CDS.
(B–F) FP coverage tracks for selected (black; bait protein indicated on top of the left axis) and unselected 80S FP (gray; right axis) across mRNAs encoding target proteins within the yeast multifactor complex (MFC): eIF3b (B and D), eIF3g (C), eIF3a (E), and eIF2β (F). Domain structures of target MFC subunits are shown under the graphs. Dotted lines indicate the selected 80S FP coverage increase. Tracks from replicate 1 are shown, and average fold induction from 4 or 2 replicates (replicates 1–4 or 1+2) with standard deviation is given for eIF3a-FLAG or eIF3c-FLAG as bait, respectively. Fold induction of the 80S Sel-TCP-seq coverage was normalized to that seen with unselected 80S.
(G) Speculative MFC assembly model. Question marks indicate an unknown mode of factor assembly.
With yeast eIF3a as the FOI, we observed an ∼58-fold increase in the 80S FP coverage along the eIF3b (PRT1) mRNA for eIF3a::80S FPs versus unselected 80S FPs starting near nucleotide 580 (Figure 7B). The onset of this increase is ∼30 codons downstream of the CDS region that encodes the eIF3b RNA recognition motif (RRM; nucleotides 237–477), which is one of the two main interacting domains for eIF3a in the eIF3 complex (Figure S1A; Valásek et al., 2002, Zeman et al., 2019). Because the ribosome exit tunnel accommodates ∼24 amino acids in extended conformation and ∼38 amino acids in α-helical conformation (Bhushan et al., 2010), the nascent RRM should have just emerged from the ribosome when an increase in FP coverage is seen. Unexpectedly, we also detected evidence of co-assembly of eIF3a with eIF3g because eIF3a::80S FP coverage increased ∼6-fold above unselected 80S FPs downstream of the eIF3g CDS region encoding its eIF3i-binding domain (Asano et al., 1998; Figure 7C). eIF3g and eIF3i have been shown previously to interact directly with the C-terminal region of eIF3b but not with eIF3a (Asano et al., 1998). Given the modest increase in eIF3a::80S FP coverage on eIF3g mRNA and the fact that we did not detect any enhanced eIF3a::80S FP coverage within eIF3i mRNA, we posit that co-translational assembly of eIF3g by eIF3a occurs indirectly, bridged by the seven-bladed WD40 β-propeller of eIF3i already bound to eIF3b, when the nascent eIF3i binding domain in eIF3g emerges from the exit tunnel on 80S translating eIF3g mRNA (Figure S7D).
Using yeast eIF3c as the FOI, we detected an increase in eIF3c::80S FP coverage within the eIF3b mRNA just downstream of the eIF3b RRM coding region (Figure 7D), as noted above for the eIF3a::80S FPs. However, this increase was much less pronounced (∼12-fold versus ∼58-fold), again suggesting that it is indirect, mediated by eIF3a, with which eIF3c directly interacts (Phan et al., 1998, Valásek et al., 2002). In accord with previous biochemical analyses, we detected a second, more robust increase (∼24-fold) in eIF3c::80S FP coverage within the sequences encoding the eIF3b C-terminal domain (Figure 7D), shown to interact directly with eIF3c (Valásek et al., 2002). Additionally, we see a very similar eIF3c::80S FP coverage pattern across the eIF3g mRNA CDS as described for eIF3a::80S data, further suggesting that co-translational assembly of eIF3g must await association of the eIF3b/eIF3i heterodimer with eIF3a late in the pathway (compare Figures 7C and S7C).
Furthermore, the eIF3c::80S FP library revealed a strong (∼60-fold) increase in 80S FP coverage within eIF3a mRNA just downstream of the sequence encoding the PCI domain (Figure 7E), which is the only known eIF3c interaction domain within eIF3a (Figure S1A; Valásek et al., 2002). The fact that we did not find evidence of co-translational assembly of eIF3c with eIF3a in the eIF3a::80S data (data not shown) suggests uni-directional co-translational binding of the nascent PCI domain in eIF3a to fully synthesized eIF3c. This type of uni-directional co-translational assembly has been suggested to prevent increased propensity for aggregation/misfolding of proteins prone to these expression pathologies (Shiber et al., 2018). Based on these findings, we propose a co-assembly pathway for yeast eIF3 that is described in detail in Figure S7E and conclude that eIF3 belongs to the growing class of multiprotein complexes that have been shown to undergo co-translational assembly by a ribosome profiling-based technique called selective ribosome profiling (SeRP) (Shiber et al., 2018, Shieh et al., 2015).
Strikingly, the eIF3c::80S data also provide evidence for co-translational assembly of the β subunit of eIF2 with eIF3 soon after the first of three lysine stretches (dubbed K-boxes) of eIF2β emerges from the exit tunnel (Figure 7F). Because the eIF2β K-boxes have been shown to interact selectively with the ε subunit of eIF2B or the C-terminal domain of eIF5 (Asano et al., 2001b), but not with eIF3c (Asano et al., 2001b), this co-translational assembly is most likely bridged by eIF5, a well-known interacting partner of eIF3c (Phan et al., 1998, Valásek et al., 2004) and a lynchpin in MFC assembly. An indirect mode of co-translational assembly is also consistent with the modest ∼3-fold increase in eIF3c::80S FPs on eIF2β mRNA. Hence, we propose that eIF3c is the main nucleation core not only of eIF3 but also of the entire MFC, fortifying the idea that eIF1, eIF3, eIF5, and eIF2-TC preassemble prior to joining the 40S subunit in the 43S PIC formation (Asano et al., 2001a; Figure 7G).
Interestingly, the recent SeRP analysis of eIF2 revealed that eIF2γ co-translationally assembles with eIF2β (Shiber et al., 2018). Because the onset of the eIF2γ-targeted 80S FP coverage increase mapped in that study to the eIF2γ–binding domain in the last third of the eIF2β CDS, it seems that the eIF3c-eIF5 dimer co-translationally interacts with eIF2β prior to its binding to eIF2γ; i.e., prior to assembly of eIF2 per se. Whether it is only a transient, stabilizing interaction before eIF2 assembles as a trimeric complex or whether the entire MFC, and perhaps other higher-order protein complexes, can assemble co-translationally, independent of the integrity of their individual multi-subunit components, remains to be addressed. In any case, our findings nicely correlate with a recent report demonstrating that translation factor mRNAs are localized to distinct granules in growing yeast cells, where they are actively translated, in contrast to other messenger RNP granules, such as P bodies and stress granules, containing translationally repressed mRNAs (Pizzinga et al., 2019).
Overall, Sel-TCP-seq appears to be a versatile technique for co-assembly discoveries because the third-party-bridged co-translational interactions we observed here were not detected with previous approaches (Shiber et al., 2018). We speculate that it is the initial, rapid formaldehyde in vivo cross-linking step that stabilizes multiprotein assemblies well enough to detect even co-translational assemblies that are bridged by one or more proteins, which are characterized by a less robust 80S FP coverage increase than that seen for direct co-assemblies.
Conclusion
By modifying yeast TCP-seq and adopting it for human cell lines, we revealed that, although eukaryotic protein synthesis is evolutionary conserved, particular differences exist, such as in factor occupancy of PICs at various initiation stages and in the AUG recognition kinetics, perhaps reflecting an increased complexity of translational control in higher eukaryotes. Furthermore, we demonstrated that the Sel-TCP-seq technique presented in this study as well as in Bohlen et al. (2020) provides researchers with a unique opportunity to dive deep into not only molecular details of assembly and general functioning of mRNA-associated ribosomal complexes in vivo but also into regulatory mechanisms governing the actual translational output. In other words, with this technique, we can now ask when and where along the mRNA 5′ UTR interactions among eIFs (beyond those investigated here), other auxiliary factors, mRNA, and ribosomal subunits are made and broken during individual phases of translation and how these processes are regulated in response to changing environmental conditions.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-eIF3b (for CoIP) | Santa Cruz | sc16377 (discontinued) |
| Rabbit polyclonal anti-eIF3a | Cell signaling | 2538 |
| Mouse polyclonal anti-eIF3b (for western blot) | Thermo Fisher | PA5-23278 |
| Rabbit polyclonal anti-eIF3c | Santa Cruz | sc28858 |
| Rabbit polyclonal anti-eIF3e | Abcam | ab36766 |
| Rabbit polyclonal anti-eIF3i | Sigma | HPA029939 |
| Rabbit polyclonal anti-eIF3l | Bethyl | A304-753A-T |
| Mouse Monoclonal anti-eIF2alpha | Santa Cruz | sc133132 |
| Rabbit polyclonal anti-eIF5 | Santa Cruz | sc282 |
| Rabbit polyclonal anti-RPS14 | Santa Cruz | sc68873 |
| Rabbit polyclonal anti-TIF32 | Dr. Jiří Hašek | N/A |
| Rabbit polyclonal anti-PRT1 | Cigan et al., 1991 | N/A |
| Rabbit polyclonal anti-GCD11 | Dr. Ernie Hanning | N/A |
| Rabbit polyclonal anti-SUI5 | Dr. Alan Hinnebusch | N/A |
| Rabbit polyclonal anti-ASC1 | Dr. Chris Browne | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Sigma | D6429 |
| FBS | Sigma | F7524 |
| Cycloheximide | Merck | 01810 |
| Formaldehyde solution | Sigma | F8775 |
| Glycine | Serva | 23395.03 |
| protease inhibitor cocktail cOmplete mini EDTA-free | Roche | 11836170001 |
| protease inhibitor cocktail cOmplete EDTA-free | Roche | 11873580001 |
| RNase I | Invitrogen | AM2294 |
| SUPERaseIN | Invitrogen | AM2694 |
| Protein A/G Agarose | Pierce | 20421 |
| ANTI-FLAG M2 affinity gel | Sigma | A2220 |
| FLAG peptide solution | Sigma | F3290 |
| Glycogen | Thermo Fisher | AM5910 |
| RNA fragmentation reagent | Thermo Fisher | AM8740 |
| T4 Polynucleotide kinase PNK | NEB | M0201L |
| Universal miRNA cloning linker | NEB | S1315S |
| T4 RNA ligase 2 | NEB | M0242L |
| SuperScript III | Thermo Fisher | 18080044 |
| CircLigase ssDNA Ligase | Epicenter | CL4115K |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher | 65001 |
| Phusion Polymerase HF | NEB | M0530S |
| 10 bp DNA ladder | Thermo Fisher | 10821-015 |
| Sybr Gold | Thermo Fisher | S11494 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | 74104 |
| Poly(A)Purist MAG Kit | Thermo Fisher | AM1922 |
| Deposited Data | ||
| Raw and analyzed data | this paper | GEO: GSE139132 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae SY182 | this paper | N/A |
| S. cerevisiae SY183 | this paper | N/A |
| S. cerevisiae SY194 | this paper | N/A |
| S. cerevisiae H25 | this paper | N/A |
| Oligonucleotides | ||
| CAAGCAGAAGACGGCATACGAGATTGAAG CGTGACTGGAGTTCAGACGTGTGCTCTTCCG |
This paper | reverse primer, index: GCTTCA, barcode A |
| CAAGCAGAAGACGGCATACGAGATCTACTA GTGACTGGAGTTCAGACGTGTGCTCTTCCG |
This paper | reverse primer, index: TAGTAG, barcode B |
| CAAGCAGAAGACGGCATACGAGATAGTCGT GTGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: ACGACT, NI-NI-3 |
| CAAGCAGAAGACGGCATACGAGATACTGATG TGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: ATCAGT, NI-NI-5 |
| CAAGCAGAAGACGGCATACGAGATATGCTG GTGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: CAGCAT, NI-NI-6 |
| CAAGCAGAAGACGGCATACGAGATACGTCG GTGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: CGACGT, NI-NI-7 |
| CAAGCAGAAGACGGCATACGAGATAGCTGC GTGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: GCAGCT, NI-NI-11 |
| CAAGCAGAAGACGGCATACGAGATATCGTA GTGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: TACGAT, NI-NI-13 |
| CAAGCAGAAGACGGCATACGAGATGACTA CGTGACTGGAGTTCAGACGTGTGCTCTTCCG |
Ingolia et al., 2012 | reverse primer, index: GTAGTC, NI-NI-22 |
| AATGATACGGCGACCACCGAGATCTACAC | Ingolia et al., 2012 | forward primer, NI-NI-2 |
| 5′-(Phos)-AGATCGGAAGAGCGTCGTGTAGGGAAAGAGT GTAGATCTCGGTGGTCGC-(SpC18)-CACTCA- (SpC18)-TTCAGACGTGTGCTCTTCCGA TCTATTGATGGTGCCTACAG |
Ingolia et al., 2012 | split adaptor primer, NI-NI-9 |
| AUGUACACGGAGUCGAG CUCAACCCGCAACGCGA-(Phos)-3′ |
Ingolia et al., 2012 | 34nt size marker oligo, RNA |
| AUGUACACGGAGUCGACCCAACGCGA-(Phos)-3′ | Ingolia et al., 2012 | 26nt size marker oligo, RNA |
| 5′ -biotin-TEG-GGGGGGATGCGTGCATTTATCAGATCA | Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-TTGGTGACTCTAGAT AACCTCGGGCCGATCGCACG |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GAGCCGCCTGGATAC CGCAGCTAGGAATAATGGAAT |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-TCGTGGGGGGCCCA AGTCCTTCTGATCGAGGCCC |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GCACTCGCCGAAT CCCGGGGCCGAGGGAGCGA |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GGGGCCGGGCCG CCCCTCCCACGGCGCG |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GGGGCCGGGC CACCCCTCCCACGGCGCG |
(Ingolia et al., 2012) | human rRNA depletion oligos |
| 5′ -biotin-TEG-CCCAGTGCGCCCCGGGCGTCG TCGCGCCGTCGGGTCCCGGG |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-TCCGCCGAGGGC GCACCACCGGCCCGTCTCGCC |
Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-AGGGGCTCTCGCTTCTGGCGCCAAGCGT | Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GAGCCTCGGTTGGCCCCGGATAGCCGGGTCCCCGT | Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GAGCCTCGGTTGGCCTCGGATAGCCGGTCCCCCGC | Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-TCGCTGCGATCTATTGAAAGTCAGCCCTCGACACA | Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-TCCTCCCGGGGCTACGCCTGTCTGAGCGTCGCT | Ingolia et al., 2012 | human rRNA depletion oligos |
| 5′ -biotin-TEG-GGTGCACAATCGACCGATC | Ingolia et al., 2012 | yeast rRNA depletion oligo |
| 5′ -biotin-TEG-GTTTCTTTACTTATTCAATGAAGCGG | Ingolia et al., 2012 | yeast rRNA depletion oligo |
| 5′ -biotin-TEG-TATAGATGGATACGAATAAGGCGTC | Ingolia et al., 2012 | yeast rRNA depletion oligo |
| 5′ -biotin-TEG-TTGCTCGAATATATTAGCATGGAATAATAGAATA | This paper | yeast rRNA deple-tion oligo, y sub 5 |
| 5′ -biotin-TEG-GAGATGTATTTATTAGATAAA | This paper | yeast rRNA deple-tion oligo, y sub 6 |
| 5′ -biotin-TEG-TGGTATAACTGTGGTAATTCTAGAGCTAATACAT | This paper | yeast rRNA deple-tion oligo, y sub 7 |
| 5′ -biotin-TEG-TTTTACTTTGAAAAAATTAGAGTGTTCAAAGCAGG | This paper | yeast rRNA deple-tion oligo, y sub 8 |
| 5′ -biotin-TEG-ATGATTAATAGGGACGGTCGGGGG | This paper | yeast rRNA deple-tion oligo, y sub 9 |
| 5′ -biotin-TEG-GCCAAGGACGTTTTCATTAATCAAGAACGAA | This paper | yeast rRNA deple-tion oligo, y sub 10 |
| 5′ -biotin-TEG-TTCCAGGGGGCATGCCTGTTTGAGCGTCATT | This paper | yeast rRNA deple-tion oligo, y sub 11 |
| 5′ -biotin-TEG-TCCACGTTCTAGCATTCAAGGTCC | This paper | yeast rRNA deple-tion oligo, y sub 12 |
| Software and Algorithms | ||
| Bowtie | Langmead et al., 2009 | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| Tophat | N/A | |
| FASTX-Toolkit | N/A | |
| Python 2.7 | van Rossum (1995) | N/A |
| HTSeq | Anders et al., 2015 | https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btu638 |
| matplotlib | Hunter (2007) | N/A |
| R | R Core Team (2018) | https://www.R-project.org/ |
| SAMtools | Li et al., 2009 | [PMID: 19505943] |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Leoš Shivaya Valášek (valasekl@biomed.cas.cz).
Materials Availability
Plasmids and yeast strains generated in this study are available upon request.
Data and Code Availability
The datasets generated during this study were deposited in the GEO database with the following accession number: GSE139132. All custom Python scripts used for the analyses in this paper are available upon request.
Experimental Model and Subject Details
Human cell line
The cell line HEK293T was used. Cells were grown in Dulbecco’s modified Eagle’s media supplemented with 10% fetal bovine serum at 37°C and 5% CO2. For experiments, 3.5 Mio cells in 20 mL of media were seeded into a Ø 15 cm dish and were grown for 48h before harvesting at approximately 80% confluency.
S. cerevisiae strains
Yeast strains SY182, SY183, LMY61 (SY194) and H25 were used in this study (Table S2). To generate SY182 and SY183, YBS52 (Munzarová et al., 2011) was transformed with pRS315-a/TIF32-FLAG-L (Klaus Nielsen) and Yep181-a/TIF32-L (Leoš Valášek), respectively, and YCp-a/TIF32-HIS-U was evicted by growth on 5-FOA. For experiments, yeast strains were grown at 30°C in 1 L of YPD up to an OD600 of 0.7-0.8.
Method Details
Formaldehyde cross-linking and preparation of whole cell extracts
HEK293T
Two Ø 15 cm dishes were processed together at any time. Dishes were transferred to a cold room and 600 μl of a 10 % (w/v) formaldehyde solution (freshly diluted in H2O at room temperature) were added to the media, resulting in a ∼0.3 % final concentration of formaldehyde. To accomplish fast and uniform mixing of the formaldehyde with the media, dishes were tilted and the formaldehyde solution was added into the pool of media, mixed, then dishes were turned back to even level and media was further mixed with the formaldehyde solution by gentle swirling, followed by a 5 min incubation in the cold room, during which the initial temperature of the media/formaldehyde mixture, being close to room temperature, gradually decreased. To inactivate any remaining formaldehyde and the reactive derivatives of fixation, 600 μl of a chilled 2.5 M Glycine solution (dissolved in H2O) was added to the media/formaldehyde mixture, the same way as described before, followed by another 5 min incubation. Liquid was then aspirated, and cells were washed with 25 mL ice cold PBS. The PBS was carefully aspirated and 500 μl of buffer A (10 mM HEPES pH 7.5, 62.5 mM KCl2, 2.5 mM MgCl2) supplemented with 1 mM DTT, 1 % (w/v) Triton X-100 and 1x protease inhibitor cocktail (cOmplete mini EDTA-free) was used for in-dish lysis of each two dishes. The lysis buffer was added to the first dish, cells were scraped off quickly and transferred into the 2nd dish together with the lysis buffer. Cells of the 2nd dish were also scraped into the lysis buffer which was then transferred into a 2 mL microcentrifuge tube, resulting in a total volume of around 1.8 ml. Cell lysis was allowed to proceed for 10 min more on ice with occasional vortexing. The lysate was clarified by a 5 min centrifugation at 14,000 rpm at 4°C. The supernatant typically had a concentration of around 10 AU OD260 per ml. Aliquots of 10 AU (1 ml) were flash frozen and stored at −80°C.
Yeast
Formaldehyde cross-linking was performed according to Valásek et al. (2007). Culture flasks (no more than two at a time) were put into ice and 250 g of ice were added to the culture followed by swirling to achieve a quick cool down. 27 mL of a 37 % w/v formaldehyde solution were added and promptly mixed with the cells and media, resulting in a total concentration of around 0.8 % w/v, taking the ice volume into account. After 1h of incubation on ice, 50 mL of a chilled 2.5 M glycine solution was added and mixed in. All subsequent steps were performed on ice or 4°C. Cells were pelleted by centrifugation, washed with ice-cold buffer B (20 mM Tris/HCl pH 7.4, 200 mM KCl, 5 mM MgAc) and pelleted again. The resulting cell pellet was weighed and resuspended in 1 mL ice-cold complemented buffer B (1 mM DTT, 10 mM PMSF, 1 ug/ml Pepstatin, 1 ug/ml Aprotinin, 1 ug/ml Leupeptin, 1x protease inhibitor cocktail (cOmplete EDTA-free) per 1 g of cells. Cell lysis was performed with a bead beater (MP - FastPrep-24). A settled bead volume of 500 μl ice-cold acid washed glass beads together with 1 mL cell suspension were jiggled for 40 s at speed 5. The supernatant was clarified twice by centrifugation at 14000 rpm for 5 and 10 min. The resulting cell lysate would have a concentration of around 150 U (OD260) per ml. Aliquots of 50 U (330 μl) were flash frozen and stored at −80°C.
Note, that formaldehyde crosslinking unavoidably introduced some variability, especially with HEK293T cells, where even small changes in formaldehyde concentration noticeably affected the polysome profile (Figure S2A). This means that some differences in recorded FP sizes, quantity and distribution might have partly technical origins.
Footprint generation and 40S/80S separation
HEK293T
1 mL of cell lysate (10 U OD260) was incubated with 70 U of RNase I (Invitrogen, cat.no. AM2294) at 24°C for 30 min with gentle shaking. 28 U of SUPERaseIn (Invitrogen, cat.no. AM2694) were then added and the mixture was loaded onto a 5-45 % (w/v) linear sucrose density gradient (12 ml, in buffer A supplemented with 1 mM DTT). After a high velocity spin (39,000 rpm, 2.5 h, 4°C, SW41 Ti rotor Beckman-Coulter) the gradients were fractionated, and fractions collected. Fractions corresponding to the small ribosomal subunit or the full ribosome, as determined by the UV absorbance traces, were pooled separately.
Yeast
330 μl of cell lysate (50 U OD260) was incubated with 750 U of RNase I (Invitrogen, cat.no. AM2294) at 24°C for 1 h with gentle shaking. 100 U of SUPERaseIn (Invitrogen, cat.no. AM2694) were added and 2 aliquots (100 U OD260) were loaded onto a 5-30 % (w/v) linear sucrose density gradient (38 ml, in buffer B supplemented with 1 mM DTT). After a high velocity spin (32,000 rpm, 3 h 44 min, 4°C, SW32 Ti rotor Beckman-Coulter) fractions were collected. Fractions corresponding to the small ribosomal subunit (40S) or the full ribosome (80S) were determined by the UV absorbance profiles and pooled separately.
Co-immunoprecipitation (CoIP)
Collected pools of 40S fractions or 80S fractions were divided into a 95 % aliquot, used for the CoIP, and a 5% aliquot that was flash-frozen and used later for TCP-seq library preparation from unselected pools of 40Ss or 80Ss.
HEK293T
150 U OD260 of cell lysate (corresponding to 15 ‘regular’ aliquots) were used per sample. After RNase I digestion, all aliquots were pooled, loaded onto 12 gradients and run in two the SW41 Ti rotors at the same time. After separation, each pool (40S and 80S) was adjusted to 0.07 % (w/v) Triton X-100 with a 20 % stock solution and split into two. To one half, 100 μl of a 50 % slurry of anti-eIF3b coated protein A/G agarose was added. To the other half, beads without antibody coating were added, to control for the background. Tubes were rotated overnight at 4°C. Then, beads were collected by spinning at 500 rpm for 3 min at 4°C and the supernatant was removed. Beads were washed 3 times with 5 mL buffer A supplemented with 0.07 % (w/v) Triton X-100, and once with buffer A lacking detergent. All supernatant was carefully removed after the last washing step and 600 μl of buffer A were added, before proceeding with TCP-seq library preparation.
Yeast
To control for the CoIP background, strain SY183 was used, which does not express FLAG-tagged protein. 300 U OD260 of cell lysate (corresponding to 6 ‘regular’ aliquots) were used per sample. After RNase I digestion, all aliquots were pooled and loaded onto 3 gradients for the SW28 Ti rotor. After separation, each pool (40S and 80S) was adjusted to 0.2 % Triton X-100 (w/v) with a 20 % stock solution. 150 μl of a 50 % slurry of ANTI-FLAG M2 affinity gel (Sigma, cat.no. A2220) in buffer B was added and tubes were rotated over night at 4°C. Beads were spun at 500 rpm for 3 min at 4°C and supernatant was removed. Beads were washed 4 times with 5 mL buffer B supplemented with 0.2 % (w/v) Triton X-100 and once with buffer B lacking detergent. Supernatant was carefully removed after the last washing step and elution was carried out by adding 200 μl of a 150 ng/μl FLAG peptide solution, incubating the resultant mixtures for 30 min at 4°C with gently shaking to keep the beads swirling. Supernatant was used to prepare TCP-seq libraries.
Polysome profile analysis
Fifteen A260 units of whole cell extract were separated by high-velocity sedimentation through a 5 % to 45 % sucrose gradient at 39,000 rpm for 2.5 h using the SW41 Ti rotor. The absorbance at 254 nm throughout the gradient was recorded to visualize the ribosomal species. These profiles were recorded on paper, scanned, and further edited to remove the grid and differently color the traces for better visualization.
TCP-seq library preparation
For both TCP-seq and Sel-TCP-seq protocols, RNA was extracted with hot acid phenol method, generally as described by Ingolia (2010), from the eluates (yeast) or directly from the beads suspended in 600 ul of buffer A (HEK293T) and the unselected pools of 40S and 80S. Briefly, samples were brought to room temperature and then incubated at 65°C for 5 min. SDS was added up to a final concentration of 1 % (w/v) using a stock solution of 20 %. One volume of acid phenol-chloroform mix (7:1, v/v) was added, followed by a 20 min incubation at 65°C with frequent intermittent vortexing. Samples were then placed on ice for 5 min, followed by a 10 min spin at 14,000 rpm in a benchtop Eppendorf centrifuge at 4°C. A second acid phenol-chloroform (7:1, v/v) extraction was performed at room temperature for 5 min with frequent vortexing. Finally, an extraction with chloroform-isoamylalcohol mix (24:1, v/v) was performed at room temperature and constant vortexing for 1 min. The aqueous phase was recovered by centrifugation and supplemented with 1 μl glycogen (5 mg/ml). Nucleic acids were then precipitated by adjusting to 300 mM sodium acetate (using a 3 M stock solution, pH 5.2) and adding 1 volume of isopropanol. After incubation at −20°C overnight, the RNA was pelleted by centrifugation, washed with ice-cold 75 % ethanol and air-dried. Pellets were dissolved in 10 μl 10 mM Tris/HCl (pH 8.0). Sequencing libraries were prepared according to Ingolia et al. (2012), with some adjustments. Importantly, we allowed for a broader size selection which included RNA fragments from around 18 nt up to around 85 nt, except for yeast replicates 3 and 4 used for 80S Sel-TCP-seq (see Table S1) where the size selection was restricted to around 20 - 50 nt. Further, for yeast a custom set of rRNA depletion oligos was used according to predominant rRNA fragments found in test libraries.
mRNA-seq library preparation
First, total RNA was extracted with the RNeasy Mini Kit. The Poly(A)Purist MAG Kit was further used to extract polyadenylated RNA which following was fragmented with RNA fragmentation reagent for 8 min at 70°C, wherefore 1μg of RNA was mixed with 1.1 μl of the RNA fragmentation reagent to obtain a final volume of 11.1 μl. The fragmentation was terminated by adding the stop buffer. Subsequent library preparation was performed as for TCP-seq samples according to Ingolia et al. (2012), with some adjustments.
High-throughput sequencing
All libraries of a replicate and species were multiplexed and sequenced together (see Table S1 for an overview of the used libraries). The human replicate 1 and 3 and the yeast replicates 1 and 2 were sequenced with a single end layout and 100 bp read length on the Illumina platform HiSeq 2500. The yeast replicates 3 and 4 were sequenced with a single end layout and 50 bp read length on the Illumina platform HiSeq 2500. The human replicate 2 was sequenced with a paired end layout and 100 bp read length on the Illumina platform HiSeq Xten, but only the first read of the pair was used.
RNA sequencing read mapping
First, the adaptor sequences were removed from reads, and reads without adaptor, adaptor-only reads, reads shorter than 18 nt after adaptor removal and reads of low quality were discarded, using fastx_clipper and fastq_quality_filter from the FASTX-Toolkit. Reads were further filtered by successive alignment to ribosomal RNA (using bowtie), a variety of ncRNAs and tRNA (using Bowtie2), retaining unaligned reads for further analyses. tRNA sequences were retrieved from the tRNA database of the University Leipzig (http://trna.bioinf.uni-leipzig.de). Using the default settings of Tophat, the resulting filtered pool of reads was first aligned to the appropriate transcriptome; reads that did not align, were mapped to the appropriate genome. For S. cerevisiae, the genome assembly R64-1-1 was used and a custom annotation file was created containing all entries with the gene_biotype protein_coding of the Ensembl annotation R64-1-1.80 and further 5′ and 3′ UTR annotation as well as 5′ UTR intron annotation retrieved from the SGD database in November 2017 (whereby the longest annotated UTRs per gene were added to the transcriptome). For genes without annotated 5′ or 3′ UTRs, 50 nt of the adjacent genomic sequence were added as generic UTRs. For H. sapiens, the genome assembly GRCh38 and the Ensembl annotation GRCh38.80 were used.
Please note that stacks of identical reads that took up at least 0.1 % of all aligned reads were removed. Also, for all further analysis only unique alignments were used, except for the tRNA enrichment analysis where primary alignments were used.
tRNA enrichment analysis
Primary alignments of reads that mapped to the tRNA reference were used. Counts for all tRNA isoforms decoding for the same amino acid were combined with the distinction of initiator and elongator methionine tRNAs. The ratio of counts derived from FOI::40S libraries versus counts from unselected 40S libraries were plotted in Figures 3J, 3K, and S6D.
Metagene start and stop codon aligned analysis
Only start or stop codons were used which are the sole annotated start or stop codon in all annotated transcripts tagged ‘protein_coding’ of a gene in the Ensembl annotations GRCh38.80 and R64-1-1.80 for human and yeast, respectively, resulting in 6,639 start sites and 6,514 stop sites for yeast. For human, further, only start sites were considered whose corresponding genes have a minimal annotated 5′ UTR and CDS length of 80 nt and 30 nt, respectively, which corresponds to the depicted 5′ UTR and CDS range. Likewise, only stop sites were considered whose corresponding genes have a minimal annotated CDS and 3′ UTR length of 80 nt and 30 nt, respectively. To increase the number of used start codons, in case a gene has more than one start site annotated, the mainly used start site was included if it could be reliably appointed from the unselected 40S libraries (A start codon got appointed the mainly used one if its FP coverage comprised 75% of the sum of the coverage of all annotated start codons of a gene or it comprised 60% of all but was at least 3 times higher than the second highest. The FP coverage in the area of −3 to +5 nucleotides from the A of the start codon was used (with A = 0). This resulted in altogether 10,778 start sites and 16,803 stop sites for human.
Heatmaps
The color represents the read count at a certain position relative to the start or stop codon and is plotted in log10 scale. The labels at the color bar are given in linear scale.
Assigning reads to a transcript feature
To reliably determine the feature of a transcript only genes were considered with a sole start and stop codon annotated for all transcripts tagged ‘protein_coding’ of that gene in the Ensembl annotations GRCh38.80 and R64-1-1.80 for human and yeast, respectively, yielding 9,241 genes for human and 5,818 genes for yeast (whereby for yeast also genes tagged ‘dubious open reading frame’ and ‘unlikely to encode a functional protein’ in the SGD gene description (August 2018) were excluded). In general, 10 nt were added upstream and downstream of the 5′ UTR and 3′ UTR, respectively, owing to our observation that FPs often extend some nucleotides beyond the annotated transcript ends. If no UTRs were annotated for a gene, 50 nt for yeast and 100 nt for human upstream of the start codon and/or downstream of the stop codon were used as 5′ and 3′ UTRs, respectively. Assigning FPs to transcript areas: 5′ UTR: The FP 3′ end should map upstream of +5 nt of the A of the start codon and the FP 5′ end should map on or downstream the transcript start. Start codon: The 5′ end should map on or upstream of −3 nt of the A of the start codon and the 3′ end should map on or downstream of +5. CDS: The 5′ end should map downstream of −3 nt of the A of the start codon and the 3′ end should map upstream of −6 of the first nucleotide of the stop codon. Stop codon: The 5′ end should map on or upstream of −6 nt of the first nucleotide of the stop codon and the 3′ end should map on or downstream of +2. 3′ UTR: The FP 5′ end should map downstream of +2 of the first nucleotide of the stop codon. Always, both ends should map to an exon. For the violin plots in Figures 3G–3I, 3L, S6B, S6C, S5E, and S5F, the log2(ratio) was plotted with default settings of the function violinplot() from the Python library seaborn, Python Software Foundation. Python Language Reference, version 2.7. Available at https://www.python.org/). For violin plots in Figures 3G–3I, S6B, and S6C, only mRNAs were considered with a minimal coverage of 100 reads and a pseudo count was added for every transcript area. For violin plots in Figures 3I, S6E, and S6F, only mRNAs were considered with a minimal read count of 10 in both plotted transcript areas, the 5′ UTR and the start. The number of mRNAs which made the cut offs is given in the individual figure legends.
Footprint 3′ end distributions in relation to the start codon
Start sites were considered if they are the sole annotated start site of all transcripts tagged ‘protein_coding’ of a gene and if this gene has a minimal annotated 5′ UTR of 50 nt; genes tagged ‘dubious’ were not used (11,174 start sites for human and 5,994 start sites for yeast). FPs whose 5′ end map within −14 to −12 of a start site (with A = 0) were included for Figures 2D, 2E, and S8E–S8G. FPs assigned to a start codon (see Assigning reads to a transcript feature) were considered for Figure 4A. 1,000 iterations of random resampling were performed (see Quantification and Statistical Analysis for details).
Count per gene for replicate correlation plots
Counting was performed with htseq-count (mode ‘union’, feature type ‘exon’ and id attribute ‘gene_id’). For human, the Ensembl annotation GRCh.38.80 was used. For yeast, Ensemble annotation R64-1-1.80 was used including further the 5′ and 3′ UTR annotation as well as the 5′ UTR intron annotation from the SGD database in November 2017 (whereby the longest annotated UTRs per gene were added to the transcriptome). For genes without annotated 5′ or 3′ UTRs, 50nt of the adjacent genomic sequence were added as generic UTRs. Generally, only genes tagged ‘protein_coding’ were included. A pseudo count of 1 was added for every gene and log10(count) was plotted.
Count per gene’s 5′ UTR + start for correlation plots of selected versus unselected
Reads were assigned to a transcript area as described in Assigning reads to a transcript feature. The log10(count) of the sum of reads assigned to the 5′ UTR and the start codon per gene was plotted. Before the log transformation a pseudo count of 1 was added.
Confirming transcript starts for metagene plots with cap alignment
Our own mRNA datasets were used. Transcripts with a minimal total count of 100 FPs should have an average coverage of maximal 3 in the region from −30 to −10 of the annotated transcript start and an average coverage of minimal 20 in the region from +10 to +30 of the annotated transcript start to be considered a confirmed transcript start. Further, the annotated 5′ UTR should have a minimal length of 80 nt to avoid interference of the start codon associated FPs with long 5′ extensions. This yielded 198 and 1,046 transcript starts for yeast and human, respectively.
Transcriptome-wide screen for co-translational assembly in the 80S Sel-TCP-seq data
The average coverage of two intervals within the CDS of a gene was compared. Interval 1 was located in the beginning of the CDS from +30 up to +90 nt (with A of the start codon set = 0). Interval 2 was located in the end of the CDS from −90 up to −30 nt (with the first nucleotide of the stop codon set = 0). A gene was identified as candidate for co-translational assembly, if the average coverage of interval 2 was at least 5 times higher than that of interval 1. Nine candidates were found. Five of those were dropped because further investigation revealed a FLAG like epitope in the protein sequence of the gene in the right distance to the onset of the increase in coverage (RPA34, RTF1, RNH203, CDC16, LEU1). Coverage tracks of unselected and FOI bound 80S FPs for the other four candidates were analyzed further and are shown in Figures 7 and S7D.
Calculation of the fold induction to estimate the co-translational binding
The average footprint coverage after the onset of induction to the end of the CDS was divided by the average coverage before the onset starting from the beginning of the CDS. The first and last 30 nt of the CDS were not regarded as well as 30 nt ± the onset position. The fold induction seen in the FOI::80S Sel-TCP-seq data was normalized by the fold induction seen in the unselected 80S TCP-seq data. The normalization was done to account for features in the mRNA which might slow down or stall translating ribosomes, leading to a coverage increase not attributable to co-translational binding.
Quantification and Statistical Analysis
Statistical details of experiments can be found in the figure legends and figures. One and Two sample t tests were performed using the R function t test. A p value larger than 0.05 is referred to as not significant (n.s.). Random gene-wise resampling was performed for data shown in Figures 2D and 2E to estimate the influence of individual genes on the density profiles. This was done by randomly re-selecting genes, allowing duplications, n times (where n is the original sample size i.e., the number of genes used, as indicated in each figure legend). The mean and 95th percentiles of such iterations are shown in the figures. A custom Python script was used.
Acknowledgments
We are grateful to Nickolas Ingolia for sequencing and analysis of our first Sel-TCP-seq libraries as well as mentoring mainly at the outset of this study. This work was supported by a Grant of Excellence in Basic Research (EXPRO 2019) provided by the Czech Science Foundation (19-25821X to L.S.V.), a Wellcome Trust grant (090812/B/09/Z to L.S.V.), an Australian Research Council Discovery Project grant (DP180100111 to T.P. and N.E.S), National Health and Medical Research Council research fellowships (APP116999 to R.D.H. and APP1135928 to T.P.), and a Cancer Council ACT Project grant (APP1120469 to T.P. and R.D.H.).
Author Contributions
S.W., T.P., and L.S.V. conceived and designed the project. S.W. carried out all experiments with collaborative contributions from A.H., V.H., S.G., N.D.S., and N.E.S. S.W. performed the data analysis. S.W., T.P., and L.S.V. interpreted the results and wrote the paper with input from A.G.H., A.H., N.E.S., and R.D.H.
Declaration of Interests
The authors declare no competing interests.
Published: June 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.06.004.
Contributor Information
Susan Wagner, Email: susan.wagner@anu.edu.au.
Thomas Preiss, Email: thomas.preiss@anu.edu.au.
Leoš Shivaya Valášek, Email: valasekl@biomed.cas.cz.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S.K., Shirokikh N.E., Beilharz T.H., Preiss T. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 2016;535:570–574. doi: 10.1038/nature18647. [DOI] [PubMed] [Google Scholar]
- Asano K., Phan L., Anderson J., Hinnebusch A.G. Complex formation by all five homologues of mammalian translation initiation factor 3 subunits from yeast Saccharomyces cerevisiae. J. Biol. Chem. 1998;273:18573–18585. doi: 10.1074/jbc.273.29.18573. [DOI] [PubMed] [Google Scholar]
- Asano K., Phan L., Valásek L., Schoenfeld L.W., Shalev A., Clayton J., Nielsen K., Donahue T.F., Hinnebusch A.G. A multifactor complex of eIF1, eIF2, eIF3, eIF5, and tRNA(i)Met promotes initiation complex assembly and couples GTP hydrolysis to AUG recognition. Cold Spring Harb. Symp. Quant. Biol. 2001;66:403–415. doi: 10.1101/sqb.2001.66.403. [DOI] [PubMed] [Google Scholar]
- Asano K., Shalev A., Phan L., Nielsen K., Clayton J., Valásek L., Donahue T.F., Hinnebusch A.G. Multiple roles for the C-terminal domain of eIF5 in translation initiation complex assembly and GTPase activation. EMBO J. 2001;20:2326–2337. doi: 10.1093/emboj/20.9.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznosková P., Cuchalová L., Wagner S., Shoemaker C.J., Gunišová S., von der Haar T., Valášek L.S. Translation initiation factors eIF3 and HCR1 control translation termination and stop codon read-through in yeast cells. PLoS Genet. 2013;9:e1003962. doi: 10.1371/journal.pgen.1003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznosková P., Wagner S., Jansen M.E., von der Haar T., Valášek L.S. Translation initiation factor eIF3 promotes programmed stop codon readthrough. Nucleic Acids Res. 2015;43:5099–5111. doi: 10.1093/nar/gkv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan S., Gartmann M., Halic M., Armache J.P., Jarasch A., Mielke T., Berninghausen O., Wilson D.N., Beckmann R. α-Helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 2010;17:313–317. doi: 10.1038/nsmb.1756. [DOI] [PubMed] [Google Scholar]
- Bohlen J., Fenzi K., Kramenr G., Bukau B., Teleman A.A. Selective 40S Footprinting Reveals Cap-Tethered Ribosome Scanning in Human Cells. Mol. Cell. 2020;79:561–574. doi: 10.1016/j.molcel.2020.06.005. [DOI] [PubMed] [Google Scholar]
- Cate J.H. Human eIF3: from ‘blobology’ to biological insight. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372:20160176. doi: 10.1098/rstb.2016.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigan A.M., Foiani M., Hannig E.M., Hinnebusch A.G. Complex formation by positive and negative translational regulators of GCN4. Mol. Cell. Biol. 1991;11:3217–3228. doi: 10.1128/mcb.11.6.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis M.D., Person M.D., Browning K.S. Phosphorylation of plant translation initiation factors by CK2 enhances the in vitro interaction of multifactor complex components. J. Biol. Chem. 2009;284:20615–20628. doi: 10.1074/jbc.M109.007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Duarte A., Lacerda R., Menezes J., Romão L. eIF3: a factor for human health and disease. RNA Biol. 2018;15:26–34. doi: 10.1080/15476286.2017.1391437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant C.M., Miller P.F., Hinnebusch A.G. Requirements for intercistronic distance and level of eukaryotic initiation factor 2 activity in reinitiation on GCN4 mRNA vary with the downstream cistron. Mol. Cell. Biol. 1994;14:2616–2628. doi: 10.1128/mcb.14.4.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunišová S., Valášek L.S. Fail-safe mechanism of GCN4 translational control--uORF2 promotes reinitiation by analogous mechanism to uORF1 and thus secures its key role in GCN4 expression. Nucleic Acids Res. 2014;42:5880–5893. doi: 10.1093/nar/gku204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunišová S., Beznosková P., Mohammad M.P., Vlčková V., Valášek L.S. In-depth analysis of cis-determinants that either promote or inhibit reinitiation on GCN4 mRNA after translation of its four short uORFs. RNA. 2016;22:542–558. doi: 10.1261/rna.055046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunišová S., Hronová V., Mohammad M.P., Hinnebusch A.G., Valášek L.S. Please do not recycle! Translation reinitiation in microbes and higher eukaryotes. FEMS Microbiol. Rev. 2018;42:165–192. doi: 10.1093/femsre/fux059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh N.R., Green R. Dom34 rescues ribosomes in 3′ untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen C.U.T. Translation Termination and Ribosome Recycling in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2018;10:a032656. doi: 10.1101/cshperspect.a032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch A.G. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014;83:779–812. doi: 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G. Structural Insights into the Mechanism of Scanning and Start Codon Recognition in Eukaryotic Translation Initiation. Trends Biochem. Sci. 2017;42:589–611. doi: 10.1016/j.tibs.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Hronová V., Mohammad M.P., Wagner S., Pánek J., Gunišová S., Zeman J., Poncová K., Valášek L.S. Does eIF3 promote reinitiation after translation of short upstream ORFs also in mammalian cells? RNA Biol. 2017;14:1660–1667. doi: 10.1080/15476286.2017.1353863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- Ingolia N.T. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Hussmann J.A., Weissman J.S. Ribosome Profiling: Global Views of Translation. Cold Spring Harb. Perspect. Biol. 2019;11:a032698. doi: 10.1101/cshperspect.a032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau L.F., Hite D.H., Hogan G.J., Brown P.O. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3:e01257. doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Li F., Huang L., Duan H., Fang J., Sun L., Xin X., Tian G., Cheng Y., Yang X., Wolf D.A. eIF3 associates with 80S ribosomes to promote translation elongation, mitochondrial homeostasis, and muscle health. Mol. Cell. 2020;79:575–587. doi: 10.1016/j.molcel.2020.06.003. [DOI] [PubMed] [Google Scholar]
- Llácer J.L., Hussain T., Marler L., Aitken C.E., Thakur A., Lorsch J.R., Hinnebusch A.G., Ramakrishnan V. Conformational Differences between Open and Closed States of the Eukaryotic Translation Initiation Complex. Mol. Cell. 2015;59:399–412. doi: 10.1016/j.molcel.2015.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llácer J.L., Hussain T.1., Gordiyenko Y., Lorsch J.R., Hinnebusch A.G., Ramakrishnan V. The beta propellers of eIF3b and eIF3i relocate together to the ribosomal intersubunit interface during translation initiation. bioRxiv. 2018 doi: 10.1101/453688. [DOI] [Google Scholar]
- Lu P.D., Harding H.P., Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy N.J., Ingolia N.T. Transcriptome-wide measurement of translation by ribosome profiling. Methods. 2017;126:112–129. doi: 10.1016/j.ymeth.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad M.P., Munzarová Pondelícková V., Zeman J., Gunišová S., Valášek L.S. In vivo evidence that eIF3 stays bound to ribosomes elongating and terminating on short upstream ORFs to promote reinitiation. Nucleic Acids Res. 2017;45:2658–2674. doi: 10.1093/nar/gkx049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzarová V., Pánek J., Gunišová S., Dányi I., Szamecz B., Valášek L.S. Translation reinitiation relies on the interaction between eIF3a/TIF32 and progressively folded cis-acting mRNA elements preceding short uORFs. PLoS Genet. 2011;7:e1002137. doi: 10.1371/journal.pgen.1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen K.H., Valásek L., Sykes C., Jivotovskaya A., Hinnebusch A.G. Interaction of the RNP1 motif in PRT1 with HCR1 promotes 40S binding of eukaryotic initiation factor 3 in yeast. Mol. Cell. Biol. 2006;26:2984–2998. doi: 10.1128/MCB.26.8.2984-2998.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.S., Himmelbach A., Browning K.S., Hohn T., Ryabova L.A. A plant viral “reinitiation” factor interacts with the host translational machinery. Cell. 2001;106:723–733. doi: 10.1016/s0092-8674(01)00487-1. [DOI] [PubMed] [Google Scholar]
- Phan L., Zhang X., Asano K., Anderson J., Vornlocher H.P., Greenberg J.R., Qin J., Hinnebusch A.G. Identification of a translation initiation factor 3 (eIF3) core complex, conserved in yeast and mammals, that interacts with eIF5. Mol. Cell. Biol. 1998;18:4935–4946. doi: 10.1128/mcb.18.8.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev A.V., Hellen C.U.T., Pestova T.V. Recycling of eukaryotic posttermination ribosomal complexes. Cell. 2007;131:286–299. doi: 10.1016/j.cell.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzinga M., Bates C., Lui J., Forte G., Morales-Polanco F., Linney E., Knotkova B., Wilson B., Solari C.A., Berchowitz L.E. Translation factor mRNA granules direct protein synthetic capacity to regions of polarized growth. J. Cell Biol. 2019;218:1564–1581. doi: 10.1083/jcb.201704019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2018. R: A language and environment for statistical computing. [Google Scholar]
- Robichaud N., Sonenberg N. Translational control and the cancer cell response to stress. Curr. Opin. Cell Biol. 2017;45:102–109. doi: 10.1016/j.ceb.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Shiber A., Döring K., Friedrich U., Klann K., Merker D., Zedan M., Tippmann F., Kramer G., Bukau B. Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature. 2018;561:268–272. doi: 10.1038/s41586-018-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh Y.W., Minguez P., Bork P., Auburger J.J., Guilbride D.L., Kramer G., Bukau B. Operon structure and cotranslational subunit association direct protein assembly in bacteria. Science. 2015;350:678–680. doi: 10.1126/science.aac8171. [DOI] [PubMed] [Google Scholar]
- Shirokikh N.E., Preiss T. Translation initiation by cap-dependent ribosome recruitment: Recent insights and open questions. Wiley Interdiscip. Rev. RNA. 2018;9:e1473. doi: 10.1002/wrna.1473. [DOI] [PubMed] [Google Scholar]
- Shirokikh N.E., Archer S.K., Beilharz T.H., Powell D., Preiss T. Translation complex profile sequencing to study the in vivo dynamics of mRNA-ribosome interactions during translation initiation, elongation and termination. Nat. Protoc. 2017;12:697–731. doi: 10.1038/nprot.2016.189. [DOI] [PubMed] [Google Scholar]
- Shirokikh N.E., Dutikova Y.S., Staroverova M.A., Hannan R.D., Preiss T. Migration of Small Ribosomal Subunits on the 5′ Untranslated Regions of Capped Messenger RNA. Int. J. Mol. Sci. 2019;20:E4464. doi: 10.3390/ijms20184464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti A., Brito Querido J., Myasnikov A.G., Mancera-Martinez E., Renaud A., Kuhn L., Hashem Y. eIF3 Peripheral Subunits Rearrangement after mRNA Binding and Start-Codon Recognition. Mol. Cell. 2016;63:206–217. doi: 10.1016/j.molcel.2016.05.033. [DOI] [PubMed] [Google Scholar]
- Skabkin M.A., Skabkina O.V., Hellen C.U., Pestova T.V. Reinitiation and other unconventional posttermination events during eukaryotic translation. Mol. Cell. 2013;51:249–264. doi: 10.1016/j.molcel.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.D., Arake-Tacca L., Nitido A., Montabana E., Park A., Cate J.H. Assembly of eIF3 Mediated by Mutually Dependent Subunit Insertion. Structure. 2016;24:886–896. doi: 10.1016/j.str.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokabe M., Fraser C.S., Hershey J.W.B. The human translation initiation multi-factor complex promotes methionyl-tRNAi binding to the 40S ribosomal subunit. Nucleic Acids Res. 2012;40:905–913. doi: 10.1093/nar/gkr772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szamecz B., Rutkai E., Cuchalová L., Munzarová V., Herrmannová A., Nielsen K.H., Burela L., Hinnebusch A.G., Valásek L. eIF3a cooperates with sequences 5′ of uORF1 to promote resumption of scanning by post-termination ribosomes for reinitiation on GCN4 mRNA. Genes Dev. 2008;22:2414–2425. doi: 10.1101/gad.480508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valásek L.S. ‘Ribozoomin’--translation initiation from the perspective of the ribosome-bound eukaryotic initiation factors (eIFs) Curr. Protein Pept. Sci. 2012;13:305–330. doi: 10.2174/138920312801619385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valásek L., Nielsen K.H., Hinnebusch A.G. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 2002;21:5886–5898. doi: 10.1093/emboj/cdf563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valásek L., Nielsen K.H., Zhang F., Fekete C.A., Hinnebusch A.G. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 2004;24:9437–9455. doi: 10.1128/MCB.24.21.9437-9455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valásek L., Szamecz B., Hinnebusch A.G., Nielsen K.H. In vivo stabilization of preinitiation complexes by formaldehyde cross-linking. Methods Enzymol. 2007;429:163–183. doi: 10.1016/S0076-6879(07)29008-1. [DOI] [PubMed] [Google Scholar]
- Valášek L.S., Zeman J., Wagner S., Beznosková P., Pavlíková Z., Mohammad M.P., Hronová V., Herrmannová A., Hashem Y., Gunišová S. Embraced by eIF3: structural and functional insights into the roles of eIF3 across the translation cycle. Nucleic Acids Res. 2017;45:10948–10968. doi: 10.1093/nar/gkx805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum G. 1995. Python Tutorial, CWI Report CS-R9526.https://gvanrossum.github.io/Publications.html [Google Scholar]
- Vattem K.M., Wek R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S., Herrmannová A., Malík R., Peclinovská L., Valášek L.S. Functional and biochemical characterization of human eukaryotic translation initiation factor 3 in living cells. Mol. Cell. Biol. 2014;34:3041–3052. doi: 10.1128/MCB.00663-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S., Herrmannová A., Šikrová D., Valášek L.S. Human eIF3b and eIF3a serve as the nucleation core for the assembly of eIF3 into two interconnected modules: the yeast-like core and the octamer. Nucleic Acids Res. 2016;44:10772–10788. doi: 10.1093/nar/gkw972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.C., Zinshteyn B., Wehner K.A., Green R. High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell. 2019;73:959–970.e5. doi: 10.1016/j.molcel.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D.J., Makeeva D.S., Zhang F., Anisimova A.S., Stolboushkina E.A., Ghobakhlou F., Shatsky I.N., Dmitriev S.E., Hinnebusch A.G., Guydosh N.R. Tma64/eIF2D, Tma20/MCT-1, and Tma22/DENR Recycle Post-termination 40S Subunits In Vivo. Mol. Cell. 2018;71:761–774.e5. doi: 10.1016/j.molcel.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman J., Itoh Y., Kukačka Z., Rosůlek M., Kavan D., Kouba T., Jansen M.E., Mohammad M.P., Novák P., Valášek L.S. Binding of eIF3 in complex with eIF5 and eIF1 to the 40S ribosomal subunit is accompanied by dramatic structural changes. Nucleic Acids Res. 2019;47:8282–8300. doi: 10.1093/nar/gkz570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Hinnebusch A.G. An upstream ORF with non-AUG start codon is translated in vivo but dispensable for translational control of GCN4 mRNA. Nucleic Acids Res. 2011;39:3128–3140. doi: 10.1093/nar/gkq1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study were deposited in the GEO database with the following accession number: GSE139132. All custom Python scripts used for the analyses in this paper are available upon request.